

Abstract
Kinetics studies with structurally varied aldehydes and ketones in aqueous buffer at pH 7.4 reveal that carbonyl compounds with neighboring acid/base groups form hydrazones at accelerated rates. Similarly, tests of a hydrazine with a neighboring carboxylic acid group show that it also reacts at an accelerated rate. Rate constants for the fastest carbonyl/hydrazine combinations are 2–20 M−1sec−1, which is faster than recent strain-promoted cycloaddition reactions.
A broad effort1 to develop new bond-forming reactions having improved selectivity, lower interference and cross-reactivity with biological molecules, and enhanced rates, has resulted in the development of important classes of reactions such as Cu-catalyzed azide-alkyne cycloadditions,2 strain-driven cycloadditions,3,4 photo-click reactions,5 and Staudinger ligations.6 Relative to earlier, less-selective reactions, these new bond-forming strategies greatly enhance the ability to construct conjugates of biomolecules, particularly under challenging aqueous conditions at pH 7.4, at low concentrations, and in cellular settings.
One of the earliest reactions used for bioconjugations is that of hydrazone/oxime formation (Fig. 1), involving the stable imine formation of aldehydes and ketones with α-nucleophiles such as hydrazines and aminooxy groups. This venerable reaction7 has been widely useful in bioconjugation,8 due to its biomolecular orthogonality and because carbonyl and hydrazine functional groups are readily installed into small molecules. Early mechanistic studies of the reaction were performed by Jencks in the 1960's,7a and work by Dawson8d and Tam8a has highlighted the utility of the reaction in peptide labeling. Very recent studies by our laboratory9 and by Raines,10 Distefano,11 and Canary12 are also contributing to the utility of the reaction, which is employed not only in bioconjugations but also in other fields, such as polymer chemistry13 and dynamic combinatorial chemistry.8d,14
Figure 1.
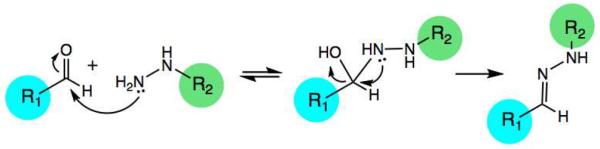
The mechanism of hydrazone formation. Breakdown of the tetrahedral intermediate is typically rate limiting at neutral pH.7a,b
However, there is a significant limitation of hydrazone and oxime formation that hinders its broader use: the slow rate of reaction of most substrates at neutral pH. This can be inconvenient for reactions in vitro (sometimes requiring hours to days8b,15), and can be strongly limiting in vivo, where concentrations of reactants are low. Although aniline can be used as nucleophilic catalyst,7c high concentrations are required, and the rate acceleration is moderate.8b,14a New-generation catalysts such as anthranilic acids and phosphonates have improved upon aniline catalysis,9b,c but in some applications the use of a catalyst is undesirable, adding complexity and in some cases, toxicity.16
Thus the identification of structural features that might speed hydrazone/oxime formation without the addition of a catalyst could significantly enhance the utility of this reaction. Surprisingly, there exist few general studies of what structural features in carbonyl substrates affect reaction rates, particularly at pH 7.4.15 Early mechanistic studies were typically carried out at acidic pH, where rates are much more rapid.17 However, the modern expansion of biological chemistry has greatly increased the relevance of reactivity at pH values near neutral.
Here we have performed kinetics studies on a range of aldehyde and ketone substrates, to examine how structure affects reactivity at pH 7.4 in aqueous buffer. We find that substrate structures can have marked effects on reaction rate, and we have identified specific structural features that yield reactions with rates that rival many modern cycloaddition reactions.
Reaction rates were measured for a range of commercially available carbonyl reactants (Tables 1,2) dissolved in phosphate-buffered saline (137 mM Na+, 2.7 mM K+, 12 mM phosphate, pH 7.4, 25 °C). A small amount of organic cosolvent (10% DMF) was added to ensure solubility of both reactants. To evaluate structural effects, we tested a range of aldehydes and ketones, both aryl- and alkyl-substituted. Electron-rich and -poor cases were included, and steric substitution near the carbonyl was varied. Finally, carbonyl compounds with proximal acid/base groups were examined as well. A standard hydrazine (phenylhydrazine) was chosen to react with these; in limited cases we tested other hydrazines as well. Rates were measured under pseudo-first-order conditions by following changes in absorption, with aldehydes at 125 μM - 1 mM and hydrazines at 2.5 – 20 μM. In general, fits were very good (see SI), although a few examples showed some curvature at later time points, indicating a deviation from strictly second-order behavior.
