

Abstract
Receptor tyrosine kinases (RTKs) are cell surface receptors that initiate signal cascades in response to ligand stimulation. Abnormal expression and dysregulated intracellular trafficking of RTKs have been shown to be involved in tumorigenesis. Recent evidence shows that these cell surface receptors translocate from cell surface to different cellular compartments, including the Golgi, mitochondria, endoplasmic reticulum (ER) and the nucleus, to regulate physiological and pathological functions. Although some trafficking mechanisms have been resolved, the mechanism of intracellular trafficking from cell surface to the Golgi is not yet completely understood. Here we report a mechanism of Golgi translocation of epidermal growth factor receptor (EGFR) in which EGF-induced EGFR travels to the Golgi via microtubule-dependent movement by interacting with dynein and fuses with the Golgi through syntaxin 6-mediated membrane fusion. We also demonstrate that the microtubule- and syntaxin 6-mediated Golgi translocation of EGFR is necessary for its consequent nuclear translocation and nuclear functions. Thus, together with previous studies, the microtubule- and syntaxin 6-mediated trafficking pathway from cell surface to the Golgi, ER and the nucleus defines a comprehensive trafficking route for EGFR to travel from cell surface to the Golgi and the nucleus.
Keywords: EGF receptor, Golgi and nuclear translocation, microtubules, syntaxin 6
INTRODUCTION
Receptor tyrosine kinases (RTKs) are cell surface receptors that initiate signal cascades in response to ligand stimulation, and intracellular trafficking of RTKs is important for the regulation of ligand-induced signaling transduction. Oncogenic activation of RTKs has been widely investigated and become the therapeutic target in cancer treatment.1–4 Recent studies showed that RTKs localized on subcellular components have specific functions. For example, vascular endothelial growth factor receptor 1 (VEGFR1) localizes on the Golgi to balance the level of VEGFR1 and VEGFR2 on the plasma membrane and dictate endothelial signaling to influence vascular physiology.5 Perinuclear accumulation of c-Met is required for the downstream signaling,6 and epidermal growth factor receptor (EGFR), transferrin receptor and G-protein-coupled proteins have been detected in the Golgi with unknown functions.7–9 In addition, EGFR or EGFRvIII transported to the mitochondria can either modulate the mitochondrial function via modification of cytochrome c oxidase subunit II (Cox-II)10 or cause drug resistance.11 Many studies have indicated that cell surface receptors translocate into the nucleus and have important roles, including the EGFR family, VEGFR family, fibroblast growth factor receptor (FGFR), c-Met and insulin-like growth factor 1 receptor.6,12–16 Specifically, EGFR1–4, VEGFR2 and FGFR1 or FGFR2 in the nucleus are involved in the transcriptional regulation of target genes, such as CCND1, iNos, b-Myb and cyclooxygenase-2.17–22 Nuclear EGFR also regulates DNA replication and repair.23–27 More recently, nuclear EGFR is shown to contribute to resistance to cetuximab and gefitinib,28–30 and nuclear ErbB2 has also been reported to associate with β-actin and RNA polymerase I to enhance the rRNA transcription.31 Taken together, these studies all suggest that cell surface receptors in different cellular locations associate with specific functions and raise the importance of studying the mechanism of intracellular trafficking of cell surface receptors to different cellular compartments.
A trafficking pathway of membrane-associated EGFR and ErbB2 from the cell surface to the nucleus through endocytosis, trafficking from the Golgi to endoplasmic reticulum (ER) and inner nuclear membrane has recently been proposed,32–34 but this pathway is missing elements that link endocytic vesicles to Golgi. Therefore, it is timely to further dissect the mechanism of Golgi translocation of cell surface receptors and to identity these missing links.
Intracellular transport of cargo proteins requires the cytoskeleton, including microtubules and motor proteins.35,36 Recent studies demonstrated that microtubule-dependent intracellular trafficking is used for EGFR degradation trafficking.37,38 In this study, we provide a link between microtubule-dependent intracellular trafficking to Golgi and nuclear translocation and functions of EGFR. Based on these results and together with previous studies,9,32–34 we propose a model demonstrating a comprehensive pathway for cell surface EGFR trafficking to Golgi/ER/the nucleus. So far, accumulated evidence indicates that many of the RTKs and other cell surface molecules are detected in the nucleus with specific functions.39,40 This model may serve as a general mechanism by which cell surface receptors move to their next destination such as the ER or the nucleus to carry out their nuclear functions.
RESULTS
EGF induces EGFR translocation to the Golgi
To address how EGFR moves from cell surface to the Golgi, we first asked whether EGF could stimulate EGFR transport to the Golgi. Upon stimulation by EGF, EGFR colocalized with syntaxin 6, a marker of the Golgi, as indicated by the merged signals observed under confocal microscopy (Figure 1a). The results from fluorescence resonance energy transfer (FRET) assay also support the EGF-stimulated transport of EGFR to the Golgi (Figure 1b). FRET pseudoimage showed that EGF stimulation enhanced energy transfer from fluorescein isothiocyanate-labeled EGFR (the donor) to Alexa Flour 555-labeled syntaxin 6 (the acceptor), suggesting that EGFR localizes near syntaxin 6 upon EGF stimulation. We then performed immune-electron microscopy to examine EGF-stimulated EGFR to transport to the Golgi apparatus. As expected, we did observe a gold particle that was present near the Golgi, which also supports that EGF stimulation induced Golgi translocation of EGFR (Supplementary Figure 1a). Using two Golgi markers, syntaxin 6 and Vti1b, we found that fraction 9 isolated from density gradient ultracentrifugation was the Golgi-enriched fraction (Figure 1c) and EGF stimulation enhanced EGFR expression in this fraction (Figure 1d). We also observed EGFR in various cellular fractions, in particular the Golgi fraction, via a time-course treatment of EGF (Supplementary Figure 1b). We quantified the protein levels of EGFR in these fractions from cell fractionation (Supplementary Figure 3a) and density gradient assay (Supplementary Figure 1b) by densito-metric analysis. As shown in Supplementary Figure S1b, approximately 19% (Supplementary Table 2) of cytosolic EGFR proteins was in the Golgi-enriched fraction upon EGF stimulation for 20 min. Cell fractionation results also showed that about 45.6% (Supplementary Table 1) of total EGFR was in the cytosolic fraction. Therefore, about 9% (19% × 45.6%) EGFR was in the Golgi-enriched fraction (Supplementary Figure 1d). Z-stack confocal images also showed that EGF induced the colocalization of EGFR with the Golgi as indicated by the merged signals of EGFR and a transfected Golgi marker GalNac T241 (Figure 1e and Supplementary Movie S1). To rule out the possibility that EGF stimulation induces EGFR synthesis in the ER and post-translational modification at the Golgi, we used cycloheximide (CHX) to inhibit protein synthesis and found that EGF still can enhance colocalization of EGFR and GalNac T2 (Supplementary Figure 1e; the colocalization of EGFR with the Golgi was quantitated by counting the number of yellow spots representing merged green (EGFR) and red (GalNac T2) channels in about 30 cells), suggesting that EGF-induced EGFR on the Golgi is not from protein synthesis. This notion was further supported by another observation that the EGFR protein and mRNA levels did not change significantly upon EGF treatment (Supplementary Figure 1f). To further rule out the potential overlapping of Golgi and endosomal EGFR localized on perinuclear areas, we performed the triple staining of EGFR, endogenous Golgi marker (GM130) and recycling endosome marker (Rab11) (Figure 1f, top). Our results indicate that EGFR does not colocalize with the recycling endosomal marker (Rab11) upon EGF stimulation. When we tried triple staining with other two endosomal markers (EEA1 and Rab9), we encountered some difficulties finding a set of workable antibodies against these markers from different species. So we performed double staining of transfected Golgi marker (GalNacT2) and endogenous marker (GM130) to rule out false-positive results caused by overexpressed Golgi marker. Double staining of GalNacT2 and GM130 showed that they overlap completely, suggesting that the transfected Golgi marker GalNac T2 can be used as an endogenous marker (Supplementary Figure 1c). Furthermore, triple staining of EGFR, endogenous endosomal markers (EEA1, Rab9) and transfected GalNacT2 indicates that some EGFR colocalized with GalNacT2 and endomosmal markers (EEA1 and rab9) upon EGF stimulation but some EGFR colocalized only with GalNacT2 (Figure 1f, middle). These results suggest that the colocalization signals of EGFR at the Golgi we observed are in addition to its interaction with the perinuclear endosomes.
Figure 1.

EGF induces translocation of EGFR to the Golgi. (a) HeLa cells were transfected with pDsRed-syntaxin 6. Cells were serum starved overnight and then treated with EGF (50 ng/ml) for 20 min. EGFR was labeled with the indicated antibodies. The boxed areas are shown in detail in the insets. Insets 2–1 and 2–2 show representative colocalizations of EGFR and syntaxin 6. Scale bar, 10 μm. (b) Cells were serum starved overnight and then treated without or with EGF (50 ng/ml) for 20 min. Endogenous EGFR and syntaxin 6 were labeled with a primary antibodies and secondary fluorescein isothiocyanate (donor, green) and Texas-Red (acceptor; red) antibody. An Fc image was obtained using the Zeiss ZEN software. Scale bar, 20 μm. Quantitation of the FRET intensity is shown in the right. (c) Cell lysate was loaded onto the 0–30% OptiPrep density gradient medium and subjected to ultracentrifugation, and fractions were separated using the Gradient Station. The early endosome, the Golgi and ER markers were used to analyze fractions. S, short expose; L, long expose. (d) HeLa cells were treated with or without EGF (50 ng/ml) for 20 min after starvation overnight. The EGFR levels in the Golgi-enriched fraction (fraction 9) were analyzed using immunoblotting. (e) Cells were serum starved overnight and then treated with EGF (50 ng/ml) for 20 min. One cell was used for z-stack scanning. Representative images were shown. The boxed areas are shown in detail in the insets. Scale bar, 10 μm. (f) Cells were transfected with GalNac T2 for 48 h or direct staining of endogenous marker, GM130. Cells were maintained in serum-free media overnight and treated without or with EGF (50 ng/ml) for indicated time and analyzed using confocal microscope. Scale bar, 20 μm. Quantitation of colocalization of EGFR and endosomal markers is shown in the bottom. (g) HeLa cells were transfected with EGFP-GalNac T2. Cells were exposed to serum-free media overnight following treatment without or with EGF (50 ng/ml) for indicated time. Scale bar, 20 μm. The boxed areas are shown in the insets. Quantitation of colocalization of phospho-EGFR and total EGFR with the GalNac T2 is shown in the bottom. (h) HeLa cells were serum-starved overnight before EGF stimulation for indicated time. Total lysate and the Golgi-enriched fractions were performed with sodium dodecyl sulfate–polyacrylamide gel electrophoresis and western blot to examine the phospho-1086 of EGFR and total EGFR levels.
