

Abstract
Kisspeptin receptor (KISS1R) signaling plays a critical role in the regulation of reproduction. We investigated the role of kisspeptin-stimulated KISS1R internalization, recycling, and degradation in the modulation of KISS1R signaling. Kisspeptin stimulation of Chinese hamster ovary or GT1–7 cells expressing KISS1R resulted in a biphasic increase in intracellular Ca2+ ([Ca2+]i), with a rapid acute increase followed by a more sustained second phase. In contrast, stimulation of the TRH receptor, another Gq/11-coupled receptor, resulted in a much smaller second-phase [Ca2+]i response. The KISS1R-mediated second-phase [Ca2+]i response was abolished by removal of kisspeptin from cell culture medium. Notably, the second-phase [Ca2+]i response was also inhibited by dynasore, brefeldin A, and phenylarsine oxide, which inhibit receptor internalization and recycling, suggesting that KISS1R trafficking contributes to the sustained [Ca2+]i response. We further demonstrated that KISS1R undergoes dynamic ligand-dependent and -independent recycling. We next investigated the fate of the internalized kisspeptin-KISS1R complex. Most internalized kisspeptin was released extracellularly in degraded form within 1 hour, suggesting rapid processing of the internalized kisspeptin-KISS1R complex. Using a biotinylation assay, we demonstrated that degradation of cell surface KISS1R was much slower than that of the internalized ligand, suggesting dissociated processing of the internalized kisspeptin-KISS1R complex. Taken together, our results suggest that the sustained calcium response to kisspeptin is dependent on the continued presence of extracellular ligand and is the result of dynamic KISS1R trafficking.
Our understanding of the central neuroendocrine regulation of reproductive development and function has undergone major advances since the discovery of the important role of kisspeptin and its receptor, KISS1R, in the control of GnRH secretion. KISS1R-deficient mice display a phenotype of hypogonadotropic hypogonadism, parallel to the phenotype observed in patients harboring mutations in KISS1R (1). The major ligand for KISS1R is a 54-amino acid peptide (referred to as kisspeptin-54 [KP54]), corresponding to residues 68 to 121 of the KISS1 gene product (2). Further proteolytic processing of KP54 results in the production of shorter peptides, namely KP14, KP13, and KP10, which retain biologic activity (2, 3). KISS1R, also known as GPR54, is a G protein–coupled receptor (GPCR) coupled to Gq/11, stimulating phospholipase C to cleave phosphatidylinositol 4,5-bisphosphate into inositol 1,4,5-trisphosphate and diacylglycerol and leading to increased [Ca2+]i (4–6). KISS1R activation also stimulates GnRH neuronal depolarization by activation of a transient receptor potential cation channel and inhibition of an inwardly rectifying potassium channel (Kir) (7, 8). Although KISS1R signaling has begun to be decoded, precise information on the signal transduction pathways, regulation, and desensitization remains incomplete. Similarly, the nature and molecular mechanisms of KISS1R trafficking and degradation are largely unknown.
GnRH secretion is the consequence of increases in intracellular calcium concentrations ([Ca2+]i) in GnRH neurons (9). Spontaneous [Ca2+]i oscillations present in prenatal GnRH neurons derived from mouse nasal explants were increased by KP10 (10). This response was not completely abolished by either tetrodotoxin, a voltage-activated sodium channel inhibitor, or cadmium, a nonselective calcium channel blocker, suggesting that intracellular calcium release contributes to the kisspeptin-induced increases in [Ca2+]i (10). Intriguingly, sustained responses to kisspeptin, recorded by either membrane depolarization or increases in [Ca2+]i, were observed in the persistent presence of KP10 or in some cases even after its removal (8, 10–12). Consistent with these in vitro studies, more recent human studies have shown that iv infusion of KP10 in healthy men stimulates a sustained increase in pulsatile LH secretion (13, 14). The underlying mechanisms, however, remain unclear.
