

Abstract
Introduction
Synthetic cannabinoids are an emerging illicit drug class. The variety of available substances is large and ever-changing, making it difficult for laboratories to remain current. We present a qualitative LC–MS/MS method identifying urinary metabolites of JWH-018, JWH-073, JWH-081, JWH-122, JWH-200, JWH-210, JWH-250, RCS-4, and AM2201 and the parent compounds JWH-018, JWH-073, JWH-081, JWH-122, JWH-210, JWH-250, RCS-4, AM2201, and MAM2201.
Methods
After enzymatic hydrolysis, urinary proteins were precipitated with acetonitrile. Chromatography utilized a 10 min gradient on a Kinetex XB-C18 column with 0.1% formic acid in water and acetonitrile. Scheduled multiple reaction monitoring “survey scans” were followed by information-dependent acquisition-enhanced product ion scan experiments on an ABSciex 5500 QTRAP mass spectrometer. Analytes were identified by software-assisted library searching against reference spectra.
Results
The method was fully validated, including proof of selectivity (no exogenous or endogenous interferences were observed), assessment of matrix effects (95–122%) and recovery (53–95%), determination of limits of detection (0.5–10 ng/mL), carry-over studies (thresholds between 100 and 1000 ng/mL), and determination of autosampler stability (samples were stable for at least 3 days). Hydrolysis efficiency was thoroughly investigated for a wide range of glucuronides and for the reference standard, JWH-018 5-hydroxypentyl glucuronide
Synthetic cannabinoids were originally developed as pharmacological tools for investigating the endocannabinoid system, as they interact with CB1 and CB2 cannabinoid receptors. In contrast to Δ9-tetrahydrocannabinol, which is the primary psychoactive compound in Cannabis and a partial agonist at both receptors, synthetic cannabinoids may be full agonists and selective for one receptor subtype. Synthetic cannabinoids are now major illicit designer drugs that are deceptively marketed as herbal incenses, room deodorizers, or air fresheners. Different structural subclasses are available: naphthoylindoles (JWH-015, JWH-018, JWH-019, JWH-073, JWH-081, JWH-122, JWH-200, JWH-210, and JWH-398), phenylacetylindoles (JWH-203, JWH-250, JWH-251, and RCS-8), benzoylindoles (RCS-4), and cyclohexylphenols (CP 47,497 C7 and C8 analogs). In Europe, almost all countries scheduled one or more compounds or structural classes, ranging from none in The Netherlands to more than 30 in Italy, Luxembourg,1and Switzerland.2 Since July 2012, in the United States not only are JWH-018, JWH-073, JWH-200, and the C7 and C8 homologues of CP 47,497 classified as Schedule I controlled substances but also JWH-019, JWH-081, JWH-122, JWH-203, JWH-210, JWH-250, JWH-398, AM694, AM2201, MAM2201, RCS-4, and RCS-8, as well as their analogs.3
In response to scheduling efforts, new analogs are introduced, requiring constant surveillance and inclusion of newly emerging substances into analytical assays. Identification and quantification are limited by the commercial availability of reference standards for parent synthetic cannabinoids and, even more relevant for identification and monitoring, their human urinary metabolites. Synthetic cannabinoids are extensively metabolized, and rarely are parent compounds identified in urine; exceptions were reported for AM2201 and UR-144.4 Metabolites of JWH-018 and JWH-073, among the first synthetic cannabinoids on the market, were identified from several in vitro and in vivo studies.5–10 Further studies with urine samples of intoxicated patients were performed for JWH-250,11 RCS-4,12 AM2201, and UR-144.4 Grigoryev et al. carried out metabolism studies for AB-00113 and AM69414, with urine samples obtained after self-experiments. The most comprehensive report identified metabolites of seven synthetic cannabinoids: JWH-018, JWH-073, JWH-081, JWH-122, JWH-210, JWH-250, and RCS-4.7 The primary metabolism route in humans is hydroxylation of the alkyl side chain and the indole ring and hydroxylation of the adamantyl structure or aromatic naphthyl or phenyl systems and side chain carboxylation. For halogenated compounds like AM2201 and AM694, enzymatic dehalogenation was observed.4,14 Currently, there are no reported RCS-4 metabolites exclusively showing side chain hydroxylation or carboxylation, despite the presence of these metabolites from JWH-018 and JWH-073.7,12 Hydroxyl and carboxyl metabolites can be excreted in urine as ether and ester glucuronides.6,8,15
There are several qualitative6,9,11 and quantitative15–18 liquid chromatography tandem mass spectrometry (LC–MS/MS) methods for synthetic cannabinoids in urine, primarily for JWH-018 and JWH-073. To the best of our knowledge, only two assays included more than two compounds: Hutter et al. performed a study of seven compounds7 and de Jager et al. quantified eleven urinary metabolites from eight synthetic cannabinoids.19 Without exception, all reported methods for synthetic cannabinoid metabolite analysis included a deconjugation step. Our goal was to establish a rapid qualitative confirmation with simple sample preparation, facilitating updates when a new reference standard becomes available. As practiced in forensic toxicology20–24 and doping control laboratories,25 as well as in the pharmaceutical industry,26 we chose the approach of a confirmatory LC–MS/MS assay with a library search, a methodology that is useful when identification is essential but precise quantification unnecessary. To date, there are no established cutoffs for synthetic cannabinoids, as little to nothing is known about dose–effect relationships or detection windows, and there are no established regulatory limits. Therefore, qualitative confirmation is the current standard procedure for military, public, and private testing. The procedure evolved over time to build and search LC–MS spectral libraries for rapidly identifying “unknown” compounds. Currently, the procedure of choice is a two-step scheme: First, multiple reaction monitoring (MRM) “survey scans” monitor a single transition for each analyte of interest. Second, an information-dependent acquisition (IDA) enhanced product ion scan (EPI) is collected, yielding a full spectrum of the selected compound. Full-scan EPI spectra showing the range of all fragments increased our confidence for correctly identifying these structurally closely related compounds. The scheduled MRM mode increases dwell time within a fixed cycle time and increases analyte sensitivity. In our assay, MS spectra were compared to an in-house developed library database using the recently launched LibraryView (ABSciex, Foster City, CA). Identification was based on the quality of the fit of the software's search algorithm, correct retention time, and visual spectral inspection.
This is the first synthetic cannabinoid LC–MS/MS assay with library searching monitoring 9 synthetic cannabinoids and 20 metabolites in urine. Instead of developing a quantitative MRM method, we chose an IDA full-scan approach because reliable identification of structurally related synthetic cannabinoids is critical, while also having the ability to quickly incorporate new analytes as reference standards become available. Revalidating a qualitative approach for a new compound can be accomplished more quickly. At the same time, we aimed for simple sample preparation and a short run time, prerequisites for high-throughput routine analysis.