Next, we asked whether EGFR retains its kinase activity while it localizes at the Golgi. To this end, we then examined the phosphorylation level of EGFR by using a phosphorylation-specific p-1086 antibody. Double staining of total EGFR and phospho-EGFR with time course of EGF stimulation indicated that EGFR is still phosphorylated while it is in the Golgi, suggesting that it continues to function in signaling transduction (Figure 1g). Colocalization of phospho-EGFR or total EGFR with Golgi was quantified by the ZEN software as shown at the bottom of Figure 1g, indicating that about 5% of colocalization of EGFR occurred with Golgi marker at 20 min EGF stimulation. We also detected the highest phosphorylation level of EGFR in Golgi-enriched fraction at 20 min after EGF stimulation (Figure 1h), which is consistent with the confocal imaging results. Taken together, our data indicate that EGF stimulation enhances EGFR translocation to the Golgi and that EGFR remains phosphorylated, which suggest that it could potentially induce downstream signaling while it is localized in the Golgi.
Syntaxin 6 is required for the Golgi translocation of EGFR
Syntaxin 6 is a well-known soluble N-ethylmaleimide-sensitive fusion protein attachment protein receptor (SNARE) protein that localizes mainly on the Golgi and regulates endosomal trafficking by membrane fusion.41,42 Next, we asked whether syntaxin 6 regulates the Golgi translocation of EGFR. When we knocked down syntaxin 6 by siRNAs, we found the decrease of EGF-induced colocalization of EGFR with GalNac T2 (Figure 2a) and EGFR protein level in Golgi-enriched fractions (Figure 2b) by two different syntaxin 6 siRNAs. To further explorer the function of syntaxin 6 in the Golgi translocation of EGFR, a dominant-negative mutant of syntaxin 6, coiled-coil domain (CCD)43 also blocked EGF-induced colocalization of EGFR and GalNac T2 (Figure 2c). In addition, we first disrupted syntaxin 6 expression by using a short hairpin RNA (shRNA) against its 3′-untranslated region (UTR) and then restored syntaxin 6 expression to determine whether the EGFR on the Golgi can be rescued. As shown in Figure 2d, ectopically expressed syntaxin 6 increased the Golgi-localized EGFR that was decreased by shRNA. Next, we investigated the effect of syntaxin 6 knockdown on the intracellular distribution of EGFR by performing triple staining of EGFR, Golgi markers and endosomal markers. Interestingly, triple staining of EGFR, Golgi marker and early endosomal (EE1A) or late endosomal markers (Rab9) show that the colocalization of EGFR with the late endosomal marker (Rab9) increased but not the early endosomal marker (EEA1) (Figure 2e), implying that knockdown of syntaxin 6 caused EGFR to accumulate in late endosomes, which suggests that late endosomal trafficking is upstream of Golgi transport of EGFR. Reciprocal immunoprecipitation (IP)–western assay further supported this result as indicated by the association of EGFR with syntaxin 6 upon EGF stimulation (Figure 2f). We further performed in vitro GST pull-down assay to evaluate the association of EGFR with syntaxin 6. Indeed, we found that syntaxin 6 interacts directly with the fragment no. 3 of EGFR containing the transmembrane and the juxtamembrane domains but not with the other two fragments containing the kinase and the C-terminal domains (Figure 2g), indicating that the EGFR interacts directly with syntaxin 6 through the transmembrane or the juxtamembrane domain and to regulate the Golgi translocation of EGFR. Taken together, these results suggest that syntaxin 6 interacts with EGFR directly and has a role in Golgi transport of EGFR.
Figure 2.

Syntaxin 6 is required for the Golgi translocation of EGFR. (a) Cells were first transfected with syntaxin 6 or control (Ctrl) siRNAs for 24 h and then transfected with GalNac T2 for 48 h. Cells were then maintained in serum-free media overnight and treated without or with EGF (50 ng/ml) for 20 min and analyzed by confocal microscopy. Scale bar, 20 μm. The boxed areas are shown in detail in the insets. Results of quantitation of colocalization of EGFR and Golgi marker are shown in the right panel. (b) Cells were transfected with syntaxin 6 or control siRNAs. After 72 h transfection, cells were maintained in serum-free media overnight and treated without or with EGF (50 ng/ml) for 20 min. The EGFR levels in the Golgi-enriched fraction were analyzed using immunoblotting. (c) Cells were transfected with CCD domain of syntaxin 6 or control vector. After 48 h transfection, cells were maintained in serum-free media overnight and treated without or with EGF (50 ng/ml) for 20 min. Cells were analyzed by confocal microscope. Scale bar, 20 μm. The boxed areas are shown in detail in the insets. Results of quantitation of colocalization of EGFR and Golgi marker are shown in the right panel. (d) Cells were transfected with syntaxin 6 shRNA targeting to the 3′-UTR region or control shRNA. Syntaxin 6 and was restored in cells with knockdown of endogenous syntaxin 6. Cells were maintained in serum-free media overnight and then treated without or with EGF (50 ng/ml) for 20 min. Cellular fractions were subjected to immunoblotting with the indicated antibodies. (e) Cells were transfected with syntaxin 6 or control siRNAs. After 24 h transfection, cells were transfected with GalNac T2 for 48 h. Cells were maintained in serum-free media overnight and treated without or with EGF (50 ng/ml) for 20 min and then analyzed by confocal microscopy. Scale bar, 20 μm. The boxed areas are shown in detail in the insets. Quantitation of colocalization of EGFR and endosomal markers is shown in the right. (f) HeLa cells were serum-starved overnight and stimulated without or with EGF (50 ng/ml) for 20 min. Cell lysates were immunoprecipitated with the indicated antibodies and subjected to immunoblot analysis as indicated. (g) In vitro transcribed and translated biotin-labeled syntaxin 6 was incubated with recombinant GST-fused EGFR fragments, pulled down using glutathione-Sepharose beads and visualized with horseradish peroxidase (HRP) conjugated streptavidin. CT, c-terminal domain; IB, immunoblot; KD, kimase domain fragment; TM, transmembrane domain fragment.
Microtubules and dynein are required for EGFR translocation from the cell surface to Golgi
While studying EGF-induced Golgi and nuclear translocation of EGFR, we also observed an accumulation of EGFR (green) at one side of the nucleus upon EGF stimulation in COS-1 cells (top two rows of Figure 3a) and HeLa and A549 cells (bottom two rows of Figure 3a). It is known that microtubules merge at the microtubule organizing center, which is usually located at one side of the nucleus. We further investigated the relationship of EGFR and microtubules, and indeed, we found that EGFR accumulated around the high-intensity tubulin staining (red; indicated by white arrows), which represents the microtubule organizing center (Figure 3a). These results suggest that the microtubule cytoskeleton is involved in the Golgi and nuclear translocation of EGFR. Previous studies have shown that Golgi translocation of EGFR takes place before its nuclear transport.9,44 Moreover, it has been demonstrated that endocytosis is required for the nuclear translocation of EGFR.33 In this study, we focused on the role of microtubules in the Golgi and nuclear translocation of EGFR after EGF-induced endocytosis.
Figure 3.

Microtubules and dynein are required for EGF-induced Golgi transport of EGFR. (a) Serum-starved cells were treated with EGF. Double staining of EGFR and α-tubulin were subjected to confocal microscopy assay. Scale bars, 20 μm. (b) HeLa cells were transfected with GFP-GalNac T2, treated with microtubules or dynein inhibitors and then stimulated with EGF. The Golgi-enriched fractions were purified and subjected to immunoblot analysis with the indicated antibodies. (c) Serum-starved HeLa cells were treated as shown in (b) and then stimulated with EGF and analyzed by a confocal microscope. Scale bars, 20 μm. The boxed areas are shown in detail in the insets. Representative colocalization of EGFR and GalNac T2 is shown in inset 2–1. Quantitation of cells with Golgi-localized EGFR is shown in the lower panel. (d) HeLa cells were transfected with GFP-GalNac T2 expression plasmid and then transfected with control (ctrl) vector or CDK1 and cyclin B plasmids, respectively. Cells were then serum starved overnight, stimulated with EGF and further analyzed under a confocal microscope. Scale bar, 20 μm. Quantitative results are shown in the right. (e) Representative frames of time-lapse confocal microscopic image of cells treated with or without nocodazole. HeLa cells were transfected with EGFP–EGFR (green) and DsRed–syntaxin 6 (red) plasmids. After serum starvation overnight and EGF stimulation, images were collected at 30-s intervals as indicated. Scale bar, 5 μm. (f) Serum-starved HeLa cells were transfected with dynein shRNAs and then stimulated with EGF. Golgi-enriched fractions were purified and subjected to immunoblot analysis with indicated antibodies. DMSO, dimethyl sulfoxide; Noc, nocodazole; PT, paclitaxel; Van, vanadate.