Persistent signaling has been observed for Gs-coupled GPCRs such as TSH receptor (TSHR) and parathyroid hormone receptor (15, 16). Earlier studies suggested that PTH- and TSH-stimulated persistent cAMP signaling was dependent on receptor internalization (15, 16), although a subsequent study suggested that sustained cAMP signaling could occur independently of TSHR internalization (17). To our knowledge, there have been no reports suggesting a relationship between persistent signaling by Gq/11-coupled GPCRs and receptor trafficking. Our previous study showed ligand- and time-dependent internalization of KISS1R (18). In light of the previous reports of sustained kisspeptin signaling in vitro (8, 10–12) and in vivo (13, 14), here we have investigated the possible coupling of kisspeptin signaling with KISS1R trafficking.
A common consequence of GPCR activation is down-regulation of the receptors. Generally, after internalization, GPCRs are sorted between divergent pathways (19). Recycling back to the cell surface results in resensitization, whereas trafficking to lysosomes is typically thought to enhance receptor down-regulation and desensitization. We hypothesized that sustained kisspeptin signaling may be the result of rapid recycling of KISS1R, slow degradation of KISS1R, and/or rapid synthesis of new KISS1R. Herein, we show that KP10 stimulates a biphasic increase in [Ca2+]i, with a rapid acute increase followed by a more sustained second-phase response. Removal of KP10 abolished the second-phase response, demonstrating that this sustained [Ca2+]i response is ligand-dependent. Our data also show that KISS1R undergoes both ligand-dependent and -independent dynamic cellular trafficking between the cell surface and intracellular pools, with rapid processing of internalized ligand but slow degradation of internalized KISS1R. Importantly, inhibition of receptor internalization abolished sustained KISS1R calcium signaling. Taken together, our data demonstrate that sustained KISS1R calcium signaling requires continued KP10 exposure and dynamic KISS1R trafficking.
Materials and Methods
Reagents
Anti-myc and horseradish peroxidase (HRP)–conjugated anti-myc antibodies were from Millipore and Invitrogen, respectively; radioisotopes were from Perkin-Elmer, and cell culture medium was from Mediatech, Inc. Kisspeptin-10 (KP10) was synthesized by the Tufts University Core Facility. All other chemicals including dynasore, phenylarsine oxide (PAO), and brefeldin A (BFA) were from Sigma-Aldrich.
Expression vectors and stable cell lines
Construction of the human KISS1R vector and the generation of Chinese hamster ovary (CHO)-KISS1R cells was described previously (1, 20). Myc-KISS1R was subcloned from the previously constructed myc-KISS1R-PCS2+ (18) into the pcDNA3 expression vector. Human embryonic kidney (HEK)-myc-KISS1R and GT1–7-myc-KISS1R stable cell lines were generated by neomycin selection of HEK and GT1–7 cells after transient transfection with myc-KISS1R-pcDNA3. CHO cells stably expressing the TRH receptor (CHO-TRHR) were a generous gift from Dr. Patricia Hinkle (University of Rochester Medical Center, Rochester, New York) (21).
Calcium assays
CHO-KISS1R and GT1–7-myc-KISS1R cells (1 × 104/well) were plated into black-walled, clear-bottomed 96-well plates. The next day, the cells were washed with DME/F12 and incubated in serum-free DME/F12 for 2 hours at 37°C. The cells were then treated with KP10 for the times indicated, and a Fluo-4 Direct Kit (Invitrogen) was used to measure intracellular calcium according to the manufacturer's protocol. CHO-TRHR cells were treated with TRH (Sigma-Aldrich). Parental CHO and GT1–7 cells were also treated with KP10 or TRH as a negative control, and there was no response to either KP10 or TRH stimulation (data not shown). The change in fluorescence intensity before and after addition of ligand was measured using a POLARstar OPTIMA multifunction plate reader (BMG Labtech). In some experiments, dynasore or PAO was added simultaneously with KP10 or TRH, whereas BFA was added 30 minutes before KP10 or TRH stimulation. In experiments in which KP10 was removed, cells were washed 5 times with assay buffer containing DME/F12 plus Fluo-4, 5 minutes after KP10 treatment. In KP10 control cells, cells were washed in the same way as described above except that KP10 remained in the washing buffer. Calcium measurements were made 5 seconds after addition of KP10 and then repeated every 10 to 30 seconds for 20 to 120 cycles. The values shown represent the ratio of the difference between KP10-stimulated and basal values divided by the basal value (relative fluorescence units [RFU]) or percentage of the maximum response at each time point.