Table 1.
Rates of reaction of varied carbonyl compounds with phenyllydrazine in aqueous buffer at pH 7.4.a
| Substrate | k1(obs) (min−1) | k2(app) (M−1sec−1) | krel | Substrate | k1(obs) (min−1) | k2(app) (M−1sec−1) | krel | ||
|---|---|---|---|---|---|---|---|---|---|
| Aryl aldehydes | |||||||||
| 1 |
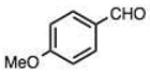
|
0.0020 (0.0005) | 0.033 (0.008) | 1 | 11 |
|
0.20 (0.06) | 3.4 (1.0) | 100 |
| 2 |
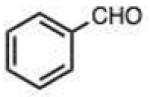
|
0.009 (0.004) | 0.15 (0.06) | 4.5 | 12 |
|
0.076 (0.032) | 1.3 (0.6) | 39 |
| 3 |
|
0.007 (0.002) | 0.12 (0.03) | 3.6 | 13 |
|
0.030 (0.002) | 0.50 (0.03) | 15 |
| 4 |
|
0.009 (0.004) | 0.15 (0.07) | 4.5 | 14 |
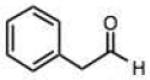
|
0.026 (0.002) | 0.43 (0.03) | 13 |
| 5 |
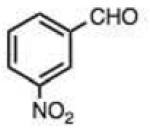
|
0.009 (0.002) | 0.15 (0.04) | 4.3 | Ketones | ||||
| 6 |
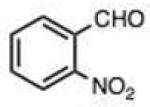
|
0.005 (0.003) | 0.09 (0.04) | 2.8 | 15 |
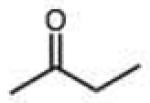
|
0.013 (0.004) | 0.22 (0.07) | 6.7 |
| 7 |
|
0.013 (0.008) | 0.22 (0.13) | 6.7 | 16 |
|
0.017 (0.006) | 0.28 (0.10) | 8.6 |
| 8 |
|
0.008 (0.001) | 0.13 (0.02) | 3.9 | 17 |
|
0.034 (0.002) | 0.57 (0.03) | 17 |
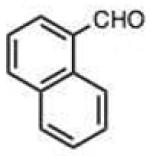
| 9 |
|
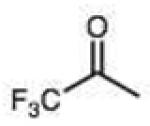
0.006 (0.002) | 0.10 (0.03) | 3.0 | 18 |
|
0.046 (0.006) | 0.77 (0.10) | 23 |
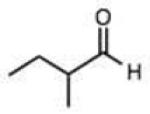
| Alkyl aldehydes | 19 |
|
0.022 (0.010) | 0.37 (0.17) | 11 | ||||
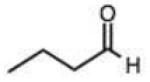
| 10 |
|
0.58 (0.14) | 9.7 (2.3) | 290 | |||||
Conditions: 137 mM NaCl, 2.7 mM KCl, 10 mM phosphate, 25 °C. Values measured 3–6 times and averaged (std. dev. in parentheses). Pseudo-first-order k(obs) normalized to standard 1000 μM aldehyde concentration.
Table 2.