To address whether trafficking of EGFR from the cell surface to Golgi requires microtubule formation, we first treated cells with microtubule inhibitors (nocodazole and paclitaxel) and found that this treatment reduced EGFR protein level in Golgi-enriched fraction (Figure 3b, lanes 3 and 4 vs lane 2). EGF-induced colocalization of EGFR with Golgi as indicated by the merged EGFR (green) and Golgi marker GalNac T2 (red) signals shown in yellow (Figure 3c, inset 2 vs inset 1) was also disrupted in cells pretreated with nocodazole and paclitaxel (Figure 3c, insets 3 and 4 vs inset 2 with quantitative results showing below). CDK1 has been reported to directly depolymerize microtubules through phosphorylation of the β-tubulin or microtubule-associated protein.45,46 To further test the role of microtubules in the Golgi translocation of EGFR, we disrupted the microtubule formation by ectopically expressing CDK1 and its activator, cyclin B, which reduced EGF-induced colocalization of EGFR with the Golgi marker, GalNac T2 (Figure 3d). Finally, we examined the effects of disrupting microtubules on Golgi translocation of EGFR using time-lapse live cell confocal microscopy analysis and another Golgi marker, syntaxin 6. We observed a dynamic colocalization of EGFR with syntaxin 6, which occurred about 20 min after EGF treatment (Figure 3e (top panel) and Supplementary Movie S2). This colocalization was blocked in cells pretreated with nocodazole (Figure 3e (lower panel) and Supplementary Movie S3). Taken together, these results suggest that microtubule formation is required for EGF-induced EGFR trafficking from the cell surface to the Golgi.
The microtubule-dependent movement toward the cell center requires the microtubule motor protein, dynein. As the ATPase functional domain of dynein, which utilizes ATP to generate energy for this movement, is usually used as a target to inhibit dynein's function, we used dynein inhibitors to determine if dynein has a role in EGF-induced EGFR trafficking to Golgi. Indeed, EGF-induced translocation of EGFR to Golgi was attenuated by treatment with dynein inhibitors, EHNA (erythro-9-(2-hydroxy-3-nonyl)-adenine) and vanadate, as indicated by the decreased EGFR protein level in Golgi-enriched fraction (Figure 3b, lanes 5 and 6 vs lane 2) and the decreased colocalization of EGFR with Golgi marker, GalNac T2 (Figure 3c, insets 5 and 6 vs inset 2). In addition, knocking down dynein expression by shRNAs decreased EGF-induced EGFR protein level in Golgi-enriched fraction (Figure 3f). Taken together, these results conclude that functional microtubules and dynein are required for the translocation of EGFR from the cell surface to Golgi and also suggest that EGFR likely travels along the microtubules to reach Golgi.
Microtubules associate with EGFR and are required for intracellular trafficking pattern of EGFR
Next, we asked whether EGF induces Golgi translocation of EGFR through the association of EGFR, microtubules and dynein. First, reciprocal IP–western blot analysis showed that EGF increased the interaction between EGFR and α-tubulin (Figure 4a). FRET assay showed that EGF stimulation enhanced energy transfer from EGFP-fused EGFR (the donor) to Alexa Flour 555-labeled microtubules (the acceptor) (Figure 4b, image 3 vs 6). Immunoelectron microscopy analysis also showed the localization of EGFR around microtubules upon EGF stimulation (Figure 4c, insets 1 and 2 vs inset 3). Therefore, IP, FRET and immunoelectron microscopy assays indicate that EGF stimulates EGFR to localize around microtubules and suggest that EGFR travels along microtubules upon EGF stimulation.
Figure 4.
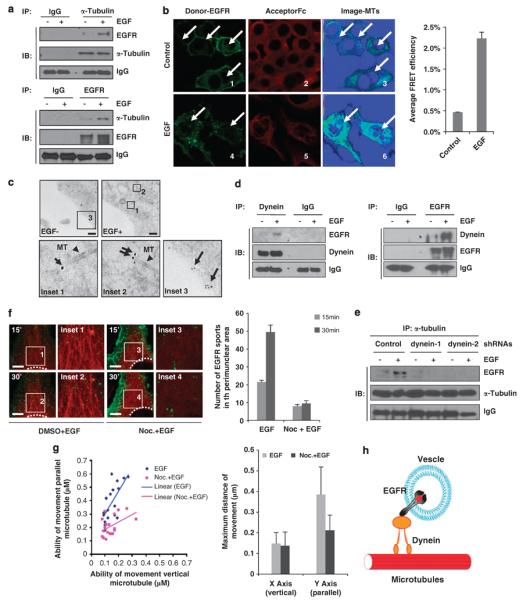
Microtubules associate with EGFR and are required for intracellular trafficking pattern of EGFR. (a) HeLa cells were serum starved overnight and stimulated with EGF for 20 min. Cell lysates were immunoprecipitated with the indicated antibodies. The molecules pulled down from immunoprecipitation were analyzed by immunoblotting. (b) HeLa cells were transfected with pEGFP–EGFR (donor; green) alone or labeled with a primary anti-α-tubulin antibody and secondary Alexa Fluor 555 (acceptor; red) antibody alone. A third condition was a combination of EGFP–EGFR and Alexa Fluor 555 with α-tubulin. An Fc image was obtained using the Zeiss ZEN software. Scale bar, 20 μm. Quantitation of the FRET efficiency is shown in the right panel. (c) HeLa cells were maintained in serum-free media overnight and treated with or without EGF for 30 min and subjected to immunoelectron microscopy as described in the Materials and methods. Scale bars, 200 nm. Arrows indicate EGFR and triangles indicate microtubules. (d) Serum-starved HeLa cells were stimulated with EGF for 20 min. Cell lysates were immunoprecipitated with the indicated antibodies and subjected to immunoblot analysis. Immunoprecipitated molecules were analyzed using immunoblotting. (e) Serum-starved HeLa cells with dynein shRNA expression plasmid were stimulated with EGF for 20 min. Cell lysates were immunoprecipitated with the indicated antibody and subjected to immunoblot analysis. IB, immunoblot. (f) HeLa cells transfected with EGFP–EGFR and mRFP-α-tubulin were maintained in serum-free media overnight. EGF-stimulated cells with or without Noc pretreatment were subjected to time-lapse confocal microscopy. Images were collected at 20-s intervals for 40 min. These images at 15 and 30 min under each condition were used to quantify particles in the perinuclear region. Scale bars, 5 μm. (g) EGF-induced particles were tracked by the AxioVision Tracking Program. Spots in the plot indicate maximum movement of these particles in two directions, in parallel or vertical with microtubule. The blue spots indicate vesicles carrying EGFR under EGF stimulation, and the pink spots indicate the vesicles carrying EGFR under pretreatment of nocodazole plus EGF stimulation. Noc, nocodazole. (h) Model of EGFR moving along the microtubules regulated by dynein.
As microtubule motor protein dynein is required for Golgi translocation of EGFR (Figures 3b, c and e) upon EGF stimulation, we further investigated whether EGF induces the association of EGFR with dynein. Both reciprocal IP–western blot analyses (Figure 4d) and confocal microscopy imaging assay (Supplementary Figure 2a) showed that EGF induced the association and colocalization of EGFR with dynein. Interestingly, knockdown of dynein by shRNAs also decreased the association of EGFR with α-tubulin, which further suggests that dynein is involved in microtubule-dependent movement of EGFR (Figure 4e).
The above results suggest that microtubules and dynein are required for EGF-induced Golgi translocation of EGFR and EGF also increases the association of EGFR/microtubules and EGFR/dynein. Next, we asked whether EGFR actually travels along microtubules. To address this question, we coexpressed EGFP–EGFR and mRFP–tubulin in cells and visualized the movement of EGFR using time-lapse live cell confocal microscopy analysis. We found that most of EGFR move forward and backward in parallel with microtubules upon EGF stimulation (Supplementary Movie S4). However, the addition of nocodazole disrupted the movement (Supplementary Movie S5). More importantly, the number of EGFR spots in the perinuclear area (cytoplasm/nucleus junction marked by white dotted line) upon EGF stimulation also decreased with nocodazole treatment (Figure 4f). Using the Zeiss AxioVision tracking application, we further measured the movement of more than 20 EGFR spots in parallel or vertical with microtubules, which were shown as the y axis or x axis in Figure 4g, respectively. Clearly, compared to EGF treatment, nocodazole treatment significantly reduced the EGF-induced movement of EGFR in parallel with microtubules. Taken together, the data suggest that the movement of EGFR from cell surface to the perinuclear area requires microtubule formation and that disruption of microtubules affects the movement of EGFR in parallel with microtubules. Collectively, these results demonstrate that dynein is critical for EGFR/α-tubulin interaction and it links EGFR with microtubules and may facilitate microtubule-dependent movement by providing power for intracellular trafficking of EGFR. These results resemble the previous studies in which transport of vesicles containing an N-methyl-d-aspartate or γ-aminobutyric acid receptor along microtubules were shown to be regulated by motor proteins or other coregulators.47,48 Thus, it is conceivable that analogous to the previous model, the endocytic vesicle containing EGFR interacts with dynein and tubulin and travels along the microtubules (Figure 4h).