Ligand internalization assay
Ligand internalization was measured in CHO-KISS1R cells using radiolabeled KP10. The methods used to measure the internalization of 125I-KP10 were adapted from assays described previously (22). In brief, determinations of the rates of internalization were performed using at least 5 different data points collected at 3- to 10-minute intervals after the addition of 0.05 nM 125I-KP10. The radioactivity of the samples was counted by a Beckman γ-counter. The endocytotic rate constant (Ke) was calculated from the slope of the line obtained by plotting the internalized radioactivity against the integral of the surface-bound radioactivity. The half-time of internalization is defined as 0.693/Ke (23–25).
KISS1R ELISA for measurement of ligand-dependent KISS1R trafficking
The use of an ELISA to study receptor trafficking was performed as described previously (26), with minor modifications. HEK-myc-KISS1R cells seeded in 0.1% gelatin-coated 24-well plates were incubated in serum-free DMEM containing 1% BSA with or without 10−7 M KP10 for 20 minutes. Some cells were then washed 5 times with DMEM containing 1% BSA and then reincubated at 37°C for 20 minutes. After incubation, the cells were washed once with PBS and fixed by incubation with 2% paraformaldehyde for 20 minutes at room temperature. The cells were then washed 3 times with PBS and preincubated in PBS containing 5% milk for 60 minutes. After preincubation, the cells were incubated with 1:2000 HRP-conjugated anti-myc antibody (Invitrogen) for 90 minutes at room temperature. The cells were then washed 3 times with PBS and incubated with 3,3′,5,5′-tetramethylbenzidine solution (Sigma-Aldrich) for 5 to 10 minutes. The reaction was terminated with stop reagent for 3,3′,5,5′-tetramethylbenzidine (Sigma-Aldrich). Then 300 μL was transferred to a 96-well plate, and the absorbance was read at 450 nm on a microplate reader (Bio-Rad Laboratories).
Measurement of constitutive KISS1R trafficking
CHO-KISS1R cells were washed with ice-cold PBS and treated with or without 0.25% trypsin at 4°C for 30 minutes. Cells were then washed 3 times with DME/F12 containing 10% FBS to inactivate the trypsin and incubated at 37°C with prewarmed DME/F12 containing 10% FBS for the times indicated. To measure the amount of cell surface KISS1R, the cells were incubated with 0.05 nM 125I-KP10 together with 1 nM cold KP10 at 4°C for 1 hour. After incubation, the cells were washed 3 times with DME/F12 containing 0.5% BSA and lysed with 1 M NaOH. The lysates were counted as described above.
Determination of the fate of internalized ligand
The fate of internalized ligand was determined using modifications of procedures published previously (24, 27). CHO-KISS1R cells were washed and incubated with 0.05 nM 125I-KP10 in DME/F12 medium containing 0.5% BSA (assay medium) for 1 hour at 37°C to allow the internalization of the 125I-KP10/KISS1R complex. The cells were then placed on ice and washed 3 times with 1-mL portions of ice-cold PBS to remove the 125I-KP10 remaining in the assay medium. Surface-bound 125I-KP10 was released by incubation of the cells in 1 mL of cold acidic wash medium containing 50 mM glycine and 150 mM NaCl, pH 3, for 2 to 4 minutes on ice (28). This buffer was removed, and the cells were washed once more with the same acidic buffer and then once with cold assay medium. The cells were placed back in 1 mL of warm assay medium containing 1 μM unlabeled KP10 (to prevent the reassociation of any intact 125I-KP10 released from the cells with cell surface KISS1R), and a second incubation at 37°C was conducted for various times to allow the cells to process the ligand that had been internalized during the first incubation. At the end of the second incubation, the dishes were placed on ice, the medium was saved, and the cells were washed once with 2 mL of cold assay medium. These 2 media collections were combined and precipitated with 20% trichloroacetic acid (TCA) to determine the amounts of intact and degraded 125I-KP10 released (28). As a positive control to confirm that KP10 is able to be precipitated by TCA, intact 125I-KP10 in binding medium (DME/F12 with 0.5% BSA) was incubated with TCA; 89% of intact 125I-KP10 was precipitated (data not shown). Finally, surface-bound 125I-KP10 was released by incubation of the cells in 1 mL of cold acidic wash medium for 2 to 4 minutes on ice and repeated once. The cells were then lysed with NaOH, and lysates were collected. The amount of radioactivity in each sample was counted using a Beckman γ-counter.