Rates of reaction of aldehydes and ketones with phenyllydrazine and other hydrazines in aqueous buffer at pH 7.4.a
| Substrate | k(obs) (min−1) | k2(app) (M−1sec−1) | krel | Substrate | k(obs) (min−1) | k2(app) (M−1sec−1) | krel | ||
|---|---|---|---|---|---|---|---|---|---|
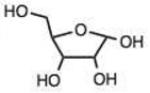
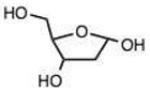
| Carbohydrates | Bulky hydrazine (ϕ2NNK2) | ||||||||
| 20 |
|
0.013 (0.003) | 0.22 (0.02) | 6.7 | 32 |
|
0.051 (0.024) | 0.85 (0.13) | 25 |
| 21 |
|
0.011 (0.001) | 0.18 (0.02) | 5.2 | 33 |
|
0.024 (0.012) | 0.40 (0.27) | 12 |
| 22 |
|
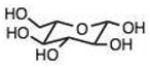
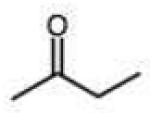
0.032 (0.002) | 0.53 (0.03) | 16 | 34 |
|
0.009 (0.003) | 0.15 (0.05) | 4.5 |
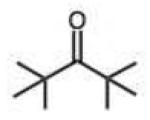
| Acid/Base substituents | 35 |
|
0.010 (0.005) | 0.17 (0.08) | 5.2 | ||||
| 23 |
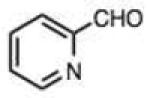
|
0.045 (0.025) | 0.75 (0.42) | 23 | 36 |
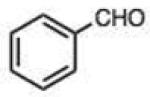
|
0.007 (0.001) | 0.12 (0.01) | 3.6 |
| 24 |
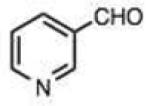
|
0.010 (0.002) | 0.16 (0.02) | 4.8 | 2-Carboxyphenylhydrazine | ||||
| 25 |
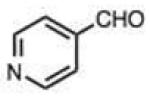
|
0.014 (0.001) | 0.24 (0.02) | 7.1 | 37 |
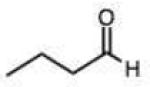
|
1.5 (0.2) | 24 (3) | 710 |
| 26 |
|
0.022 (0.006) | 0.37 (0.10) | 11 | 38 |
|
0.021 (0.004) | 0.35 (0.07) | 10 |
| 27 |
|
0.018 (0.006) | 0.30 (0.10) | 9.0 | 39 |
|
0.077 (0.006) | 1.3 (0.1) | 39 |
| 28 |
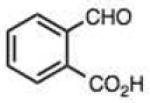
|
0.036 (0.012) | 0.58 (0.25) | 17 | 40 |
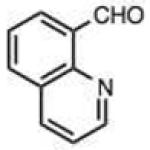
|
0.11 (0.02) | 1.9 (0.4) | 57 |
| 29 |
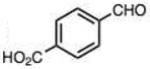
|
0.025 (0.001) | 0.42 (0.02) | 12 | 41 |
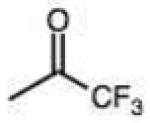
|
0.16 (0.03) | 2.7 (0.5) | 81 |
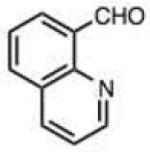
| 30 |
|
0.050 (0.004) | 0.83 (0.07) | 25 | 42 |
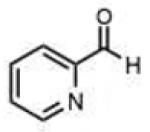
|
0.14 (0.03) | 2.4 (0.4) | 73 |
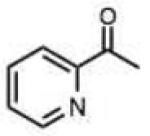
| 31 |
|
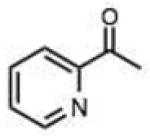
0.036 (0.010) | 0.60 (0.17) | 18 | 43 |
|
0.16 (0.02) | 2.7 (0.4) | 81 |
Conditions as in Table 1. Values measured 3–6 times and averaged (standard deviations in parentheses). Pseudo-first-ordcr k(obs) normalized to the standard 1 mM aldehyde concentration.
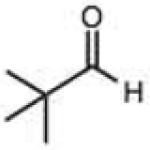
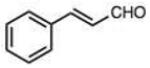
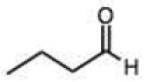
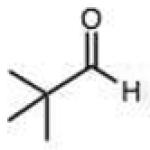
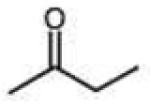
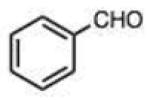
Structures and rate constants are given in Tables 1 and 2. Examination of the whole set shows some broad trends, and identifies specific features of interest that affect reaction rates very significantly. First, among all substrates, simple alkyl aldehydes are the fastest reactants. Indeed, the three fastest-reacting carbonyl compounds in the study are butyraldehyde, 2-methylbutyraldehyde and pivaldehyde (entries 10–12, Table 1). Butyraldehyde, the fastest carbonyl substrate overall, formed a hydrazone 65-fold more rapidly than did benzaldehyde (entry 2). It seems likely that the faster reaction of alkyl carbonyls compared to aryl substrates is due to the conjugation in the aryl cases, which is disrupted upon formation of the tetrahedral intermediate. This is also consistent with the observation that cinnamaldehyde reacts 6-fold more slowly than phenylacet-aldehyde. A second general observation is that aldehydes are in most cases faster than comparable ketones (e.g. butyraldehyde is faster than 2-butanone by 44-fold and pivaldehyde is 2.3-fold faster than di-t-butylketone (entries 17, 19). One exception to this general rule is acetophenone (entry 20), which reacted 2.4-fold more rapidly than benzaldehyde. Overall, the two fastest-reacting ketones in the study were 2-acetylpyridine and 1,1,1-trifluoroacetone (entries 18, 31), which may be due in part to their more electron-deficient character.