Methods
Chemicals and Reagents
JWH-018, JWH-018 N-(5-hydroxypentyl), JWH-018 5-hydroxyindole, JWH-018 6-hydroxyindole, JWH-018 N-pentanoic acid, and JWH-018 N-(5-hydroxypentyl) glucuronide, JWH-073, JWH-073 N-(4-hydroxybutyl), JWH-073 5-hydroxyindole, JWH-073 6-hydroxyindole, and JWH-073 N-butanoic acid, JWH-081, and JWH-081 N-(5-hydroxypentyl), JWH-122 and JWH-122 N-(5-hydroxypentyl), JWH-200 5-hydroxyindole, and JWH-200 6-hydroxyindole, JWH-210, JWH-210-N-(4-hydroxypentyl), JWH-210 N-(5-hydroxypentyl), JWH-210 5-hydroxyindole, and JWH-210 N-pentanoic acid, JWH-250, JWH-250 N-(4-hydroxypentyl), JWH-250 N-(5-hydroxypentyl), JWH-250 5-hydroxyindole, and JWH-250 N-pentanoic acid, AM2201, AM2201 N-(4-hydroxypentyl), and AM2201 5-hydroxyindole, MAM2201, RCS-4, RCS-4 N-(5-hydroxypentyl), and RCS-4 N-pentanoic acid as well as the internal standards, D5-JWH-200 and D9-JWH-081, were purchased from Cayman Chemicals (Ann Arbor, MI). The structures of all parent compounds are included in Figure 1. Formic acid for LC–MS and methanol HPLC grade were obtained from Fisher Scientific (Fair Lawn, NJ). HPLC and LC–MS grade acetonitrile and ammonium acetate were acquired from Sigma Aldrich (St. Louis, MO). Abalone beta-glucuronidase powder from Campbell Science containing 1500000 units/gram beta-glucuronidase and 150000 units/g sulfatase was diluted with distilled water to contain 100000 units/mL beta-glucuronidase and 10000 units/mL sulfatase activity for enzymatic hydrolysis (Campbell Science, Rockton, IL). Water was purified in-house with an ELGA Purelab Ultra Analytic purifier (Siemens Water Technologies, Lowell, MA).
Figure 1.

Structures of synthetic cannabinoid parent compounds included in the method.
Instrumentation
Chromatography was performed on a Shimadzu Prominence high-performance liquid chromatography system (Shimadzu Corp, Columbia, MD) consisting of two LC-20AD XR pumps, a DGU-20A3 degasser, a SIL-20AC XR autosampler, and a CTO-20 AC column oven. As a detector, an ABSciex 5500 QTRAP triple quadrupole/linear ion trap mass spectrometer with a TurboIonSpray source was employed. ABSciex Analyst, version 1.5.1, and LibraryView, version 1.0, were used for data acquisition and analysis, respectively. Samples were centrifuged in an Eppendorf 5804 R centrifuge (Eppendorf, Hamburg, Germany).
Blank Urine, Analyte Working Solutions, and Internal Standard Solution
Blank urine from ten volunteers was verified to be negative for analytes prior to use. Primary analyte stock solutions were prepared in methanol. A 2000 ng/mL solution containing synthetic cannabinoids and their metabolites was prepared and diluted to obtain 1.25, 2.5, 5.0, 10.0, 25, 50, and 100 ng/mL working solutions. The internal standard solution contained D5-JWH-200 and D9-JWH-081 in methanol at concentrations of 800 and 100 ng/mL, respectively. All primary and working solutions were stored at −20 °C.
Sample Preparation of Controls and Authentic Specimens
For quality control samples, 100 μL of urine was fortified with 10 μL of internal standard solution and 10 μL of the corresponding methanolic working solution. After adding 50 μL of 0.4 M ammonium acetate buffer (pH 4.0) for pH adjustment and 2000 units beta-glucuronidase (20 μL solution), samples were vortexed and incubated for 2 h at 55 °C. Samples were cooled at room temperature, precipitated with 190 μL acetonitrile, vortexed, and centrifuged for 10 min at 15000g and 4 °C. Supernatant (150 μL) was transferred to glass inserts in autosampler vials. In order to verify limits of detection, controls at 0.125, 0.25, 0.5, 1.0, 2.5, 5.0, and 10.0 ng/mL were prepared. Authentic specimens were prepared as described, with the only difference being that analyte working solutions were replaced with 10 μL methanol.
Assessment of Hydrolysis Efficiency
In each run, we analyzed two additional controls to assess hydrolysis efficiency. The first contained a high concentration of JWH-018 N-(5-hydroxypentyl) glucuronide that is available as a reference standard. Blank urine was fortified to a final concentration of 2.5 μg/mL glucuronide (“hydrolysis control I”).
The second control was a pool created from 10 fresh authentic urine samples prescreened positive and containing a variety of ether- and ester-linked glucuronides. This pool was prepared as an authentic specimen (“hydrolysis control II”). Hydrolysis control I and II samples underwent hydrolysis under the above-mentioned conditions in each batch and were analyzed in a scheduled MRM mode to monitor peak areas. After optimizing hydrolysis during method development, we required peak area ratios of the remaining glucuronide to aglycone to be <2.5% in the hydrolysis control I and peak areas of all aglycones >80% of the initially established baseline in the hydrolysis control II. The peak area ratios of hydrolyzed to nonhydrolyzed samples were calculated. A percentage higher than 100% indicated that a conjugated metabolite was successfully hydrolyzed. The baseline serving as 100% was the average peak area of each metabolite in the nonhydrolyzed samples (n = 4).
Chromatographic Conditions
Chromatographic separation was achieved with a Kinetex XB-C18 column (50 mm × 3.0 mm, 2.6 μm) fitted with a KrudKatcher Ultra HPLC in-line filter (0.5 μm × 0.1 mm ID, Phenomenex, Torrance, CA). Autosampler temperature was 4 °C and column oven temperature 40 °C. Injection volume was 5 μL for all samples except for the first hydrolysis control that only required 1 μL to avoid detector saturation. Gradient elution was performed with 0.1% formic acid in water (mobile phase A) and 0.1% formic acid in acetonitrile (mobile phase B) at an initial flow rate of 0.5 mL/min. Gradient conditions were 10% B for 0.5 min to 90% B over 5.50 min, held for 0.9 min, increased to 98% at 7.0 min, and held for 2 min before column re-equilibration at 10% for 1.7 min. At 6.5 min, flow rate was increased from 0.5 to 1.0 mL/min, held for 3.9 min, and returned to the initial rate of 0.5 mL/min at 10.6 min. The total run time was 10.7 min. Before and after aspiration, the autosampler was rinsed with 1600 μL of acetonitrile/isopropanol/water (45:45:10, v/v/v), containing 0.1% formic acid.