Syntaxin 6 is required for the nuclear translocation of EGFR
The above results indicated that syntaxin 6-, microtubule-and dynein-mediated intracellular trafficking is required for EGF-induced Golgi translocation of EGFR. Therefore, we asked whether disruption of syntaxin 6 affects downstream trafficking pathway of EGFR to the nucleus. As shown in Figure 5a, upon EGF stimulation, EGF-induced nuclear localization of EGFR (Figure 5a, insets 1 and 2; green channel merged with the blue channel) was reduced in cells with syntaxin 6 knockdown (cells that do not have red color surrounding the nucleus) compared with cells expressing syntaxin 6 (Figure 5a, insets 3 and 4; cells in red color surrounding the nucleus). We counted 100 cells and quantitated the percentage of cells with nuclear EGFR under different conditions (shown in the lower panel of Figure 5a). In the nuclear fraction, EGFR level was also significantly decreased by knocking down syntaxin 6 (Figure 5b). To further confirm the role of syntaxin 6 in nuclear translocation of EGFR, we first disrupted syntaxin 6 expression by using an shRNA against its 3′-UTR and then examine levels of nuclear EGFR after syntaxin 6 was re-expressed. Knockdown of syntaxin 6 and re-expression of empty vector reduced nuclear EGFR, but ectopically expressed syntaxin 6 restored the nuclear levels of EGFR (Figure 5c). Similarly, the dominant-negative mutant (CCD) of syntaxin 6 also decreased the localization of EGFR in the nucleus (Figure 5d, insets 2 and 4; green channel (EGFR) merged with the blue channel, which indicates the nucleus) as well as the level of nuclear EGFR (Figure 5e). Thus, we concluded that syntaxin 6-regulated Golgi translocation of EGFR is critical for EGF-induced nuclear translocation of EGFR.
Figure 5.
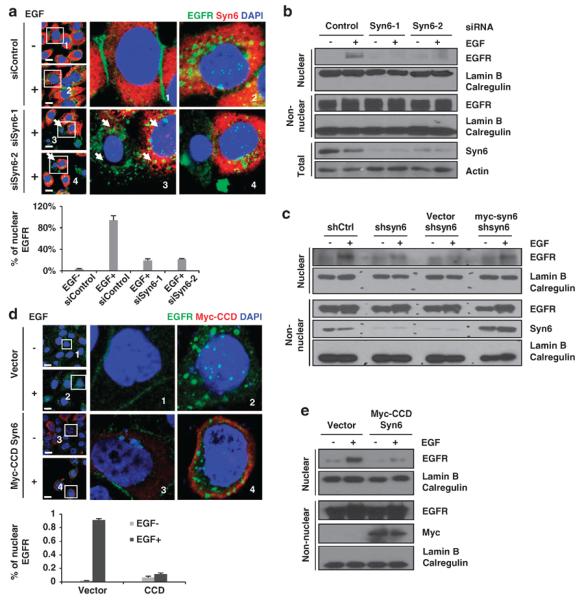
Syntaxin 6 is required for EGFR nuclear translocation. (a) HeLa cells were transfected with syntaxin 6 or control siRNAs and maintained in a serum-free media overnight and treated with EGF (50 ng/ml) for 30 min. Quantitation of positive cells with nuclear EGFR is shown in the lower panel. Scale bar, 20 μm. (b) Cells were transfected with syntaxin 6 or control siRNA and maintained in serum-free media overnight and then treated with EGF (50 ng/ml) for 30 min. Cellular fractions were subjected to immunoblotting with the indicated antibodies. (c) Cells were transfected with syntaxin 6 shRNA targeting to the 3′-UTR region or control shRNA. Syntaxin 6 and vector control were restored in cells with knockdown of endogenous syntaxin 6. Cells were maintained in serum-free media overnight and then treated with EGF (50 ng/ml) for 30 min. Cellular fractions were subjected to immunoblotting with the indicated antibodies. (d) HeLa cells were transfected with a control vector and syntaxin 6 CCD and maintained in serum-free media overnight, and then stimulated with EGF. Quantitation of positive cells with nuclear EGFR is shown in the lower panel. Scale bar, 20 μm. (e) HeLa cells were transfected with a control vector and syntaxin 6 CCD and maintained in serum-free media overnight, and then stimulated with EGF. Nuclear and non-nuclear fractions were subjected to immunoblot analysis with the indicated antibodies. DAPI, 4′,6-diamidino-2-phenylindole.
Microtubules and dynein are required for the nuclear translocation of EGFR
To further investigate the function of the microtubule-dependent trafficking in EGF-induced Golgi and nuclear translocation of EGFR, we next examined whether microtubule-dependent movement of EGFR is required for its downstream nuclear translocation. We found that the EGF-induced colocalization of EGFR with the nucleus (Figure 6a) and the nuclear level of EGFR (Figure 6b) decreased significantly in the presence of nocodazole and paclitaxel. In addition, to test whether depolymerizing microtubules affects nuclear translocation of EGFR, we overexpressed CDK1 and its activator cyclin B to disrupt microtubule formation. We found that overexpression of CDK1 decreased the nuclear level of EGFR (Figure 6c) and also decreased the nuclear localization of EGFR (Figure 6d, inset 4 vs inset 2). Finally, the role of microtubules in nuclear translocation of EGFR was examined using time-lapse live cell confocal microscopy analysis in which EGFR was labeled with EGFP and the inner nuclear membrane protein lamin B (representing the boundary of the nucleus) was labeled with RFP. A comparison between Supplementary Movie S6 and Supplementary Movie S7 showed that EGFR translocation into the nucleus occurs at the 23- and 23.5-min time points after EGF stimulation (Figure 6e, upper panels and Supplementary Movie S6). However, in the presence nocodazole, microtubules were disrupted and cannot serve as a trafficking route. Endocytic EGFR was blocked on the cell surface membrane and EGFR signal in the nucleus cannot be detected (Figure 6e, lower panels and Supplementary Movie S7). In addition, we separated plasma membrane, cytosol, nuclear membrane and nuclear plasma using cellular fractionation and investigated whether nocodazole affects the cellular localization of EGFR. As shown in Supplementary Figure 3a, nocodazole treatment reduced the EGF-induced localization of EGFR on nuclear membrane and nuclear plasma, but had no significant effect on the levels of cytosolic or membrane EGFR under EGF stimulation. Taken together, these results suggest that microtubules are required for the nuclear translocation of EGFR, and this is likely through the regulation of Golgi translocation.
Figure 6.
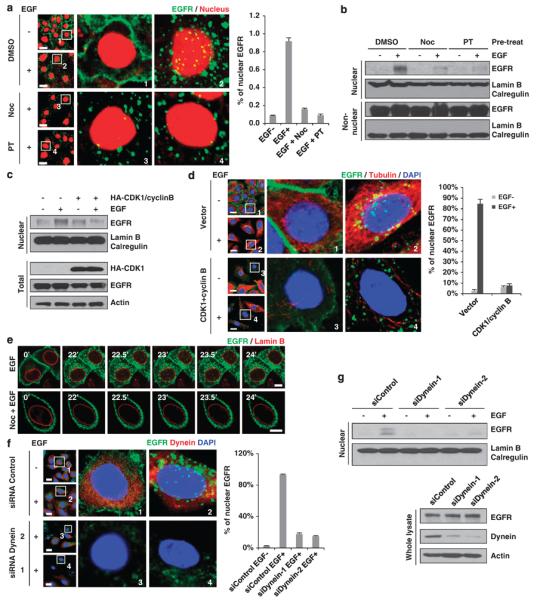
Disruption of microtubules and dynein blocks EGFR nuclear translocation. (a) HeLa cells maintained in serum-free media overnight were treated with microtubule inhibitors and stimulated with EGF (50 ng/ml) and assayed using a confocal microscope. Positive cells with nuclear EGFR were quantitated as shown in the right panel. Scale bar, 20 μm. (b) HeLa cells were treated under the same conditions as in (a). Non-nuclear and nuclear fractions were separated using cellular fractionation and then subjected to immunoblotting with the indicated antibodies. (c) HeLa cells were transfected with a control vector or HA-CDK1 and cyclin B for 48 h. Before EGF stimulation, cells were maintained in serum-free media overnight. Nuclear and non-nuclear fractions were subjected to immunoblotting with the indicated antibodies. (d) HeLa cells were treated as described in (c) and assayed using confocal microscopy. Positive cells with nuclear EGFR were quantitated as shown in the right panel. Scale bar, 20 μm. (e) Representative frames of time-lapse confocal microscopic images showing blockage of nuclear translocation of EGFR by pretreatment with nocodazole. EGFP–EGFR (green channel) and RFP–lamin B (red channel) were transfected into HeLa cells. After EGF stimulation, images were obtained at 30-s intervals as indicated. Scale bar, 10 μm. (f) HeLa cells were transfected with a control siRNA or dynein siRNAs for 48 h. Serum-starved cells were stimulated with EGF and analyzed by confocal microscopy. Scale bars, 20 μm. (g) HeLa cells were treated as described in (f). Serum-starved cells were stimulated with EGF and non-nuclear and nuclear fractions were separated using cellular fractionation and then subjected to immunoblotting with the indicated antibodies. DAPI, 4′,6-diamidino-2-phenylindole; DMSO, dimethyl sulfoxide; Noc, nocodazole; PT, paclitaxel.
To determine whether dynein also regulates the nuclear trafficking of EGFR, we treated cells with its inhibitors, vanadate and EHNA, and found that disruption of dynein ATPase activity decreased EGF-induced nuclear localization of EGFR and protein level of EGFR in the nucleus (Supplementary Figures 3b and c). To rule out possible nonspecific effects of these inhibitors, we knocked down dynein expression using siRNAs and found that depletion of dynein expression also reduced EGF-induced nuclear translocation of EGFR as shown by confocal microscopy analyses (Figure 6f) and western blot (Figure 6g). Thus, as expected, dynein is required for EGF-induced nuclear translocation of EGFR.