On this topic, it has long been known that electron-with-drawing groups can increase reactivity of aldehydes in hydrazone formation.7d,17 However, the overall effect has been found to be small (Jencks observed a Hammett ρ value of 0.91 at pH 1.75).17 In our experiments at pH 7.4 we observed a general trend favoring electron-deficient aldehydes and ketones over electron-rich ones. For example, the slowest-reacting compound in the study was 4-methoxybenzaldehyde (entry 1), while 4-nitrobenzaldehyde reacted 4.5-fold more rapidly. Similarly, trifluoroacetone formed a hydrazone 3.4-fold more rapidly than 2-butanone, which could also be attributed to the electron deficiency of the former. However, this effect did not always hold; for example, dimethoxyacetaldehyde reacted 8-fold more slowly than 2-methylbutyraldehyde (compare entries 11,13).
Carbohydrates are of special interest for hydrazone formation because they naturally contain reactive ketone or aldehyde functional groups, which can be useful in labeling and bioconjugation.18 Interestingly, the three aldoses tested here – ribose, deoxyribose, and glucose - reacted at reasonably good rates despite the fact that they exist largely in the hemiacetal state in solution. Ribose and deoxyribose displayed rates similar to that of benzaldehyde, while glucose reacted 3.5-fold more rapidly, and was similar in reactivity to an electron-deficient alkyl aldehyde (entry 13). Reactions of anomeric carbons in deoxyribose are of interest in the development of reagents for detecting abasic damage in DNA,18a and reactions of hexoses are useful in making bioconjugates,18b and the current results show that such reactions can proceed with moderate to good rates.
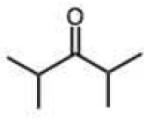
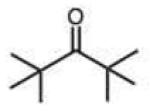
Steric effects could potentially have significant effects on the rate of hydrazone formation, by blocking approach of the hydrazine nucleophile to the carbonyl carbon. However, the rate of breakdown of the tetrahedral intermediate might well be enhanced by relief of steric crowding. We are unaware of any prior literature studies of steric effects on hydrazone or oxime formation. In the current study, steric effects with the phenylhydrazine standard reactant are found to be small and inconsistent. For example, we observed a 7.5-fold decrease in rate for the reaction of pivaldehyde relative to butyraldehyde (Table 1). Interestingly, however, in the ketone series, di-t-butyl-ketone reacts faster than diisopropyl ketone, which in turn reacts faster than 2-butanone (entries 15–17), showing a significant enhancement by alkyl substitution. Use of the more bulky diphenylhydrazine (entries 32–36) did not result in magnified effects on rate. Overall we conclude that steric effects are generally moderate for this reaction, and that increased substitution is not always detrimental to rate, especially for ketones.
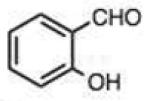
Finally, we explored the effects of substituting acid/base groups near the reactive center on the rate of reaction. It was observed several decades ago that ortho-substitution in benzaldehydes could speed hydrazone formation, although a satisfactory explanation for the effect was elusive.19 Pyridoxal (which has an ortho hydroxy group) has long been known as an efficient reactant for imine and hydrazone formation,20 and a recent report employed rapidly-reacting pyridoxal phosphoramide derivatives in bioconjugations.12 In our recent study, we observed that 2-carboxybenzaldehyde reacted more rapidly than the 4-isomer;9c we proposed that the acid group might donate a proton at the transition state, acting as an intramolecular catalyst in the reaction.