Mass Spectrometry
Positive electrospray ionization (ESI) was employed. MRM parameters were optimized via direct infusion of analytes (Table 1). Source settings were as follows: source temperature, 500 °C; ionspray voltage, 5500 V; ion source gas-1, 60 psi and gas-2 50 psi; and curtain gas, 50 psi. Scheduled MRM transitions with a detection window of 20 s were monitored for each analyte (Table 1). Up to three of the most intense peaks were evaluated by IDA scan experiments. If an MRM signal exceeded 1000 cps, EPI scans were triggered. These scans had a range from 80 to 600 amu, a scan rate of 20000 Da/s, and a 30 eV collision energy with ±15 eV spread. The declustering potential was 100 V and entrance potential was 10 V. Previously identified target MRMs were not excluded in subsequent MRM scan cycles to acquire multiple spectra across each peak. The trap was filled dynamically with a maximum fill time of 250 ms.
Table 1. Optimized MS Parameters for the Monitored Transitions in the Survey Scan, Retention Times, Limits of Detection, Matrix Effects, Recovery, and Carryover Threshold for All Analytes Listed in Order of Retention Time (RT = Retention Time, DP = Declustering Potential, EP = Entrance Potential, CE = Collision Energy, CXP = Collision Cell Exit Potential, and LOD = Limit of Detection).
| analyte | RT (min) | Q1 mass m/z | Q3 mass m/z | DP (V) | EP (V) | CE (V) | CXP (V) | LOD (ng/mL) | matrix effect (%) | recovery (%) | carryover threshold (ng/mL) |
|---|---|---|---|---|---|---|---|---|---|---|---|
| number of different urine samples in validation experiments | |||||||||||
|
| |||||||||||
| 17 | 10 | 10 | 4 | ||||||||
| JWH-200 5-hydroxyindole | 2.80 | 401.1 | 155.0 | 31 | 10 | 29 | 14 | 5 | 14 | 91 | 400 |
| JWH-200 6-hydroxyindole | 3.00 | 401.1 | 155.0 | 120 | 10 | 29 | 12 | 2.5 | 4 | 94 | 400 |
| RCS-4 pentanoic acid | 4.30 | 352.1 | 135.0 | 51 | 10 | 31 | 14 | 10 | 19 | 94 | 400 |
| RCS-4 5-hydroxypentyl | 4.40 | 338.2 | 135.0 | 141 | 10 | 27 | 12 | 5 | 13 | 92 | 400 |
| JWH-250 pentanoic acid | 4.50 | 366.1 | 121.1 | 56 | 10 | 27 | 14 | 0.5 | 16 | 95 | 400 |
| JWH-250 4/5-hydroxypentyl | 4.60 | 352.1 | 121.0 | 50 | 10 | 27 | 12 | 1 | 22 | 94 | 400 |
| JWH-073 butanoic acid | 4.60 | 358.1 | 155.0 | 61 | 10 | 31 | 14 | 5 | 11 | 92 | 400 |
| JWH-073 4-hydroxybutyl | 4.70 | 344.1 | 155.0 | 36 | 10 | 29 | 12 | 2.5 | 15 | 87 | 400 |
| JWH-018 pentanoic acid | 4.80 | 372.2 | 155.0 | 106 | 10 | 31 | 14 | 2.5 | 7 | 92 | 400 |
| JWH-018 5-hydroxypentyl | 4.90 | 358.1 | 155.1 | 56 | 10 | 29 | 12 | 0.5 | 11 | 87 | 1000 |
| AM2201 4-hydroxypentyl | 4.80 | 376.1 | 155.0 | 46 | 10 | 33 | 14 | 2.5 | 9 | 90 | 400 |
| AM2201 6-hydroxyindole | 5.00 | 376.2 | 155.0 | 66 | 10 | 33 | 16 | 5 | 11 | 89 | 400 |
| JWH-081 5-hydroxypentyl | 5.00 | 388.2 | 185.1 | 56 | 10 | 29 | 16 | 2.5 | 12 | 90 | 400 |
| JWH-122 5-hydroxypentyl | 5.10 | 372.1 | 169.0 | 51 | 10 | 29 | 12 | 0.5 | 14 | 86 | 400 |
| JWH-073 5/6-hydroxyindole | 5.20 | 344.1 | 155.0 | 51 | 10 | 33 | 12 | 2.5 | 11 | 82 | 400 |
| JWH-250 5-hydroxyindole | 5.20 | 352.1 | 121.0 | 66 | 10 | 27 | 14 | 2.5 | 11 | 84 | 400 |
| JWH-210 pentanoic acid | 5.30 | 400.1 | 183.0 | 40 | 10 | 33 | 14 | 2.5 | 8 | 87 | 400 |
| JWH-210 4/5-hydroxypentyl | 5.40 | 386.2 | 183.1 | 30 | 10 | 31 | 16 | 1 | 7 | 85 | 400 |
| JWH-018 5/6-hydroxyindole | 5.50 | 358.1 | 155.1 | 91 | 10 | 33 | 16 | 1 | 7 | 80 | 400 |
| AM2201 | 5.80 | 360.1 | 155.1 | 106 | 10 | 33 | 10 | 1 | 9 | 69 | 100 |
| RCS-4 | 5.90 | 322.1 | 135.1 | 56 | 10 | 31 | 12 | 2.5 | 0 | 53 | 400 |
| JWH-210 5-hydroxyindole | 6.00 | 386.2 | 183.0 | 51 | 10 | 35 | 14 | 10 | 8 | 80 | 400 |
| MAM2201 | 6.00 | 374.1 | 169.0 | 30 | 10 | 35 | 14 | 2.5 | 2 | 68 | 200 |
| JWH-250 | 6.00 | 336.1 | 121.2 | 50 | 10 | 27 | 12 | 5 | −2 | 56 | 400 |
| JWH-073 | 6.10 | 328.0 | 155.2 | 51 | 10 | 31 | 12 | 5 | 2 | 54 | 200 |
| JWH-018 | 6.40 | 342.1 | 155.2 | 46 | 10 | 33 | 12 | 1 | 3 | 54 | 200 |
| JWH-081 | 6.50 | 372.1 | 185.1 | 181 | 10 | 33 | 16 | 10 | −5 | 66 | 400 |
| JWH-122 | 6.60 | 356.1 | 169.0 | 51 | 10 | 33 | 16 | 2.5 | 10 | 55 | 200 |
| JWH-210 | 6.70 | 370.1 | 183.1 | 51 | 10 | 33 | 18 | 10 | 21 | 56 | 400 |
| glucuronide standard | |||||||||||
| JWH-018 5-hydroxypentyl glucuronide | 4.10 | 534.2 | 358.1 | 36 | 10 | 27 | 24 | ||||
| internal standards | |||||||||||
| d5-JWH-200 | 3.30 | 390.1 | 155.1 | 41 | 10 | 29 | 14 | – | 6 | 88 | – |
| d9-JWH-081 | 6.40 | 381.2 | 185.0 | 51 | 10 | 35 | 18 | – | −1 | 64 | – |
Data Analysis
Analyte peak areas were calculated with Analyst, while library searching and analyte identification were performed with the LibraryView. Precursor mass range 0.4 amu and mass tolerance range 0.4 amu were used for analyte LibraryView spectral searching. The intensity factor determining impact of spectral intensity differences between the acquired and the reference spectrum on the purity percentage was adjusted to 5. The intensity threshold utilized to remove small peaks under a specific intensity was set to 5.