Measurement of KISS1R degradation
HEK-myc-KISS1R cells plated in 6-well plates were incubated in serum-free DMEM for 2 hours before treatment. Cells were pretreated with 10 μM cycloheximide (CHX) for 10 minutes and then treated with 10−7 M KP10 for the indicated times. The cells were then placed on ice, washed 3 times with ice-cold PBS, and lysed at 4°C by radioimmunoprecipitation assay buffer containing 50 mM Tris-HCl, pH 8.0, with 150 mM sodium chloride, 1.0% NP-40 (Igepal CA-630), 0.5% sodium deoxycholate, and 0.1% SDS plus protease inhibitor (cOmplete, EDTA-free; Roche). Then 10 μg of each of the lysates was resolved on SDS gels, followed by electrophoretic transfer to nitrocellulose membranes. After blocking with 5% milk for 1 hour, the blots were incubated overnight with HRP-conjugated anti-myc antibody (1:5000; Invitrogen), and the proteins were visualized using the SuperSignal West Femto maximum sensitivity system (Thermo Fisher Scientific).
Analysis of KISS1R turnover using a surface biotinylation assay
HEK-myc-KISS1R cells plated in gelatin-coated 100-mm wells were washed 3 times with ice-cold PBS, pH 8.0, and then were biotinylated during a 30-minute incubation at 4°C with a freshly prepared 0.5 mg/mL solution of EZ-Link Sulfo-NHS-LC-LC-Biotin (Thermo Fisher Scientific) in the same PBS buffer. After incubation, the cells were washed once with DMEM and then incubated with DMEM for 15 minutes on ice to quench the free biotin (29). Some cells were saved and processed immediately (t = 0 samples), whereas others were incubated in DMEM at 37°C for the times indicated. The cells were then placed on ice, washed with ice-cold PBS, lysed in radioimmunoprecipitation assay buffer, and immunoprecipitated with an anti-myc antibody (Millipore) prebound to protein G–agarose (Invitrogen). The immunoprecipitates were resolved on SDS-PAGE gels, and the resolved proteins were electrophoretically transferred to nitrocellulose membranes. After blocking with 5% BSA in Tris-buffered saline/0.05% Tween 20 (pH 7.4), the blots were incubated for 1 hour with 100 ng/mL HRP-conjugated streptavidin (Thermo Fisher Scientific) at room temperature, and the proteins were visualized using the SuperSignal West Femto maximum sensitivity system.
Statistical analyses
Statistical analyses were performed using Prism software (GraphPad Software, Inc.). One-way or two-way ANOVA followed by post hoc Bonferroni multiple comparison tests was performed to determine statistical significance. Significant difference is defined as a value of P < .05.