Disruption of syntaxin 6 and microtubules inhibits nuclear functions of EGFR
As shown above, our results indicated that syntaxin 6 and microtubules are involved in Golgi translocation and consequent nuclear transport of EGFR. Next, we asked whether disruption of syntaxin 6 and microtubule-dependent trafficking affects the nuclear function of EGFR. As shown in Figure 7a, disruption of microtubule and dynein by nocodazole and EHNA, respectively, blocked EGF-induced binding of EGFR to the CCND1 promoter, a known target of nuclear EGFR18 by chromatin-IP (ChIP) analysis (Figure 7a). We also observed similar results in which the DNA binding ability of EGFR is reduced when the syntaxin 6 was knocked down by siRNAs (Figure 6b). Furthermore, knockdown of syntaxin 6 also reduced EGF-induced CCND1 mRNA level (Figure 7c) and EGF-induced luciferase activity of the CCND1 promoter (Figure 7d). These findings indicate that syntaxin 6 and microtubule-dependent trafficking are critical for the transcriptional activity of nuclear EGFR. As nuclear EGFR is critical for cell proliferation,18,23 we further asked whether knockdown of syntaxin 6 affects cell growth. As shown in Figure 7e, knocking down syntaxin 6 significantly reduced cell growths. Using bromodeoxyuridine (BrdU) incorporation assay, we observed decreased cell proliferation when syntaxin 6 was disrupted (Figure 7f). A recent study showed that nuclear EGFR contributes tyrosine kinase inhibitor resistance through upregulated gene expression of the breast cancer resistance protein.30 In breast cancer cell line, BT20, and ovarian cancer cell line, OVCAR3, we found that when syntaxin 6 was knocked down, cells were more sensitive to the tyrosine kinase inhibitor treatment (Figures 7g and h). Taken together, these results support the notion that syntaxin 6 and microtubule-dependent trafficking are critical for nuclear localization of EGFR and its nuclear functions and also provide another layer of support for the importance of syntaxin 6-, microtubule- and dynein-mediated Golgi translocation for EGF-induced nuclear translocation of EGFR. In summary, we identified a novel trafficking mechanism of nuclear EGFR in which EGF induces EGFR to travel along microtubule cytoskeleton to Golgi via syntaixn 6-mediated membrane fusion and to the nucleus to control the nuclear functions of EGFR (Figure 7i).
Figure 7.

Nuclear function of EGFR requires syntaxin 6 and microtubules. (a) After overnight serum starvation, cells were pretreated with the indicated inhibitors for 30-min treatment and then stimulated with EGF for 30 min, followed by chromatin-IP assay. For IgG control, lysate of cells without EGF stimulation was used. (b) Cells were transfected with siRNAs of syntaxin 6. After 72 h transfection, cells were serum starved overnight and then stimulated with EGF for 30 min, followed by chromatin-IP assy. For IgG control, lysate of cells without EGF stimulation was used. (c) Cells were transfected with siRNAs of syntaxin 6. After 72 h transfection, cells were serum starved overnight and then stimulated with EGF for indicated time. Quantitative reverse transcription–polymerase chain reaction (RT–PCR) was used to analyze the mRNA level. (d) HeLa cells transfected with control siRNAs and siRNAs for syntaxin 6 were transfected with reporter plasmids containing CCND1 promoter. Then, after 24 h transfection, cells were maintained in serum-free media overnight and treated with EGF for indicated time. Total lysates were used for luciferase assay. Error bars were derived from three independent experiments. (e) HeLa cells were transfected with control siRNAs and siRNAs for syntaxin 6. After transfection, 4 × 105 cells were seeded in a six-well plate, incubated for 72 h and then counted. (f) HeLa cells were transfected with control siRNAs and siRNAs for syntaxin 6. After 48 h transfection, cells were treated with BrdU (100 μm) for 1 h. Cells were assayed for BrdU incorporation by flow cytometry. (g) BT20 cells were transfected with control siRNAs and siRNAs for syntaxin 6. After 24 h transfection, 2 × 105 cells were seeded in a 12-well plate overnight, treated with 0.1, 1 and 10 μm of gefitinib for 72 h and then counted. (h) OVCAR3 cells were transfected with control siRNAs and siRNAs for syntaxin 6. After 24 h transfection, 2 × 105 cells were seeded in a 12-well plate overnight, treated with 0.1, 1 and 10 μm of gefitinib for 72 h and then counted. (i) A schematic model of syntaxin 6- and microtubule-mediated Golgi and nuclear transport of EGFR.
DISCUSSION
Cellular distribution of cell surface receptors is considered to be more complicated than degradation and recycling. After receptor-regulated endocytosis, RTKs continue to activate downstream signals.3,49 Recent studies have reported that RTKs, such as EGFR, fibroblast growth factor receptor 1 and VEGFR1,5,10,40 transport to Golgi, mitochondrial and the nucleus, implying that cell surface receptors may have non-canonical functions at different cellular compartments.
Our results suggest that a SNARE protein, syntaxin 6, regulates EGFR fusion with the Golgi, and EGFR consequently transports to the nucleus. In addition, trafficking of EGFR from the cell surface to Golgi is not a random process. Rather, EGFR moves along microtubules powered by the motor protein, dynein. Several unique observations led to these conclusions: (1) syntaxin 6 is involved in Golgi and nuclear translocation of EGFR in response to EGF stimulation, indicating that membrane fusion may be a critical trafficking event for the Golgi and nuclear translocation of EGFR, and during these trafficking events, EGFR remains membrane-bound; (2) microtubules and dynein are both required for the translocation of EGFR to the Golgi and the nucleus. Microtubules have been reported to function in endosomal trafficking to the lysosome and membrane trafficking between the Golgi and ER,50 and our study further shows that the intracellular trafficking of cell surface receptors from endosome to Golgi also requires microtubules, indicating that microtubules may serve for most intracellular trafficking. As the dynein complex contains four subunits, it would be interesting to further investigate, which subunit(s) might directly or indirectly interact with EGFR in the future; (3) disruption of syntaxin 6 and microtubules attenuated nuclear functions of EGFR in cell proliferation and drug resistance, indicating that Golgi translocation of EGFR is a critical trafficking step for EGFR to function in the nucleus. Taken together with the previous studies,9,33,34,44 our study reveals a model showing schematic representation of Golgi translocation of EGFR. In conjunction with the previous studies showing both transport of EGFR from Golgi to ER via COPI-regulated retrograde trafficking and Sec61-mediated trafficking of EGFR, our study provides a clear mechanism outlining the Golgi and nuclear translocation of EGFR.
Since the discovery of nuclear translocation of EGFR more than 20 years ago,15,51 many studies have identified important biological functions of nuclear EGFR, including cell proliferation, DNA synthesis, DNA repair and drug resistance.16,28,30,52,53 However, the field of nuclear RTKs has progressed slowly partly because of a lack of clear trafficking mechanism of cell surface receptors to the nucleus. Although the partial mechanism of nuclear translocation of EGFR has been shown to include endocytosis,32,33,54 nuclear localization signals (NLS), importin α1/β1,18,32,33,55–58 COPI-mediated retrograde trafficking from Golgi to ER9 and translocon protein Sec61-regulated nuclear translocation of EGFR,34,44 experimental data demonstrating Golgi translocation of EGFR is still absent. Our present findings filled the gap regarding how EGFR is transported to Golgi/ER after endocytosis and then into the nucleus.
While Golgi trafficking model depicted in Figure 7i is attractive, a few questions still require further investigation. For example, it is not known whether EGFRs have specific functions at Golgi in addition to docking at Golgi for further intracellular trafficking. It would also be interesting to investigate whether other groups of molecules, such as small GTPase Rabs, which are involved in the retrograde trafficking pathway,59 regulate the transport of EGFR after endocytosis to Golgi. It is worthwhile to mention that EGF induces EGFR degradation through receptor-mediated endocytosis and endosomal trafficking to the lysosomes. How EGFR bypasses this degradation pathway or whether different populations of EGFR have different fates either into the lysosome or other cellular compartments such as the mitochondria60 or the nucleus remain to be investigated.
MATERIALS AND METHODS
Chemicals and antibodies
All chemicals were obtained from Sigma-Aldrich Co. (St Louis, MO, USA). The antibodies used in this study are as follows: anti-EGFR (Santa Cruz Biotechnology Inc., Santa Cruz, CA, USA and Neomarkers, Fremont, CA, USA); anti-dynein IC (Santa Cruz Biotechnology); anti-tubulin (Abcam, Cambridge, MA, USA and Sigma-Aldrich Co); anti-syntaxin 6 (BD Bioscience, San Jose, CA, USA); anti-lamin B (Calbiochem, San Diego, CA, USA); anti-calregulin (Santa Cruz Biotechnology); anti-Vti1b (BD Bioscience); and anti-actin, anti-myc and anti-HA (Roche, Indianapolis, IN, USA). All fluorescence-labeled secondary antibodies were obtained from Invitrogen (Carlsbad, CA, USA).
Plasmid constructs and siRNA oligonucleotides
The syntaxin 6 full-length plasmid was purchased from OriGene (Rockville, MD, USA), which was subcloned into pDsRedC1 (Clontech, Mountain View, CA, USA) for fluorescence staining. The coil-coiled domain of syntaxin6 was subcloned into the pCDNA6His-myc A (Invitrogen). The GalNAc-T2-GFP plasmid was a gift from Dr B Storrie (University of Arkansas for Medical Sciences). The plasmids expressing CDK1 (no. 1888), cyclin B (no. 10911) and RFP-tubulin (no. 21041) were obtained from Addgene (Cambridge, MA, USA). For siRNA transfection, siRNA oligonucleotides targeting dynein IC (siRNA ID: SASI_Hs01_00129737 and SASI_Hs01_00129739), syntaxin 6 (siRNA ID: SASI_Hs01_00129146 and SASI_Hs01_00129147) and nonspecific siRNA control (containing the sequences 5′-AUCACAUCUGUCAAAUUAUU-3′, 5′-GAACGUGGCUCUCAAAGUUU-3′, 5′-AAAGGAAAUCGACACUGAUU-3′ and 5′-GCCCUGGGAUUUAUGAUGAUU-3′) and shRNAs targeting dynein IC (TRCN0000116797 and TRCN0000116799) were purchased from Sigma-Aldrich. The shRNA targeting 3′-UTR of syntaxin 6 (V3LHS_411430) was purchased from Thermo Fisher Scientific Inc. (Waltham, MA, USA). The siRNAs were transfected into cells using the cationic liposome SN (stabilized non-viral) or Lipofectamine 2000 (Invitrogen) as described previously.2 Briefly, cells were grown overnight and incubated with plasmid/liposome complexes in Opti-MEM medium for 4 h, followed by replacement of complete medium and incubation at 37 °C for 24–48 h pEGFP-EGFR was constructed by subcloning full-length EGFR into the pEGFP-N1 (Clontech) with HindIII and KpnI. GST-fusion EGFR fragments were constructed by subcloning EGFR fragments described in the text into the pGEX-6p-1 vector (GE Healthcare Biosciences, Pittsburgh, PA, USA).