Identification criteria were as follows: a library match with a purity score higher than 60%, relative retention time within ±0.05 min of the expected retention time, and presence of three characteristic masses, including the molecular ion. Statistical calculations were performed with Prism, version 5.02 (GraphPad Software, La Jolla, CA).
Optimization of Hydrolysis
Initial conditions were incubation at 55 °C for 60 min with 2000 units enzyme at pH 4.0. The incubation temperature was varied from 37, 55, and 70 °C, duration of incubation from 30, 60, and 120 min, amount of enzyme from 1000, 2000, and 4000 units, and pH from 3.0, 4.0, and 6.0. Optimal hydrolysis conditions were determined in a set of experiments with four replicates of a pool created from 10 authentic urine samples that had been prescreened positive for synthetic cannabinoids. For data acquisition, scheduled MRM experiments without EPI spectra acquisition were performed to obtain reliable peak areas for all metabolite aglycones, as well as JWH-018 hydroxypentyl glucuronide. One-way ANOVA (analysis of variance) was performed to test for differences between hydrolysis conditions on observed peak areas. Follow-up Bonferroni's multiple comparisons test compared specific conditions to determine optimal hydrolysis conditions.
Validation
Limits of Detection
Limits of detection (LODs) were determined in 17 different blank urine samples, each fortified to yield 0.125, 0.25, 0.5, 1.0, 2.5, 5.0, and 10.0 ng/mL samples for each analyte. The limit of detection was defined as the lowest concentration producing an acceptable identification by library search in at least 16 out of 17 samples. Since spectral quality may vary with analyte concentration, we also evaluated whether high concentrations produced positive spectral matches. Therefore, six urine samples from different sources were fortified to 200 ng/mL and analyzed.
Selectivity
Potential endogenous interferences were assessed by analyzing 10 urine specimens from different individuals. In addition, potential interferences from commonly used drugs were evaluated for the production of false positive or false negative results. Blank urine as well as LOD samples were fortified to contain 1000 ng/mL of potentially interfering drugs. In summary, 98 substances were tested (see the Supporting Information). No interference was noted if blank samples failed to meet analyte identification criteria and if all analytes in the LOD samples fulfilled analyte identification criteria.
Matrix Effects and Recovery
Matrix effects and recovery were determined in 3 sets of samples prepared in 10 different urine samples, according to the procedure described by Matuszewski et al.27 Analyte concentrations were chosen to produce peak intensities around 50000 cps between 0.5 and 2.5 ng/mL. Set 1 consisted of extracted urine samples fortified with analytes and internal standards after hydrolysis and precipitation. Set 2 urine samples were fortified with analytes and internal standards prior to hydrolysis and precipitation. Set 3 contained 6 replicates of neat standards in the mobile phase.
The matrix effects for each analyte were calculated by dividing mean peak areas of Set 1 by mean peak area of neat standards. The value was converted to a percentage subtracted from 100% to represent the amount of signal suppression or enhancement caused by coeluting matrix components. Recovery, representing analyte loss during hydrolysis and precipitation and expressed as a percentage, was calculated by dividing mean peak areas of set 2 by mean peak areas of set 1.
Carryover
Carryover was evaluated by injecting blank samples fortified with internal standard after high concentration samples. Carryover was tested for analyte concentrations of 100, 200, and 400 ng/mL. A higher limit of 1000 ng/mL was additionally evaluated for JWH-018 hydroxypentyl. The highest concentration for which no peak meeting our identification criteria was observed in the following negative sample was considered the carryover limit.
Stability
We investigated autosampler stability by injecting urine samples fortified at different LOD concentrations (0.5, 1.0, 2.5, 5.0, and 10.0 ng/mL) immediately after sample preparation and again after sitting in the 4 °C autosampler for 72 h. Analytes were considered stable if peak areas did not decrease by more than 20%.
Results and Discussion
Synthetic cannabinoids have recently become an analytical challenge for forensic, clinical, and workplace drug testing laboratories, but there are few methods identifying or quantifying these compounds in urine samples.6,7,9,11,15–19 The monitoring of synthetic cannabinoid metabolites, which predominate in urine, is confounded by the fact that few studies exist, identifying which metabolites are formed in humans. Furthermore, availability of metabolite reference standards typically lags behind parent compound availability. To the best of our knowledge, only one comprehensive and fully validated confirmation method for urinary metabolites is available. This quantitative assay developed by de Jager et al.19 includes eleven metabolites, which are extracted by liquid–liquid extraction and analyzed by triple quadrupole LC–MS/MS via MRM monitoring.
The present method is the first LC–MS/MS assay with spectral library searching for qualitative confirmation of 9 synthetic cannabinoids and 20 metabolites. After hydrolysis and protein precipitation, full EPI spectra are acquired for each analyte via IDA experiments. Full EPI spectra provide more structural information than traditional MRM methodology for reliably identifying close structurally related compounds, while fulfilling forensic guidelines. After attempting to incorporate scheduled C7 and C8 analogs of CP 47,497, they were not included in the method because they are not readily ionized and yield poorly reproducible spectra that confound spectral matching.
LC–MS/MS
The total run time of 10.7 min with analytes eluting between 2.78 and 6.69 min provides an efficient analysis time for forensic, clinical, and workplace testing programs. Despite multiple attempts, complete baseline separation could not be achieved for all isomeric pairs within a reasonable run time. The isomeric pairs of the JWH-200 hydroxyindoles and the two AM2201 hydroxy metabolites could be separated, while the two JWH-018 hydroxyindoles, JWH-073 hydroxyindoles, JWH-210 hydroxypentyls, and JWH-250 hydroxypentyls coeluted to a certain extent (Figure 2). This is not a major concern for military or workplace urine testing because the presence of either isomer indicates intake. Instrument run time is critical in these laboratories and is why we opted for a shorter run time in this method. The four pairs were considered as one analyte in the following validation experiments and were simply reported as hydroxy indole/pentyl metabolites without assignment of hydroxyl position. Concentrations of each individual isomer in all reference standard solutions were reduced by 50%.