Results
Kisspeptin induces a biphasic intracellular calcium response in CHO-KISS1R cells
To evaluate kisspeptin-stimulated KISS1R signaling, we measured the dynamic [Ca2+]i response to kisspeptin stimulation in CHO-KISS1R cells. We demonstrated a dose-dependent, biphasic [Ca2+]i response to kisspeptin stimulation, with an acute first-phase [Ca2+]i response that was 5 minutes in duration, followed by a longer, more sustained second-phase response lasting more than 30 minutes (Figure 1A). Increasing concentrations of kisspeptin were associated with higher amplitudes of both phases of the [Ca2+]i response. The duration of the second-phase response was also longer in response to higher concentrations of kisspeptin. In contrast, stimulation of CHO-TRHR cells with 10−7 M TRH resulted in a much smaller second-phase response (Figure 1B).
Figure 1.

KP10 stimulates a biphasic intracellular calcium response that requires the continued presence of extracellular KP10. A, CHO-KISS1R cells were treated with increasing concentrations (10−9–10−6 M) of KP10. [Ca2+]i was measured as RFU every 30 seconds for 30 to 60 minutes. A single representative experiment is shown, which was repeated 3 times with similar results. B, CHO-KISS1R and CHO-TRHR cells were treated with 10−7 M KP10 and 10−7 M TRH, respectively. [Ca2+]i was recorded every 30 seconds for 10 minutes and is depicted as RFU. A representative experiment is shown, which was repeated 3 times with similar results. C, CHO-KISS1R cells were treated with 10−8 M KP10. After 5 minutes, some cells (marked KP10 removal and KP10 removal + KP10) were washed 5 times with the same calcium assay medium to remove ligand. After the final wash, calcium assay medium without KP10 (for the KP10 removal group) or calcium assay medium containing 10−8 M KP10 (for the KP10 removal + KP10 group) was added. The total time for the washes was less than 20 seconds. After 30 minutes, the medium in the KP10 removal and KP10 removal + KP10 was again removed and replaced with medium containing 10−8 M KP10. D, GT1–7-myc-KISS1R cells were treated with 10−8 M KP10. After 5 minutes, some cells (marked KP10 removal and KP10 removal + KP10) were washed 5 times with the same calcium assay medium in the absence or presence of KP10 and further treated as described for C, except that the interval between treatments was 10 minutes. A representative plot of 3 independent experiments with similar results is shown.
To further characterize the second phase of the Ca2+ response to kisspeptin, the effects of kisspeptin removal from the cell culture medium after acute stimulation were studied. Interestingly, removal of the exogenous ligand from CHO-KISS1R abolished the sustained second-phase [Ca2+]i response (Figure 1C). These results indicate that this second phase is dependent on the continued presence of exogenous kisspeptin. Of note, readministration of 10−8 M KP10 30 minutes after its removal stimulated another acute first-phase Ca2+ response. This response did not occur if KP10 was readministered immediately after its removal (Figure 1C). Nonetheless, readministration of KP10 resulted in further prolongation of the sustained Ca2+ response. Taken together, these findings suggest that KP10 stimulation causes acute, rapid KISS1R desensitization and that the prolonged, exogenous KP10-dependent [Ca2+]i response may be the result of recruitment of naive KISS1R to the cell surface from intracellular pools.
To exclude the possibility that the prolonged [Ca2+]i response occurs only in heterologous cell lines, we tested the [Ca2+]i response to kisspeptin in a GnRH neuronal cell line, GT1–7 cells. These cells did not respond to kisspeptin with a detectable increase in [Ca2+]i, even at high concentrations of KP10 (data not shown). Therefore, a stable GT1–7 cell line expressing moderate levels of myc-KISS1R was generated. KP10 induced a biphasic [Ca2+]i response in GT1–7-myc-KISS1R cells, similar to that observed in CHO-KISS1R cells (Figure 1D). Removal of KP10 abolished the sustained second-phase [Ca2+]i response, whereas readministration of 10−8 M KP10, 10 minutes after its removal, stimulated another acute first-phase [Ca2+]i response (Figure 1D). This response did not occur if KP10 was readministered immediately after its removal, but further prolongation of the sustained Ca2+ response was observed. These results demonstrate that in both a heterologous cell line and a GnRH neuronal cell line, a sustained [Ca2+]i response is observed, which relies on continuous KP10 exposure.