Cell culture and treatment
All cells lines were maintained in Dulbecco's modified Eagle's medium/F12 supplemented with 10% fetal bovine serum and antibiotics. Cells were serum-starved overnight or 12 h before EGF (50 ng/ml) stimulation. For experiments using inhibitors, serum-starved cells were treated with inhibitors first and then stimulated with EGF.
Cellular fractionation
Nuclear and non-nuclear fractionations were performed as described previously.34 For separated cells to four cellular fractions, treated cells were washed with ice-cold phosphate-buffered saline (PBS), swelled with hypotonic buffer (10 mM Tris-HCl, pH 8.0, 10 mM KCl, 2 mM MgCl2, 0.5 mM dithiothreitol, 1 mM phenylmethylsulfonyl fluoride and 0.15 U/ml aprotinin), cells were then homogenized using a Dounce homogenizer and nuclei were pelleted via centrifugation at 600 g for 5 min, and the supernatant was collected as a cytoplasmic fraction. The pellets were solubilized in lysis buffer (10 mM Tris–HCl, pH 8.0, 10 mM KCl, 0.5% NP-40, 2 mM MgCl2, 0.5 mM dithiothreitol, 1 mM phenylmethylsulfonyl fluoride and 0.15 μ/ml aprotinin) for 20 min on ice and centrifuged at 600 g for 5 min. Supernatant was collected as the plasma membrane fraction. The nuclei pellet was sonicated with lysis buffer and centrifuged at 16 000 g for 5 min for nuclear plasma. The pellets were finally solubilized in an NETN buffer (10 mM Tris–HCl (pH 8.0), 150 mM NaCl, 1% NP-40, 1% sodium deoxycholate, 0.1% sodium dodecyl sulfate, 0.5 mM dithiothreitol, 1 mM phenylmethylsulfonyl fluoride and 0.15 μ/ml aprotinin) for the nuclear membrane. All fractions were further centrifuged at 16 000 g for 20 min at 4 °C. The supernatant was collected.
Purification of the Golgi apparatus
The 0–30% OptiPrep continuous density gradient was constructed using the Gradient Station (BIOCOMP, Fredericton, NB, Canada). The Golgi apparatus was purified by using the OptiPrep (Sigma-Aldrich) density gradient medium according to the manufacturer's guidelines with slight modifications.
IP, immunoblotting and ChIP
For IP, lysates of HeLa cells treated as described in the text were precleared with 1 μg of mouse or rabbit IgG and 20 μl of protein G-agarose (Roche) for 1 h at 4 °C. Precleared lysates were incubated with 1 μg of primary antibodies or anti-IgG antibody at 4 °C overnight with gentle agitation. Lysates were further incubated with protein G-agarose for 30 min at 4 °C. Protein G-agarose pellets were collected and washed three times at 4 °C. The immunoprecipitants were subjected to sodium dodecyl sulfate-polyacrylamide gel electrophoresis and western blot analysis as described previously.32 ChIP was performed as described in manual of EZ ChIP. In brief, cells treated were fixed with 1% high quality of formaldehyde for 10 min at room temperature. Then, cells were lysed and sonicated to shear the genome DNA to manageable fragments. Lysates were precleared with protein G-agarose beads and then incubated with indicated antibody or anti-IgG antibody. DNA was isolated by EZ-ChIP kit (EMD Millipore, Billerica, MA, USA) for PCR and analyzed by gel electrophoresis.
Confocal microscopy analysis
For fixed cells, confocal microscopy assay was performed as described previously.32 Briefly, cells grown on chamber slides (Labtek, Scotts Valley, CA, USA) were treated as described in the text. After washing with ice-cold PBS, cells were fixed, permeabilized and incubated with primary antibodies and fluorescence-labeled secondary antibodies. Immunostained cells were examined using an Olympus FluoView FV300 confocal microscope (Olympus America, Melville, LA, USA) or Zeiss LSM 710 laser-scanning microscope (Carl Zeiss Inc., Thornwood, NY, USA) with × 63/1.4 objective. For time-lapse imaging, HeLa cells were grown in 35-mm glass-bottomed dishes (MatTek Corporation, Ashland, MA, USA). Images were obtained using the Zeiss LSM 710 microscope (Carl Zeiss Inc.) with a 37 °C incubation chamber and × 40/1.2 NA objective. Lasers (488, 561 and 630 nm) were used to acquire the images. After EGF stimulation, cells underwent 30-min time lapse and 15-s intervals or 40-min time lapse and 20-s intervals in different experiments. The ZEN and AxioVison (Carl Zeiss Inc.) and ImageJ software programs (National Institutes of Health, Bethesda, MD, USA) were used for data analysis.
Immunoelectron microscopy
Cells were fixed with 2% paraformaldehyde containing 0.1% glutaraldehyde for 1 h, permeabilized with 0.5% Triton X-100 for 15 min and then incubated with 5% bovine serum albumin for 15 min. After overnight incubation at 4 °C with primary antibodies, cells were then washed with PBS and further incubated with the gold particle-labeled secondary antibody (Electron Microscopy Science, Hatfield, PA, USA) overnight at 4 °C for immunogold labeling. After post-fixation with 2% glutaraldehyde, cells were washed and stained with 1% Millipore-filtered uranyl acetate. Samples were fixed with a solution containing 3% glutaraldehyde plus 2% paraformaldehyde in 0.1 m cacodylate buffer, pH 7.3, for 1 h. After fixation, the samples were washed, postfixed with 1% buffered osmium tetroxide for 30 min and stained en bloc with 1% Millipore-filtered uranyl acetate. The samples were dehydrated in increasing concentrations of ethanol, infiltrated and embedded in the LX-112 medium. The samples were polymerized in a 60 °C oven for 2 days. Ultrathin sections were cut in a Leica Ultracut microtome (Leica, Deerfield, IL, USA), stained with uranyl acetate and lead citrate in a Leica EM Stainer (Leica), and examined in a JEM 1010 transmission electron microscope (JEOL USA Inc., Peabody, MA, USA) at an accelerating voltage of 80 kV. Digital images were obtained using AMT Imaging System (Advanced Microscopy Techniques Corp., Danvers, MA, USA).
Fluorescence resonance energy transfer
HeLa cells were transfected with pEGFP-EGFR (donor) for 48 h. Cells were exposed to serum-free media overnight, treated with EGF (50 ng/ml) for 30 min following fixation overnight at 4 °C with 4% paraformaldehyde, and then washed three times with PBS. Cells were incubated with 0.05% Triton X-100 for 15 min and washed with PBS three times. Cells were blocked with 3% bovine serum albumin in PBS and incubated with primary mouse anti-α-tubulin antibody and secondary mouse Alexa Fluor 555 antibody (acceptor). The slides were examined with Zeiss LSM710 confocal microscope (Carl Zeiss MicroImaging Inc., Thornwood, NY, USA). For FRET data acquisition, three channels were setup: donor (GFP), acceptor (Alexa Fluor 555) and the FRET channel. The donor channel has a 488 nm excitation and 495–525 nm emission collection, and the acceptor channel has a 561 nm excitation and 575–635 nm emission collection. In contrast, the FRET channel has a 488 nm donor-excitation and 575–635 nm acceptor emission collection to image acceptor emission resulted from energy transferred from the donor. Then, optimal laser power levels and PMT settings were determined for the double-labeled sample to avoid photobleaching and saturation in all three imaging channels. After optimization, nine images were acquired for background and spectral bleed-through correction and subsequent FRET analysis from single-labeled donor sample, single-labeled acceptor sample and double-labeled sample containing donor and acceptor fluorophores. To analyze FRET efficiency quantitatively, Youvan method4 was chose to calculate the raw FRET images based on the following formula and a color-coded FRET image was then created. Fc = (fretgv – bg fret) – cfdon*(dongv – bgdon) – cfacc*(accgv – bgacc), where Fc is the FRET as calculated by the Youvan method, gv the intensity as gray value, bg the background intensity, cf the correction factor, fret the raw fret-channel image, don the donor channel image and acc the acceptor channel image.
BrdU incorporation assay
HeLa cells were transfected with siRNAs to knockdown syntaxin 6 expression. After 48 h transfection, cells were incubated with BrdU (100 μm) for 1 h and then fixed in cold 70% ethonal on ice for 30 min. Cells were washed with PBS and incubated with 2 m HCl/0.5% Triton X-100 for 30 min to denature the DNA and 0.1 m Na2B4O7, pH 8.5, was added to neutralize the acid. Cells were washed again with PBS and about 1×106 cells were resuspended in 20 μl of PBS containing 0.5% Tween-20, 1% bovine serum albumin and 10 μl of fluorescein isothiocyanate-conjugated anti-BrdU and incubated for 1 h at room temperature in the dark. Cells were washed with 50 ml of PBS containing 0.5% Tween-20 and 1% bovine serum albumin and resuspended in 200 μl propidium iodide solution (38 μm Na citrate, 69 μm PI and 19 μg/ml RNaseA), incubated for 30 min and analyzed by flow cytometry (BD Bioscience).