Figure 2.
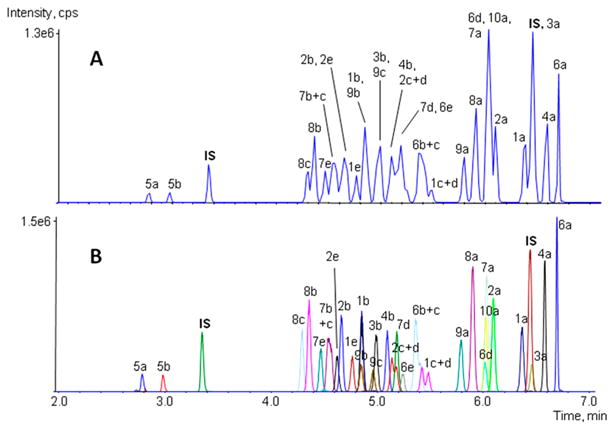
Total ion chromatograms of an LOD control at 5 ng/mL with (A) sMRM and EPI triggering and (B) unscheduled MRM. 1a, JWH-018; 1b, JWH-018 5-hydroxypentyl; 1c, JWH-018 6-hydroxyindole; 1d, JWH-018 5-hydroxyindole; 1e, JWH-018 pentanoic acid; 2a, JWH-073; 2b, JWH-073 4-hydroxybutyl; 2c, JWH-073 6-hydroxyindole; 2d, JWH-073 5-hydroxyindole; 2e, JWH-073 butanoic acid; 3a, JWH-081; 3b, JWH-081 5-hydroxypentyl; 4a, JWH-122; 4b, JWH-122 5-hydroxypentyl; 5a, JWH-200 5-hydroxyindole; 5b, JWH-200 6-hydroxyindole; 6a, JWH-210; 6b, JWH-210 5-hydroxypentyl; 6c, JWH-210 4-hydroxypentyl; 6d, JWH-210 5-hydroxyindole; 6e, JWH-210 pentanoic acid; 7a, JWH-250; 7b, JWH-250 5-hydroxypentyl; 7c, JWH-250 4-hydroxypentyl; 7d, JWH-250 5-hydroxyindole; 7e, JWH-250 pentanoic acid; 8a, RCS-4; 8b, RCS-4 5-hydroxypentyl; 8c, RCS-4 pentanoic acid; 9a, AM2201; 9b, AM2201 4-hydroxypentyl; 9c, AM2201 6-ydroxyindole; 10a, MAM2201.
Scheduled MRM increased sensitivity by maximizing dwell time. But still, coeluting compounds can complicate EPI acquisition and identification, as we observed for JWH-210 5-hydroxyindole, MAM 2201, and JWH-250 that all elute around 6.05 min. The 250 ms EPI cycle time for each coeluting compound limited our ability to obtain high-quality spectra across peaks at low concentrations, producing higher LODs than we achieved for other analytes.
We employed two internal standards that eluted early and late in our gradient to monitor retention time drift and to increase confidence in negative sample results when analyzing unknown specimens. Internal standard concentrations were selected to produce an abundant signal. In Figure 2, the total ion chromatograms of an LOD control sample at 5 ng/mL acquired in scheduled MRM mode with EPI scans, as well as unscheduled without EPI scans, are depicted. As can be seen in Figure 2A, acquisition of the product ion spectra reduces the number of MRM scans across each analyte peak, producing a ragged, asymmetric rather than smooth, Gaussian peak shape as typically observed in traditional MRM experiments (Figure 2B).
Data Analysis
Spectral library searching was performed with the recently launched LibraryView. Its searching algorithm provides scores expressed as “Purity”, “Fit”, and “Reverse fit”. Fit compares ions present in the library spectrum to those present in the acquired spectrum. Reverse fit calculations factor masses present in the acquired spectrum, which are not present in the library spectrum. Additional masses caused by impurities or coeluting compounds that do not appear in the reference spectrum produce lower Reverse fit scores. Purity is a computation that includes both Fit and Reverse fit scores for an overall spectrum match score. Purity values, like the values for the Fit and the Reverse fit, range from 0 to 100% with a perfect match, giving the score of 100%.
An analyte was confirmed positive when purity was ≥60%, the retention time was within ±0.05 min of the expected retention time, and the visual confirmation of the three characteristic masses, including the molecular ion. The latter criterion is handled differently in previously published approaches. Gergov et al. required the same number of fragments in their method for antihistamines22 but decreased the number to two masses in their screening method for 238 drugs,21 while Dresen et al. did not mention a requirement for a specific number of characteristic ions.24
Figure 3 illustrates the library search with LibraryView, using the example of AM2201 4-hydroxypentyl in the 2.5 ng/mL LOD sample and in an authentic specimen. At the top, the extracted ion chromatogram of the MRM transition is depicted where the black dash indicates the time point of the EPI spectrum acquisition. If the peak is intense and the spectra quality is good, multiple positive hits with varying purity percentages will be seen across a peak. The corresponding EPI spectrum is shown below with all masses in red; the reference library spectrum is depicted in green. At the bottom, all hits and the calculated Fit, Reverse fit, and Purity (91.7% and 85.2%) percentages are listed.
Figure 3.
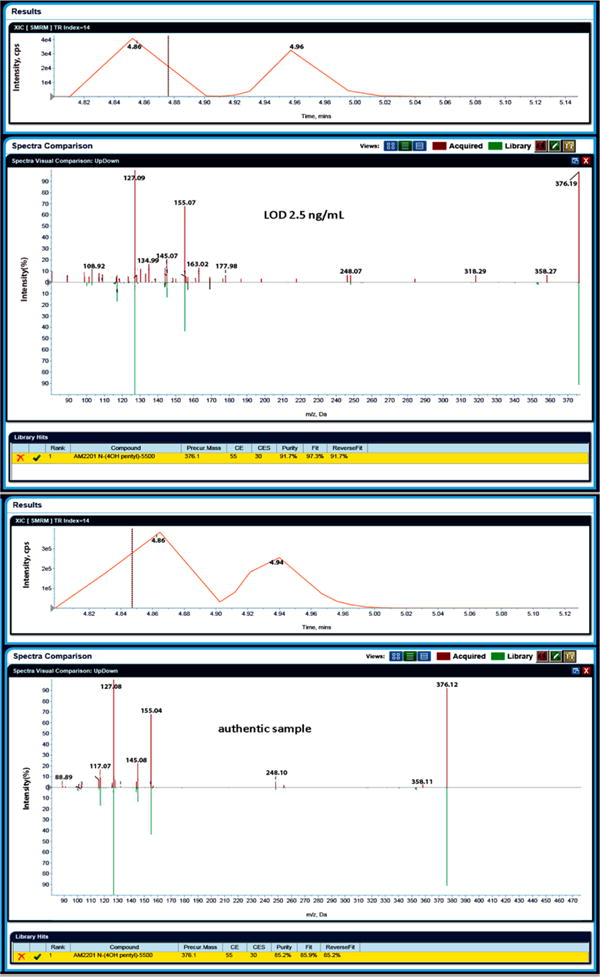
Library search with LibraryView for AM2201 4-hydroxypentyl (4.86 min retention time) in an LOD control sample at 2.5 ng/mL and in an authentic urine sample.