Given the finding that the sustained Ca2+ response is dependent on the presence of exogenous kisspeptin, we hypothesized that dynamic KISS1R trafficking and recycling resulted in KISS1R resensitization to contribute to the sustained Ca2+ response. Our group had shown previously that KISS1R undergoes rapid desensitization and internalization, but, in addition, a dynamic pool of KISS1R is maintained at the cell surface, probably derived at least in part from the recycling of desensitized/resensitized receptors (18). To test our hypothesis, we first studied the role of KISS1R internalization in Ca2+ signaling. CHO-KISS1R cells were stimulated with kisspeptin in the presence or absence of dynasore, a dynamin inhibitor. Dynasore has been shown to effectively and rapidly inhibit clathrin-coated pit–mediated endocytosis (30, 31). Dynasore abolished the second-phase calcium response to kisspeptin, with little effect on the acute first-phase calcium response (Figure 2A). In addition, PAO, an endocytosis inhibitor, also blunted the second-phase calcium response to KP10 (Figure 2A). Similar effects of both dynasore and PAO on the second-phase calcium response to kisspeptin were observed in GT1–7-myc-KISS1R cells (Figure 2B). These data suggest that KISS1R internalization contributes to the KP10-stimulated second-phase calcium response in both the heterologous CHO cell line and in the GT1–7 GnRH neuronal cell line. To demonstrate that the inhibitory effect of dynasore on KP10-stimulated second-phase intracellular calcium response is specific, CHO-TRHR cells were treated with TRH with or without dynasore. In contrast to its effect on KISS1R signaling, dynasore had no significant effect on the TRH-stimulated intracellular calcium response in CHO-TRHR cells (Figure 2C).
Figure 2.
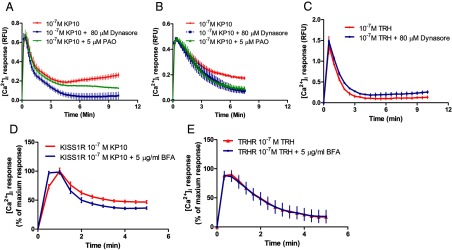
KISS1R trafficking is essential for the KP10-induced sustained second-phase calcium response. A–C, CHO-KISS1R (A), GT1–7-KISS1R (B), and CHO-TRHR (C) cells were treated with 10−7 M KP10 or 10−7 M TRH, respectively, in the presence or absence of 80 μM dynasore, a dynamin inhibitor, or 5 μM PAO, an endocytosis inhibitor, to block KISS1R internalization. The values shown are depicted as RFU. D and E, CHO-KISS1R cells were treated with 10−7 M KP10 in the presence or absence of 5 μg/mL BFA, a receptor recycling inhibitor. The [Ca2+]i responses were recorded every 10 to 30 seconds for 5 to 10 minutes. The values shown are depicted as percent maximum response. All experiments were repeated 3 times, each in triplicate. Means ± SEM are shown. Significant differences were present among/between the curves in A, B, and D, as determined by two-way ANOVA (P < .001).
To further test the contribution of receptor recycling to the sustained calcium response, BFA was used. BFA has been shown to inhibit recycling of melanocortin-2 receptor (32). Our data show that BFA inhibited the KP10-stimulated, KISS1R-mediated sustained [Ca2+]i response (Figure 2D) but had no effect on the TRH-stimulated, TRHR-mediated [Ca2+]i response (Figure 2E). As expected, KP10-stimulated KISS1R recycling was blunted by BFA, as determined by measurement of cell surface KISS1R levels in the presence vs absence of BFA (data not shown). Taken together, the findings suggest that inhibition of either KISS1R internalization or recycling inhibited the sustained [Ca2+]i response, strongly suggesting a critical role for dynamic KISS1R trafficking in the second-phase sustained [Ca2+]i response.