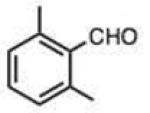
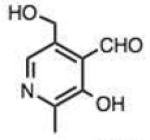
To address this hypothesized self-catalytic effect, we tested several compounds with imino, hydroxy, or carboxy groups substituted near the reactive carbonyl (Table 2, entries 23–31 and Fig. S4). Significantly, all cases were found to react more rapidly at pH 7.4 than do control compounds that either lack the group or have it substituted remote from the carbonyl. Imino groups (as pyridine derivatives) and ortho carboxy groups had a similar effects. Ortho hydroxy groups accelerate reaction by 2–4-fold (see entries 26,27 relative to benzaldehyde). Finally, the greatest effect occurred with the imino group of quinoline-8-carboxaldehyde (entry 30), which accelerates the rate by a sizeable factor of 8.3 relative to 1-naphthaldehyde (entry 9). Notably, while pyridoxal is a moderately fast reactant in the aromatic carbonyl series, both this new quinoline aldehyde substrate and 2-acetylpyridine are considerably faster, and thus these latter two are prime candidates for further development in bioconjugation reactions.
The rate-limiting step for hydrazone formation at neutral pH is the breakdown of the tetrahedral intermediate to eliminate water.7b Thus, catalysis of the reaction could occur by intramolecular transfer of a proton to the leaving hydroxy group.9c We tentatively propose that an arylhydroxy group (pKa~9–10 in the substrates tested here), a pyridine-type imino group (pKa ~5.2), or a carboxy group (pKa ~4.2) could achieve proton transfer via 5-, 6-, or 7-membered ring transition states (Fig. 2). More studies will be needed to test this mechanistic hypothesis, but the results suggest that careful tuning of pKa and geometry in the future could result in new and yet faster carbonyl substrates for this reaction.
Figure 2.
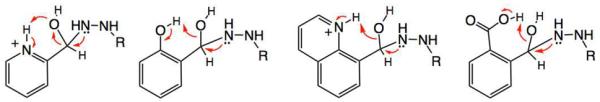
Proposed mechanism for rate enhancement by acid/base groups near the carbonyl center. Intramolecular protonation of the leaving group speeds breakdown of the tetrahedral intermediate.
Spurred by this finding, we carried out preliminary studies to investigate whether a similar self-catalytic effect might be observed in a hydrazine reactant. 2-Carboxyphenylhydrazine was reacted with selected aldehydes and ketones (Table 2 entries 37–43), and rates were compared to those with phenylhydrazine. In every case the reaction was significantly faster with the 2-carboxy-substituted hydrazine (Fig. S5). The rate enhancement by the carboxy group varied from a factor of 1.6-fold (in the reaction with 2-butanone) to 8.7-fold (benzaldehyde). With the quinoline substrate, already enhanced by the nearby imino group, we observed a further 2.3-fold rate acceleration in reaction with the 2- carboxyphenylhydrazine derivative (Fig. 3). Importantly, the rate constant for reaction of this pair of enhanced substrates was 1.9 M−1sec−1, which is faster than many recently developed bioorthogonal bond-forming reactions. Similarly high rates and enhancements were also seen for ketone substrates (entries 38,41,43). Remarkably, the rate constant for the fastest substrate in this study, butyraldehyde, with this enhanced hydrazine was 24 M−1sec−1, which is an order of magnitude more rapid than the rates of strain-promoted azide-alkyne cycloadditions and a tetrazinenorbornene addition/rearrangement.3,4b Thus although hydrazone formation has been generally characterized as slow at neutral pH, it is clear that specialized hydrazine and carbonyl substrates can undergo very rapid reactions.
Figure 3.
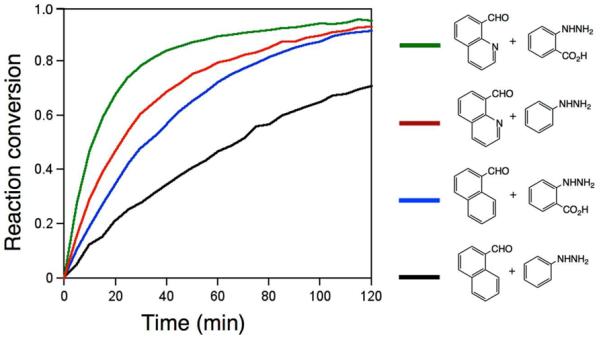
Time course plots for four related hydrazone-forming reactions, showing favorable effects of the imino group in the quinoline electrophile and of a carboxy group in the arylhydrazine nucleophile. Conditions as in Table 1, with 500 μM aldehyde and 10 μM hydrazine.