Optimization of Hydrolysis
Considering previous reports about deconjugation of Δ9-tetrahydrocannabinol metabolites,28,29 we were concerned that beta-glucuronidase may not be effective for hydrolyzing ester- and ether-linked synthetic cannabinoid glucuronide metabolites. Therefore, we conducted comprehensive hydrolysis optimization experiments. Initial experiments using commercially available JWH-018 5-hydroxypentyl glucuronide showed that incubation using 2000 units of beta glucuronidase, adjusted to pH 4 and incubated at 55 °C for two hours, achieved optimal hydrolysis.
In the authentic urine pool of presumptive positive synthetic cannabinoids, 14 metabolites were detected after hydrolysis. Since the other glucuronide metabolites were not commercially available, it was not possible to evaluate hydrolysis efficiency for the six remaining compounds. The absolute peak areas for these substances after hydrolysis under the conditions described above are given in Table 3 (see the Supporting Information). Initial conditions were incubation at 55 °C for 60 min with 2000 units enzyme at pH 4.0. A constant peak area for the parent analyte before and after hydrolysis indicated hydrolysis failure or that there was no glucuronide present in the sample. This was the case for JWH-018 pentanoic acid, JWH-073 butanoic acid, and JWH-250 pentanoic acid; peak areas with and without hydrolysis did not change.
The only ester metabolite identified with changes after hydrolysis was RCS-4 pentanoic acid. Though it remains unclear how much glucuronide was present initially, these results indicate that this analyte is glucuronidated and that we successfully hydrolyzed the conjugated species. Whether or not complete hydrolysis occurred cannot be assessed for all glucuronides, as reference standards are not available; however, evaluating peak areas after different hydrolysis conditions provided data for selecting the optimal procedure. For the duration of our hydrolysis studies, no change in JWH-018 pentanoic acid peak areas was observed. Ester-linked glucuronide species are known to be unstable;30 we may not have observed any effect of hydrolysis on JWH-018 pentanoic peak areas, since the glucuronide species may already have undergone deconjugation prior to analysis. We did try to use the freshest samples available for our hydrolysis studies, but sample handling prior to our receipt was unknown.
ANOVA confirmed a significant difference between hydrolyzed and nonhydrolyzed samples for all sets (F9,30 = 5.565 − 443.3, p < 0.05), except for JWH-018 pentanoic acid, JWH-073 butanoic acid, and JWH-250 pentanoic acid. Bonferroni's multiple comparison test could not detect a significant difference between initial conditions (1 h incubation at 55 °C with 2000 units enzyme at pH 4.0) and the other sets, except for JWH-250 hydroxypentyl. For cleaving its corresponding glucuronide, the approach using 4000 units enzyme was significantly superior to 2000 units enzyme (t = 4.96, p < 0.05). For economic reasons, we did not opt for using more enzyme but decided to increase incubation time to 2 h, which was as effective as 4000 units of enzyme. Two-hour incubation trended to yield higher peak areas for several analytes and exhibited low variability without producing large matrix effects for all analytes. The optimized hydrolysis conditions utilized for analyzing authentic specimens included incubation at 55 °C for 120 min with 2000 units of enzyme at pH 4.0. Monitoring the fortified “hydrolysis control I” in each run yielded an average peak area ratio of remaining glucuronide to aglycone of 0.37%, with a relative standard deviation of 1.5% (number of runs = 43).
Validation
Common drugs and their metabolites fortified at 1000 ng/mL in LOD samples did not interfere with identification of analytes of interest and did not produce false-positive results when fortified into blank samples. Blank urine from 17 individuals did not have any endogenous interferences. LODs were between 0.5 and 10 ng/mL for all analytes, 14 of 20 metabolites had a LOD ≤ 2.5 ng/mL (Table 1). Not all peak signals, including some intense peaks, were easily identified. For instance, RCS-4 and its metabolites have two dominant ions, even at low concentrations, while the third ion, which is required for a positive match, did not reach an acceptable intensity until a relatively high concentration, yielding higher LODs from 2.5 to 10 ng/mL. Ten nanograms per milliliter also was the LOD for JWH-210 5-hydroxyindole, one of the three coeluting substances at 6.05 min, as well as for JWH-081 and JWH-210, whose mass spectra do not contain many meaningful ions and were, thus, harder to match.
There are few published quantitative data for metabolites of synthetic cannabinoids in human urine. Moran et al. and Chimalakonda et al. report concentrations after consumption of JWH-018 and JWH-073, which were in the low nanogram per milliliter range for the main metabolites (JWH-018 5-hydroxypentyl 23 ng/mL17/26 ng/mL;15 JWH-018 pentanoic acid 16.5 ng/mL17/16 ng/mL,15 JWH-073 butanoic acid 6.2 ng/mL17). In a single sample Jager et al. found concentrations of 8.7 ng/mL JWH-018 5-hydroxypentyl, 0.89 ng/mL JWH-018 pentanoic acid, 0.55 ng/mL JWH-073 butanoic acid, 0.67 ng/mL JWH-073 hydroxybutyl, and 0.25 ng/mL JWH-250 pentanoic acid.19 Whereas some analyte concentrations were below our assay LOD, we would be able to detect the main metabolite, JWH-018 5-hydroxypentyl, demonstrating that our assay sensitivity appears to be appropriate to document consumption, and our identification criteria are highly selective.
Matrix effects were between −5 and +22% for all analytes, recovery after protein precipitation ranged from 53 to 95% (Table 1). Processed sample stability was at least 72 h when stored at 4 °C. No significant decrease in absolute peak areas was observed.