KISS1R undergoes dynamic kisspeptin-dependent and -independent trafficking
To further characterize KISS1R trafficking, the ligand-induced KISS1R internalization rate was measured and calculated using 125I-KP10 (Figure 3A). The acid-sensitive (cell surface) and -resistant (internalized) 125I-KP10 fractions were collected at various times after the addition of 125I-KP10, and the half-time (t1/2) of internalization was determined as described in Materials and Methods. The calculated t1/2 for KP10-stimulated KISS1R internalization was 9.6 ± 1.0 minutes (Figure 3A). PAO, an endocytosis inhibitor, inhibited the KISS1R internalization rate, which resulted in prolongation of the t1/2 to 30.8 ± 4.8 minutes, suggesting that ligand-dependent KISS1R internalization follows classic clathrin- and dynamin-mediated endocytosis pathways. These findings are consistent with an earlier study by Pampillo et al (6), which demonstrated that KISS1R localizes to clathrin-coated pits and undergoes internalization via clathrin-coated vesicles.
Figure 3.

KISS1R undergoes rapid ligand-dependent and -independent internalization and recycling. A, CHO-KISS1R cells were incubated with 0.05 nM 125I-KP10 at 37°C in the presence or absence of 5 μM PAO. Cell surface and internalized radioligand were separated by acidic washes after various incubation times as indicated. The ratio of internalized to surface radioligand was plotted by linear regression. The graph shown is a representative plot from 1 of 3 independent experiments with similar results, and the t1/2 values shown are the means ± SEM from the 3 experiments. B, HEK-myc-KISS1R cells were incubated with or without 10−7 M KP10 at 37°C for 20 minutes. Some cells were then washed 5 times to remove KP10 and reincubated in KP10-free medium for an additional 20 minutes. Cells were fixed, and cell surface myc-KISS1R was detected using an HRP-anti-myc antibody. Data shown are means ± SEM from 3 independent experiments. *, P < .05; ***, P < .001. C, CHO-KISS1R cells were treated with 0.25% trypsin for 30 minutes at 4°C and then incubated at 37°C for the times indicated. The amount of cell surface KISS1R was measured by incubation with 0.05 nM 125I-KP10 plus 1 nM cold KP10 at 4°C for 1 hour. Means ± SEM are shown from a representative experiment, performed with triplicate samples, of 3 independent experiments, each with similar results. Statistical analysis was performed using one-way ANOVA followed by a post hoc Bonferroni multiple comparison test. *, P < .05; **, P < .001; ***, P < .0001 compared with time 0.
To further and more directly evaluate ligand-dependent homeostasis of cell surface KISS1R, an ELISA was performed to detect and quantify cell surface receptors more directly. For this study, a HEK cell line with stable expression of myc-KISS1R was generated (HEK-myc-KISS1R). We had shown previously that the myc tag does not interfere significantly with KISS1R function (33). Furthermore, the HEK-myc-KISS1R cells displayed a KP10-stimulated biphasic [Ca2+]i response similar to that observed in CHO-KISS1R cells (data not shown). The results of the ELISA showed that after 20 minutes of treatment with 10−7 M KP10, there was a significant 13.9 ± 2.7% decrease in cell surface receptor compared with baseline levels before treatment (P < .001) (Figure 3B). The cells were then washed to remove ligand, and 20 minutes after removal of KP10, measurement of cell surface levels of KISS1R indicated that the receptor levels were replenished to close to the baseline, pretreatment levels.
Because the half-time for 125I-KP10-stimulated KISS1R internalization was less than 10 minutes, the cell surface levels of KISS1R as measured directly by ELISA were higher than initially expected after KP10 treatment for 20 minutes. These data suggest that KISS1R recruitment to the cell surface occurs simultaneously with internalization, to result in higher cell surface KISS1R levels than would be predicted if internalization alone occurred. To explore the possibility that an intracellular KISS1R pool exists from which receptor recycling to the cell surface can occur, we studied ligand-independent, constitutive KISS1R recycling. CHO-KISS1R cells were treated with trypsin to proteolytically remove cell surface receptors. Trypsin treatment removed 48 ± 3% of cell surface receptors (Figure 3C). Cell surface levels of KISS1R, as reflected by specific radioligand binding, returned to baseline, pre-trypsin treatment levels within 5 to 10 minutes (Figure 3C). These results support the existence of an intracellular KISS1R pool and suggest the occurrence of dynamic, rapid KISS1R trafficking from this pool to the cell surface that is constitutive and ligand independent. This ligand-independent KISS1R cell surface trafficking may be coupled with constitutive internalization or may be a reactive response to cell surface KISS1R depletion.