We conclude that with careful choice of carbonyl and hydrazine substrate structure, hydrazone formation can be rapid at biological pH even in the absence of a catalyst. Future studies will test whether additional structural tuning can yield yet faster hydrazone-forming reactants. With the advent of such rapid substrates, there are multiple reasons why this reaction may become more broadly useful, including ease of synthesis, water solubility of reactants, and facile introduction into biomolecules.
Supplementary Material
ACKNOWLEDGMENT
We thank the National Institutes of Health (GM068122) for support.
Footnotes
Supporting Information Available. Experimental details, kinetic traces and plots. This information is available free of charge via the internet at http://pubs.acs.org.
References
- (1).(a) Kalia J, Raines RT. Curr. Org. Chem. 2010;14:138. doi: 10.2174/138527210790069839. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Lim RK, Lin Q. Chem. Commun. 2010;46:1589. doi: 10.1039/b925931g. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Jewett JC, Bertozzi CR. Chem. Soc. Rev. 2010;39:1272. doi: 10.1039/b901970g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).(a) Kolb HC, Finn MG, Sharpless KB. Angew. Chem., Int. Ed. 2001;40:2004. doi: 10.1002/1521-3773(20010601)40:11<2004::AID-ANIE2004>3.0.CO;2-5. [DOI] [PubMed] [Google Scholar]; (b) Moses JE, Moorhouse AD. Chem. Soc. Rev. 2007;36:1249. doi: 10.1039/b613014n. [DOI] [PubMed] [Google Scholar]; (c) Mamidyala SK, Finn MG. Chem. Soc. Rev. 2010;39:1252. doi: 10.1039/b901969n. [DOI] [PubMed] [Google Scholar]; (d) El-Sagheer AH, Brown T. Chem. Soc. Rev. 2010;39:1388. doi: 10.1039/b901971p. [DOI] [PubMed] [Google Scholar]; (e) Lallana E, Riguera R, Fernandez-Megia E. Angew. Chem., Int. Ed. 2011;50:8794. doi: 10.1002/anie.201101019. [DOI] [PubMed] [Google Scholar]
- (3).(a) Baskin JM, Prescher JA, Laughlin ST, Agard NJ, Chang PV, Miller IA, Lo A, Codelli JA, Bertozzi CR. Proc. Natl. Acad. Sci. USA. 2007;104:16793. doi: 10.1073/pnas.0707090104. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Ning XH, Guo J, Wolfert MA, Boons G-J. Angew. Chem., Int. Ed. 2008;47:2253. doi: 10.1002/anie.200705456. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Jewett JC, Sletten EM, Bertozzi CR. J. Am. Chem. Soc. 2010;132:3688. doi: 10.1021/ja100014q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).(a) Blackman ML, Royzen M, Fox JM. J. Am. Chem. Soc. 2008;130:13518. doi: 10.1021/ja8053805. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Devaraj NK, Upadhyay R, Haun JB, Hilderbrand SA, Weissleder R. Angew. Chem., Int. Ed. 2009;48:7013. doi: 10.1002/anie.200903233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Gress A, Völkel A, Schlaad H. Macromolecules. 2007;40:7928. [Google Scholar]
- (6).(a) Saxon E, Bertozzi CR. Science. 2000;287:2007. doi: 10.1126/science.287.5460.2007. [DOI] [PubMed] [Google Scholar]; (b) Saxon E, Armstrong JI, Bertozzi CR. Org. Lett. 2000;2:2141. doi: 10.1021/ol006054v. [DOI] [PubMed] [Google Scholar]
- (7).(a) Jencks WP. Progr. Phys. Org. Chem. 1964;2:63. [Google Scholar]; (b) Jencks WP. J. Am. Chem. Soc. 1959;81:475. [Google Scholar]; (c) Cordes EH, Jencks WP. J. Am. Chem. Soc. 1962;84:826. [Google Scholar]; (d) Cordes EH, Jencks WP. J. Am. Chem. Soc. 1962;84:4319. [Google Scholar]