In vitro transcription and translation, GST pull-down assay
In vitro transcription and translation of EGFR, syntaxin 6 and dynactin were carried out using the TnT Reticulocyte Lysate Systems, Transcend Chemiluminescent Translation Detection System (Promega, Madison, WI, USA) and Pierce GST Protein Interaction Pull-Down Kit (Thermo Fisher Scientific, Rockford, IL, USA) according to the manufacturer's guidelines. Briefly, GST-tagged EGFR fragments were expressed in the BL-21 cells. EGFR, syntaxin 6 and dynectin were transcribed and translated in vitro with biotin-labeled lysine. Streptavidin-conjugated horseradish peroxidase was used to detect the binding proteins after sodium dodecyl sulfate–polyacrylamide gel electrophoresis.
Supplementary Material
ACKNOWLEDGEMENTS
We thank Dr B Storrie (University of Arkansas for Medical Sciences) for providing the GalNac T2-GFP plasmid. This study was partially supported by the following grants: National Institutes of Health Grants CA109311 and CA099031 (to MC Hung); Cancer Research Center of Excellence (D0H101-TD-C-111-005, Taiwan); Private University Grant (NSC99-2632-B-039-001-MY3, Taiwan); Program for Stem Cell and Regenerative Medicine Frontier Research (NSC100-2321-B-039-002, Taiwan); International Research-Intensive Centers of Excellence in Taiwan (NSC101-2911-I-002-303, Taiwan); The University of Texas MD Anderson-China Medical University and Hospital Sister Institution Fund; National Institutes of Health through MD Anderson's Cancer Center Support Grant and the Center for Biological Pathways (CA016672); Patel Memorial Breast Cancer Research Fund.
Footnotes
CONFLICT OF INTEREST The authors declare no conflict of interest.
Supplementary Information accompanies the paper on the Oncogene website (http://www.nature.com/onc)
REFERENCES
- 1.Sorkin A, Goh LK. Endocytosis and intracellular trafficking of ErbBs. Exp Cell Res. 2008;314:3093–3106. doi: 10.1016/j.yexcr.2008.07.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Yarden Y, Shilo BZ. SnapShot: EGFR signaling pathway. Cell. 2007;131:1018. doi: 10.1016/j.cell.2007.11.013. [DOI] [PubMed] [Google Scholar]
- 3.Lemmon MA. Schlessinger J. Cell signaling by receptor tyrosine kinases. Cell. 2010;141:1117–1134. doi: 10.1016/j.cell.2010.06.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Gschwind A, Fischer OM, Ullrich A. The discovery of receptor tyrosine kinases: targets for cancer therapy. Nat Rev Cancer. 2004;4:361–370. doi: 10.1038/nrc1360. [DOI] [PubMed] [Google Scholar]
- 5.Mittar S, Ulyatt C, Howell GJ, Bruns AF, Zachary I, Walker JH, et al. VEGFR1 receptor tyrosine kinase localization to the Golgi apparatus is calcium-dependent. Exp Cell Res. 2009;315:877–889. doi: 10.1016/j.yexcr.2008.12.020. [DOI] [PubMed] [Google Scholar]
- 6.Kermorgant S, Parker PJ. Receptor trafficking controls weak signal delivery: a strategy used by c-Met for STAT3 nuclear accumulation. J Cell Biol. 2008;182:855–863. doi: 10.1083/jcb.200806076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Akgoz M, Kalyanaraman V, Gautam N. Receptor-mediated reversible translocation of the G protein betagamma complex from the plasma membrane to the Golgi complex. J Biol Chem. 2004;279:51541–51544. doi: 10.1074/jbc.M410639200. [DOI] [PubMed] [Google Scholar]
- 8.Robertson BJ, Park RD, Snider MD. Role of vesicular traffic in the transport of surface transferrin receptor to the Golgi complex in cultured human cells. Arch Biochem Biophys. 1992;292:190–198. doi: 10.1016/0003-9861(92)90067-7. [DOI] [PubMed] [Google Scholar]
- 9.Wang YN, Wang H, Yamaguchi H, Lee HJ, Lee HH, Hung MC. COPI-mediated retrograde trafficking from the Golgi to the ER regulates EGFR nuclear transport. Biochem Biophys Res Commun. 2010;399:498–504. doi: 10.1016/j.bbrc.2010.07.096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Demory ML, Boerner JL, Davidson R, Faust W, Miyake T, Lee I, et al. Epidermal growth factor receptor translocation to the mitochondria: regulation and effect. J Biol Chem. 2009;284:36592–36604. doi: 10.1074/jbc.M109.000760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cao X, Zhu H, Ali-Osman F, Lo HW. EGFR and EGFRvIII undergo stress- and EGFR kinase inhibitor-induced mitochondrial translocalization: a potential mechanism of EGFR-driven antagonism of apoptosis. Mol Cancer. 2011;10:26. doi: 10.1186/1476-4598-10-26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sehat B, Tofigh A, Lin Y, Trocme E, Liljedahl U, Lagergren J, et al. SUMOylation mediates the nuclear translocation and signaling of the IGF-1 receptor. Sci Signal. 2010;3:ra10. doi: 10.1126/scisignal.2000628. [DOI] [PubMed] [Google Scholar]
- 13.Feng Y, Venema VJ, Venema RC, Tsai N, Caldwell RB. VEGF induces nuclear translocation of Flk-1/KDR, endothelial nitric oxide synthase, and caveolin-1 in vascular endothelial cells. Biochem Biophys Res Commun. 1999;256:192–197. doi: 10.1006/bbrc.1998.9790. [DOI] [PubMed] [Google Scholar]
- 14.Stachowiak MK, Maher PA, Joy A, Mordechai E, Stachowiak EK. Nuclear localization of functional FGF receptor 1 in human astrocytes suggests a novel mechanism for growth factor action. Brain Res Mol Brain Res. 1996;38:161–165. doi: 10.1016/0169-328x(96)00010-1. [DOI] [PubMed] [Google Scholar]
- 15.Marti U, Burwen SJ, Wells A, Barker ME, Huling S, Feren AM, et al. Localization of epidermal growth factor receptor in hepatocyte nuclei. Hepatology. 1991;13:15–20. [PubMed] [Google Scholar]
- 16.Wang SC, Hung MC. Nuclear translocation of the epidermal growth factor receptor family membrane tyrosine kinase receptors. Clin Cancer Res. 2009;15:6484–6489. doi: 10.1158/1078-0432.CCR-08-2813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hanada N, Lo HW, Day CP, Pan Y, Nakajima Y, Hung MC. Co-regulation of B-Myb expression by E2F1 and EGF receptor. Mol Carcinogen. 2006;45:10–17. doi: 10.1002/mc.20147. [DOI] [PubMed] [Google Scholar]
- 18.Lin SY, Makino K, Xia W, Matin A, Wen Y, Kwong KY, et al. Nuclear localization of EGF receptor and its potential new role as a transcription factor. Nat Cell Biol. 2001;3:802–808. doi: 10.1038/ncb0901-802. [DOI] [PubMed] [Google Scholar]
- 19.Lo HW, Hsu SC, Ali-Seyed M, Gunduz M, Xia W, Wei Y, et al. Nuclear interaction of EGFR and STAT3 in the activation of the iNOS/NO pathway. Cancer Cell. 2005;7:575–589. doi: 10.1016/j.ccr.2005.05.007. [DOI] [PubMed] [Google Scholar]
- 20.Ni CY, Murphy MP, Golde TE, Carpenter G. gamma -Secretase cleavage and nuclear localization of ErbB-4 receptor tyrosine kinase. Science. 2001;294:2179–2181. doi: 10.1126/science.1065412. [DOI] [PubMed] [Google Scholar]
- 21.Wang SC, Lien HC, Xia W, Chen IF, Lo HW, Wang Z, et al. Binding at and trans-activation of the COX-2 promoter by nuclear tyrosine kinase receptor ErbB-2. Cancer Cell. 2004;6:251–261. doi: 10.1016/j.ccr.2004.07.012. [DOI] [PubMed] [Google Scholar]
- 22.Xie Y, Hung MC. Nuclear localization of p185neu tyrosine kinase and its association with transcriptional transactivation. Biochem Biophys Res Commun. 1994;203:1589–1598. doi: 10.1006/bbrc.1994.2368. [DOI] [PubMed] [Google Scholar]
- 23.Wang SC, Nakajima Y, Yu YL, Xia W, Chen CT, Yang CC, et al. Tyrosine phosphorylation controls PCNA function through protein stability. Nat Cell Biol. 2006;8:1359–1368. doi: 10.1038/ncb1501. [DOI] [PubMed] [Google Scholar]
- 24.Das AK, Chen BP, Story MD, Sato M, Minna JD, Chen DJ, et al. Somatic mutations in the tyrosine kinase domain of epidermal growth factor receptor (EGFR) abrogate EGFR-mediated radioprotection in non-small cell lung carcinoma. Cancer Res. 2007;67:5267–5274. doi: 10.1158/0008-5472.CAN-07-0242. [DOI] [PubMed] [Google Scholar]
- 25.Chen DJ, Nirodi CS. The epidermal growth factor receptor: a role in repair of radiation-induced DNA damage. Clin Cancer Res. 2007;13(Part 1):6555–6560. doi: 10.1158/1078-0432.CCR-07-1610. [DOI] [PubMed] [Google Scholar]
- 26.Dittmann KH, Mayer C, Ohneseit PA, Raju U, Andratschke NH, Milas L, et al. Celecoxib induced tumor cell radiosensitization by inhibiting radiation induced nuclear EGFR transport and DNA-repair: a COX-2 independent mechanism. Int J Radiat Oncol Biol Phys. 2008;70:203–212. doi: 10.1016/j.ijrobp.2007.08.065. [DOI] [PubMed] [Google Scholar]