Our carryover experiment was designed based on the highest reported concentrations in the literature. Maximum concentrations were 845 ng/mL for JWH-018 5-hydroxypentyl, 284 ng/mL for JWH-018 pentanoic acid, 315 ng/mL for JWH-073 butanoic acid, 10 ng/mL for JWH-018 6-hydroxyindole,31 and 6.2 ng/mL for JWH-073 4-hydroxybutyl.17 Carryover thresholds were 100 ng/mL for AM2201, 200 ng/mL for MAM2201, JWH-018, JWH-073, and JWH-122, as well as 400 ng/mL for all other analytes. No carryover was observed after injecting samples that contained 1000 ng/mL JWH-018 5-hydroxypentyl (Table 1).
Application in Routine Analysis
Since new synthetic cannabinoids appear on the market regularly, adding new substances will be a constant issue. Still, addition of a new metabolite in this assay requires several steps, including a partial validation that covers the evaluation of selectivity, determination of limits of detection, assessment of matrix effects, recovery, carryover, and autosampler stability. Although this is a considerably time-consuming procedure, less effort is required than updating a quantification method.
To reduce the possibility of MS contamination, sample injection volume was limited to 5 μL, curtain gas pressure was increased to 50 psi, and the source probe housing was positioned as far from the orifice as possible. To prevent LC contamination with these highly lipophilic substances, we employed an extensive autosampler rinse procedure using a higher organic autosampler rinsing solution (acetonitrile/isopropanol/water 45/45/10 (v/v/v); 0.1% formic acid) than typically recommended by the instrument manufacturer.
We tested the suitability of the method by analyzing several authentic urine samples. Fortified samples at the limits of detection were analyzed in each run to document instrument sensitivity. Table 2 shows results for 10 deidentified urine samples. Fourteen synthetic cannabinoid metabolites were identified, including JWH-018 5-hydroxypentyl, JWH-018 pentanoic acid, JWH-018 hydroxyindole, JWH-073 butanoic acid, and JWH-073 4-hydroxybutyl, JWH-122 5-hydroxypentyl, JWH-200 6-hydroxyindole, JWH-210 5-hydroxypentyl, and JWH-210 5-hydroxyindole, JWH-250 hydroxypentyl, JWH-250 pentanoic acid, JWH-250 5-hydroxyindole, AM2201 4-hydroxypentyl, and AM 2201 6-hydroxyindole. JWH-073 hydroxyindole, JWH-200 5-hydroxyindole, and JWH-210 pentanoic acid were not detected, even when samples contained other metabolites originating from the same parent compound. These metabolites are either minor metabolites showing concentrations below our LODs, are not formed at all, or were instable. JWH-081 hydroxypentyl and the two RCS-4 metabolites were never identified either. Published metabolism studies identified no parent compounds in urine; however, these studies included few samples, low doses, and might not represent analytes present in chronic frequent synthetic cannabinoid smokers' urine. As mentioned above, parent compounds were occasionally reported for AM2201 and UR-144. In our samples, AM2201 was the only parent compound identified in a single specimen. Hydroxyindoles were rarely noted at all, and only when high signals for the other main metabolites were seen. Though we only required one metabolite for a positive confirmation, presence of a second supporting metabolite was the norm, increasing result confidence.
Table 2. Synthetic Cannabinoid Results for 10 Deidentified Urine Specimens Confirmed Positive by LC–MS/MS.
| 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 | 10 | |
|---|---|---|---|---|---|---|---|---|---|---|
| AM2201 | x | |||||||||
| AM2201 4-hydroxypentyl | x | x | x | x | x | x | x | x | ||
| AM2201 6-hydroxyindole | x | x | x | x | x | x | ||||
| JWH-018 | ||||||||||
| JWH-018 pentanoic acid | x | x | x | x | x | x | x | x | x | |
| JWH-018 5-hydroxypentyl | x | x | x | x | x | x | x | x | x | x |
| JWH-018 hydroxyindole | x | |||||||||
| JWH-073 | ||||||||||
| JWH-073 4-hydroxybutyl | x | x | x | |||||||
| JWH-073 butanoic acid | x | x | x | x | x | x | x | x | ||
| JWH-073 hydroxyindole | ||||||||||
| JWH-081 | ||||||||||
| JWH-081 5-hydroxypentyl | ||||||||||
| JWH-122 | ||||||||||
| JWH122 5-hydroxypentyl | x | x | x | x | ||||||
| JWH-200 5-hydroxyindole | ||||||||||
| JWH-200 6-hydroxyindole | x | |||||||||
| JWH-210 | ||||||||||
| JWH-210 pentanoic acid | ||||||||||
| JWH-210 5-hydroxyindole | x | x | ||||||||
| JWH-210 hydroxypentyl | x | x | ||||||||
| JWH-250 | ||||||||||
| JWH-250 pentanoic acid | x | x | ||||||||
| JWH-250 5-hydroxyindole | x | |||||||||
| JWH-250 hydroxypentyl | x | x | ||||||||
| MAM2201 | ||||||||||
| RCS-4 | ||||||||||
| RCS-4 pentanoic acid | ||||||||||
| RCS-4 5-hydroxypentyl | ||||||||||
Conclusion
We developed a LC–MS/MS qualitative method using a library search for identification of 9 parent compounds and 20 urinary metabolites of synthetic cannabinoids. In the absence of multitargeted and reliable synthetic cannabinoid immunoassays, a rapid and preferably easy-to-update qualitative confirmation method was needed for urine testing.
Sample preparation was straightforward and included enzymatic hydrolysis that was extensively investigated for a range of glucuronides and a reference standard. The method was fully validated, according to forensic guidelines, showed good sensitivity, good recoveries, and low matrix effects. Our qualitative approach collected additional spectral data to confirm analyte identities and offered the ability to add newly available substances with fewer validation steps.
Supplementary Material
Acknowledgments
This research was funded by the Intramural Research Program, National Institute on Drug Abuse, and National Institutes of Health.
Footnotes
Supporting Information: All authors contributed to the writing of the manuscript. All authors provided approval for the final version of the manuscript.
This material is available free of charge via the Internet at http://pubs.acs.org.
Notes: The authors declare no competing financial interest.