- (8).(a) Tam JP, Xu J, Eom KD. Biopolymers. 2001;60:194. doi: 10.1002/1097-0282(2001)60:3<194::AID-BIP10031>3.0.CO;2-8. [DOI] [PubMed] [Google Scholar]; (b) Cornish VW, Hahn KM, Schultz PG. J. Am. Chem. Soc. 1996;118:8150. [Google Scholar]; (c) Zeng Y, Ramya TNC, Dirksen A, Dawson PE, Paulson JC. Nat. Methods. 2009;6:207. doi: 10.1038/nmeth.1305. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Dirksen A, Dirksen S, Hackeng TM, Dawson PE. J. Am. Chem. Soc. 2006;128:15602. doi: 10.1021/ja067189k. [DOI] [PubMed] [Google Scholar]
- (9).(a) Crisalli P, Hernández AR, Kool ET. Bioconjugate Chem. 2012;23:1969. doi: 10.1021/bc300344b. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Crisalli P, Kool ET. J. Org. Chem. 2013;78:1184. doi: 10.1021/jo302746p. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Crisalli P, Kool ET. Org. Lett. 2013;15:1646. doi: 10.1021/ol400427x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Kalia J, Raines RT. Angew. Chem., Int. Ed. 2008;47:7523. doi: 10.1002/anie.200802651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Rashidian M, Mahmoodi MM, Shah R, Dozier JK, Wagner CR, Distefano MD. Bioconjugate Chem. 2013;24:333. doi: 10.1021/bc3004167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Wang X, Canary JW. Bioconjugate Chem. 2012;23:2329. doi: 10.1021/bc300430k. [DOI] [PubMed] [Google Scholar]
- (13).(a) Brinkhuis RP, de Graaf F, Hansen MB, Visser TR, Rutjes FPJT, van Hest JCM. Polym. Chem. 2013;4:1345. [Google Scholar]; (b) Lazny R, Nodzewska A, Sienkiewicz M, Wolosewicz K. J. Comb. Chem. 2005;7:109. doi: 10.1021/cc049874y. [DOI] [PubMed] [Google Scholar]
- (14).(a) Sanders JK. Philos. Trans. A. 2004;362:1239. doi: 10.1098/rsta.2004.1376. [DOI] [PubMed] [Google Scholar]; (b) Gromova AV, Ciszewski JM, Miller BL. Chem. Commun. 2012;48:2131. doi: 10.1039/c2cc17192a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).(a) Dirksen A, Dawson PE. Bioconjugate Chem. 2008;19:2543. doi: 10.1021/bc800310p. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Wu B, Wang Z, Huang Y, Liu WR. ChemBioChem. 2012;13:1405. doi: 10.1002/cbic.201200281. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Rayo J, Amara N, Krief P, Meijler MM. J. Am. Chem. Soc. 2011;133:7469. doi: 10.1021/ja200455d. [DOI] [PubMed] [Google Scholar]
- (16).Kennedy DC, McKay CS, Legault MC, Danielson DC, Blake JA, Pegoraro AF, Stolow A, Mester Z, Pezacki JP. J. Am. Chem.Soc. 2011;133:17993. doi: 10.1021/ja2083027. [DOI] [PubMed] [Google Scholar]
- (17).Anderson BM, Jencks WP. J. Am. Chem. Soc. 1960;82:1773. [Google Scholar]
- (18).(a) Kubo K, Ide H, Wallace SS, Kow YW. Biochemistry. 1992;31:3703. doi: 10.1021/bi00129a020. [DOI] [PubMed] [Google Scholar]; (b) Beaudette TT, Cohen JA, Bachelder EM, Broaders KE, Cohen JL, Engleman EG, Fréchet JM. J. Am. Chem. Soc. 2009;131:10360. doi: 10.1021/ja903984s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Wolfenden R, Jencks WP. J. Am. Chem. Soc. 1961;83:2763. [Google Scholar]
- (20).French TC, Auld DS, Bruice TC. Biochemistry. 1965;4:77. doi: 10.1021/bi00877a014. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.