- 27.Hsu SC, Miller SA, Wang Y, Hung MC. Nuclear EGFR is required for cisplatin resistance and DNA repair. Am J Transl Res. 2009;1:249–258. [PMC free article] [PubMed] [Google Scholar]
- 28.Li C, Iida M, Dunn EF, Ghia AJ, Wheeler DL. Nuclear EGFR contributes to acquired resistance to cetuximab. Oncogene. 2009;28:3801–3813. doi: 10.1038/onc.2009.234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Chen YJ, Huang WC, Wei YL, Hsu SC, Yuan P, Lin HY, et al. Elevated BCRP/ABCG2 expression confers acquired resistance to gefitinib in wild-type EGFR-expressing cells. PLoS One. 2011;6:e21428. doi: 10.1371/journal.pone.0021428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Huang WC, Chen YJ, Li LY, Wei YL, Hsu SC, Tsai SL, et al. Nuclear translocation of epidermal growth factor receptor by Akt-dependent phosphorylation enhances breast cancer-resistant protein expression in gefitinib-resistant cells. J Biol Chem. 2011;286:20558–20568. doi: 10.1074/jbc.M111.240796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Li LY, Chen H, Hsieh YH, Wang YN, Chu HJ, Chen YH, et al. Nuclear ErbB2 enhances translation and cell growth by activating transcription of ribosomal RNA genes. Cancer Res. 2011;71:4269–4279. doi: 10.1158/0008-5472.CAN-10-3504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Giri DK, Ali-Seyed M, Li LY, Lee DF, Ling P, Bartholomeusz G, et al. Endosomal transport of ErbB-2: mechanism for nuclear entry of the cell surface receptor. Mol Cell Biol. 2005;25:11005–11018. doi: 10.1128/MCB.25.24.11005-11018.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lo HW, Ali-Seyed M, Wu Y, Bartholomeusz G, Hsu SC, Hung MC. Nuclear–cytoplasmic transport of EGFR involves receptor endocytosis, importin beta1 and CRM1. J Cell Biochem. 2006;98:1570–1583. doi: 10.1002/jcb.20876. [DOI] [PubMed] [Google Scholar]
- 34.Wang YN, Yamaguchi H, Huo L, Du Y, Lee HJ, Lee HH, et al. The translocon Sec61beta localized in the inner nuclear membrane transports membrane-embedded EGF receptor to the nucleus. J Biol Chem. 2010;285:38720–38729. doi: 10.1074/jbc.M110.158659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Allan VJ, Schroer TA. Membrane motors. Curr Opin Cell Biol. 1999;11:476–482. doi: 10.1016/s0955-0674(99)80068-4. [DOI] [PubMed] [Google Scholar]
- 36.Cole NB, Lippincott-Schwartz J. Organization of organelles and membrane traffic by microtubules. Curr Opin Cell Biol. 1995;7:55–64. doi: 10.1016/0955-0674(95)80045-x. [DOI] [PubMed] [Google Scholar]
- 37.Gao YS, Hubbert CC, Yao TP. The microtubule-associated histone deacetylase 6 (HDAC6) regulates epidermal growth factor receptor (EGFR) endocytic trafficking and degradation. J Biol Chem. 2010;285:11219–11226. doi: 10.1074/jbc.M109.042754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Deribe YL, Wild P, Chandrashaker A, Curak J, Schmidt MH, Kalaidzidis Y, et al. Regulation of epidermal growth factor receptor trafficking by lysine deacetylase HDAC6. Sci Signal. 2009;2:ra84. doi: 10.1126/scisignal.2000576. [DOI] [PubMed] [Google Scholar]
- 39.Carpenter G, Liao HJ. Trafficking of receptor tyrosine kinases to the nucleus. Exp Cell Res. 2009;315:1556–1566. doi: 10.1016/j.yexcr.2008.09.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wang YN, Yamaguchi H, Hsu JM, Hung MC. Nuclear trafficking of the epidermal growth factor receptor family membrane proteins. Oncogene. 2010;29:3997–4006. doi: 10.1038/onc.2010.157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Martens S, McMahon HT. Mechanisms of membrane fusion: disparate players and common principles. Nat Rev Mol Cell Biol. 2008;9:543–556. doi: 10.1038/nrm2417. [DOI] [PubMed] [Google Scholar]
- 42.Jahn R, Scheller RH. SNAREs—engines for membrane fusion. Nat Rev Mol Cell Biol. 2006;7:631–643. doi: 10.1038/nrm2002. [DOI] [PubMed] [Google Scholar]
- 43.Kabayama H, Tokushige N, Takeuchi M, Mikoshiba K. Syntaxin 6 regulates nerve growth factor-dependent neurite outgrowth. Neurosci Lett. 2008;436:340–344. doi: 10.1016/j.neulet.2008.03.061. [DOI] [PubMed] [Google Scholar]
- 44.Liao HJ, Carpenter G. Role of the Sec61 translocon in EGF receptor trafficking to the nucleus and gene expression. Mol Biol Cell. 2007;18:1064–1072. doi: 10.1091/mbc.E06-09-0802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Fourest-Lieuvin A, Peris L, Gache V, Garcia-Saez I, Juillan-Binard C, Lantez V, et al. Microtubule regulation in mitosis: tubulin phosphorylation by the cyclin-dependent kinase Cdk1. Mol Biol Cell. 2006;17:1041–1050. doi: 10.1091/mbc.E05-07-0621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ookata K, Hisanaga S, Sugita M, Okuyama A, Murofushi H, Kitazawa H, et al. MAP4 is the in vivo substrate for CDC2 kinase in HeLa cells: identification of an M-phase specific and a cell cycle-independent phosphorylation site in MAP4. Biochemistry. 1997;36:15873–15883. doi: 10.1021/bi971251w. [DOI] [PubMed] [Google Scholar]
- 47.Setou M, Nakagawa T, Seog DH, Hirokawa N. Kinesin superfamily motor protein KIF17 and mLin-10 in NMDA receptor-containing vesicle transport. Science. 2000;288:1796–1802. doi: 10.1126/science.288.5472.1796. [DOI] [PubMed] [Google Scholar]
- 48.Heisler FF, Loebrich S, Pechmann Y, Maier N, Zivkovic AR, Tokito M, et al. Muskelin regulates actin filament- and microtubule-based GABA(A) receptor transport in neurons. Neuron. 2011;70:66–81. doi: 10.1016/j.neuron.2011.03.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Sadowski L, Pilecka I, Miaczynska M. Signaling from endosomes: location makes a difference. Exp Cell Res. 2009;315:1601–1609. doi: 10.1016/j.yexcr.2008.09.021. [DOI] [PubMed] [Google Scholar]
- 50.Caviston JP, Holzbaur EL. Microtubule motors at the intersection of trafficking and transport. Trends Cell Biol. 2006;16:530–537. doi: 10.1016/j.tcb.2006.08.002. [DOI] [PubMed] [Google Scholar]
- 51.Kamio T, Shigematsu K, Sou H, Kawai K, Tsuchiyama H. Immunohistochemical expression of epidermal growth factor receptors in human adrenocortical carcinoma. Hum Pathol. 1990;21:277–282. doi: 10.1016/0046-8177(90)90227-v. [DOI] [PubMed] [Google Scholar]
- 52.Lo HW, Hung MC. Nuclear EGFR signalling network in cancers: linking EGFR pathway to cell cycle progression, nitric oxide pathway and patient survival. Br J Cancer. 2006;94:184–188. doi: 10.1038/sj.bjc.6602941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Dittmann K, Mayer C, Rodemann HP. Nuclear EGFR as novel therapeutic target: insights into nuclear translocation and function. Strahlenther Onkol. 2010;186:1–6. doi: 10.1007/s00066-009-2026-4. [DOI] [PubMed] [Google Scholar]
- 54.Bryant DM, Wylie FG, Stow JL. Regulation of endocytosis, nuclear translocation, and signaling of fibroblast growth factor receptor 1 by E-cadherin. Mol Biol Cell. 2005;16:14–23. doi: 10.1091/mbc.E04-09-0845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Hsu SC, Hung MC. Characterization of a novel tripartite nuclear localization sequence in the EGFR family. J Biol Chem. 2007;282:10432–10440. doi: 10.1074/jbc.M610014200. [DOI] [PubMed] [Google Scholar]
- 56.Reilly JF, Maher PA. Importin beta-mediated nuclear import of fibroblast growth factor receptor: role in cell proliferation. J Cell Biol. 2001;152:1307–1312. doi: 10.1083/jcb.152.6.1307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Offterdinger M, Schofer C, Weipoltshammer K, Grunt TW. c-erbB-3: a nuclear protein in mammary epithelial cells. J Cell Biol. 2002;157:929–939. doi: 10.1083/jcb.200109033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Srinivasan R, Gillett CE, Barnes DM, Gullick WJ. Nuclear expression of the c-erbB-4/HER-4 growth factor receptor in invasive breast cancers. Cancer Res. 2000;60:1483–1487. [PubMed] [Google Scholar]
- 59.Johannes L, Popoff V. Tracing the retrograde route in protein trafficking. Cell. 2008;135:1175–1187. doi: 10.1016/j.cell.2008.12.009. [DOI] [PubMed] [Google Scholar]
- 60.Boerner JL, Demory ML, Silva C, Parsons SJ. Phosphorylation of Y845 on the epidermal growth factor receptor mediates binding to the mitochondrial protein cytochrome c oxidase subunit II. Mol Cell Biol. 2004;24:7059–7071. doi: 10.1128/MCB.24.16.7059-7071.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.