References
- 1.European Monitoring Centre for Drugs and Drug Abuse. European Database on New Drugs. 2012 [Google Scholar]
- 2.Swiss Confederation. SR 812.121.11. 2011 [Google Scholar]
- 3.U.S. Government. Synthetic Drug Abuse Prevention Act. 2012:138–139. [Google Scholar]
- 4.Sobolevsky T, Prasolov I, Rodchenkov G. Drug Test Anal. 2012;4:745–753. doi: 10.1002/dta.1418. [DOI] [PubMed] [Google Scholar]
- 5.Chimalakonda KC, Seely KA, Bratton SM, Brents LK, Moran CL, Endres GW, James LP, Hollenberg PF, Prather PL, Radominska-Pandya A, Moran JH. Drug Metab Dispos. 2012;40:2174–84. doi: 10.1124/dmd.112.047530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Grigoryev A, Saychuk S, Melnik A, Moskaleva N, Dzhurko J, Ershov M, Nosyrev A, Vedenin A, Izotov B, Zabirova I, Rozhanets V. J Chromatogr B. 2011;879:1126–1136. doi: 10.1016/j.jchromb.2011.03.034. [DOI] [PubMed] [Google Scholar]
- 7.Hutter M, Broecker S, Kneisel S, Auwärter V. J Mass Spectrom. 2012;47:54–65. doi: 10.1002/jms.2026. [DOI] [PubMed] [Google Scholar]
- 8.Möller I, Wintermeyer A, Bender K, Jübner M, Thomas A, Krug O, Schänzer W, Thevis M. Drug Test Anal. 2011;3:609–620. doi: 10.1002/dta.158. [DOI] [PubMed] [Google Scholar]
- 9.Sobolevsky T, Prasolov I, Rodchenkov G. Forensic Sci Int. 2010;200:141–147. doi: 10.1016/j.forsciint.2010.04.003. [DOI] [PubMed] [Google Scholar]
- 10.Wintermeyer A, Moller I, Thevis M, Jubner M, Beike J, Rothschild MA, Bender K. Anal Bioanal Chem. 2010;398:2141–53. doi: 10.1007/s00216-010-4171-0. [DOI] [PubMed] [Google Scholar]
- 11.Grigoryev A, Melnik A, Savchuk S, Simonov A, Rozhanets V. J Chromatogr B. 2011;879:2519–2526. doi: 10.1016/j.jchromb.2011.07.004. [DOI] [PubMed] [Google Scholar]
- 12.Kavanagh P, Grigoryev A, Melnik A, Simonov A. J Anal Toxicol. 2012;36:303–11. doi: 10.1093/jat/bks032. [DOI] [PubMed] [Google Scholar]
- 13.Grigoryev A, Kavanagh P, Melnik A. Drug Test Anal. 2012;4:519–524. doi: 10.1002/dta.350. [DOI] [PubMed] [Google Scholar]
- 14.Grigoryev A, Kavanagh P, Melnik A. Drug Test Anal. 2013;5:110–115. doi: 10.1002/dta.1336. [DOI] [PubMed] [Google Scholar]
- 15.Moran CL, Le VH, Chimalakonda KC, Smedley AL, Lackey FD, Owen SN, Kennedy PD, Endres GW, Ciske FL, Kramer JB, Kornilov AM, Bratton LD, Dobrowolski PJ, Wessinger WD, Fantegrossi WE, Prather PL, James LP, Radominska-Pandya A, Moran JH. Anal Chem. 2011;83:4228–4236. doi: 10.1021/ac2005636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Beuck S, Moeller I, Thomas A, Klose A, Schloerer N, Schaenzer W, Thevis M. Anal Bioanal Chem. 2011;401:493–505. doi: 10.1007/s00216-011-4931-5. [DOI] [PubMed] [Google Scholar]
- 17.Chimalakonda KC, Moran CL, Kennedy PD, Endres GW, Uzieblo A, Dobrowolski PJ, Fifer EK, Lapoint J, Nelson LS, Hoffman RS, James LP, Radominska-Pandya A, Moran JH. Anal Chem. 2011;83:6381–6388. doi: 10.1021/ac201377m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Yanes EG, Lovett DP. J Chromatogr B. 2012;909:42–50. doi: 10.1016/j.jchromb.2012.10.013. [DOI] [PubMed] [Google Scholar]
- 19.de Jager AD, Warner JV, Henman M, Ferguson W, Hall A. J Chromatogr B. 2012;897:22–31. doi: 10.1016/j.jchromb.2012.04.002. [DOI] [PubMed] [Google Scholar]
- 20.Liu HC, Liu RH, Ho HO, Lin DL. Anal Chem. 2009;81:9002–11. doi: 10.1021/ac901599d. [DOI] [PubMed] [Google Scholar]
- 21.Gergov M, Ojanpera I, Vuori E. J Chromatogr B. 2003;795:41–53. doi: 10.1016/s1570-0232(03)00498-7. [DOI] [PubMed] [Google Scholar]
- 22.Gergov M, Robson JN, Ojanpera I, Heinonen OP, Vuori E. Forensic Sci Int. 2001;121:108–15. doi: 10.1016/s0379-0738(01)00460-1. [DOI] [PubMed] [Google Scholar]
- 23.Herrin GL, McCurdy HH, Wall WH. J Anal Toxicol. 2005;29:599–606. doi: 10.1093/jat/29.7.599. [DOI] [PubMed] [Google Scholar]
- 24.Dresen S, Ferreiros N, Gnann H, Zimmermann R, Weinmann W. Anal Bioanal Chem. 2010;396:2425–34. doi: 10.1007/s00216-010-3485-2. [DOI] [PubMed] [Google Scholar]
- 25.Liu Y, Uboh CE, Soma LR, Li X, Guan F, You Y, Chen JW. Anal Chem. 2011;83:6834–6841. doi: 10.1021/ac2016163. [DOI] [PubMed] [Google Scholar]
- 26.Josephs JL, Sanders M. Rapid Commun Mass Spectrom. 2004;18:743–59. doi: 10.1002/rcm.1402. [DOI] [PubMed] [Google Scholar]
- 27.Matuszewski BK, Constanzer ML, Chavez-Eng CM. Anal Chem. 2003;75:3019–30. doi: 10.1021/ac020361s. [DOI] [PubMed] [Google Scholar]
- 28.Kemp PM, Abukhalaf IK, Manno JE, Manno BR, Alford DD, McWilliams ME, Nixon FE, Fitzgerald MJ, Reeves RR, Wood MJ. J Anal Toxicol. 1995;19:292–298. doi: 10.1093/jat/19.5.292. [DOI] [PubMed] [Google Scholar]
- 29.Abraham TT, Lowe RH, Pirnay SO, Darwin WD, Huestis MA. J Anal Toxicol. 2007;31:477–485. doi: 10.1093/jat/31.8.477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Shipkova M, Armstrong VW, Oellerich M, Wieland E. Ther Drug Monit. 2003;25:1–16. doi: 10.1097/00007691-200302000-00001. [DOI] [PubMed] [Google Scholar]
- 31.Rana S. Components of “SPICE”. Trends in the US and Detection in Urine. Presented at the Society of Forensic Toxicologists Conference; San Francisco, CA. 2012. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.