Internalized kisspeptin undergoes rapid processing
After the binding and internalization of 125I-KP10, the fate of the internalized ligand was studied to determine whether it was degraded intracellularly, recycled back to the cell surface plasma membrane, or released from the cells. The fate of the internalized ligand will provide direct evidence of ligand turnover and indirect evidence of receptor turnover. CHO-KISS1R cells were preincubated with 0.05 nM 125I-KP10 for 1 hour to achieve sufficient internalization of the 125I-KP10/KISS1R complex to measure its fate. The cell surface, internalized, and released 125I-KP10 was then collected at various times as indicated and as described in Materials and Methods. The 125I-KP10 that was released extracellularly was further subdivided into intact and degraded ligand by TCA precipitation. The amount of released ligand increased rapidly and reached 50% of the total ligand in 15 minutes (Figure 4), suggesting that internalized 125I-KP10 undergoes rapid processing. Furthermore, most of the released 125I-KP10 was in degraded form, suggesting that the internalized KP10 undergoes rapid degradation (Figure 4). The combined amount of 125I-KP10 released extracellularly in intact form together with that bound to the cell surface, which can be considered to be an indirect measure of KISS1R recycling, accounted for approximately 25% of the total 125I-KP10 and reached a plateau by 10 minutes. In contrast, 125I-KP10 that was released in degraded form increased progressively with increasing incubation time. To eliminate the possibility that the ligand degradation occurred during the 1-hour preincubation period, we repeated this experiment by incubating the cells with 125I-KP10 at 4°C instead of at 37°C for 1 hour. The KISS1R internalization and recycling and the ligand releasing were measured simultaneously; the results were consistent with those shown in Figure 4 (data not shown). Taken together, the results of this study suggest that only a small fraction of internalized 125I-KP10 is recycled back to the cell surface, whereas most the internalized ligand undergoes degradation. The rapid degradation of kisspeptin after internalization provides an explanation for the requirement for the continued presence of exogenous kisspeptin to maintain a sustained calcium response (Figure 1C).
Figure 4.

Internalized kisspeptin undergoes rapid processing. CHO-KISS1R cells were treated with 0.05 nM 125I-KP10 at 37°C for 1 hour to achieve significant internalization. After removal of free and cell surface 125I-KP10, the cells were incubated at 37°C for the times indicated. After this incubation, the culture medium was collected, and 125I-KP10 that was released from the cells into the medium during the incubation was separated by TCA precipitation to distinguish intact from degraded 125I-KP10. Cell surface-bound 125I-KP10 was released by an acidic wash and collected; 125I-KP10 that remained internalized intracellularly was collected after cell lysis by 0.5 M NaOH. The cell surface bound, internalized, and extracellular intact and degraded radioligand was then quantified by counting in a Beckman γ-counter. A representative plot from 3 independent experiments, each done in triplicate, is shown. Mean ± SEM percentage at each time point is shown.
Mature cell surface KISS1R undergoes slow degradation
Measurement of 125I-KP10 processing gives only an indirect assessment of KISS1R degradation. To measure the rate of KISS1R degradation directly, the HEK-myc-KISS1R cell line was used. In these cells, Western blot analysis using an anti-myc antibody (1:2000; Millipore) resulted in 3 bands with molecular sizes of 42, 50, and 73 kDa, respectively (Figure 5A). Given that the calculated molecular weight of human KISS1R is 42 kDa, the 2 larger molecular weight bands may represent KISS1R that has undergone posttranslational modification. Two hours after blockade of new protein synthesis by the translation inhibitor, CHX (34), either in the presence or absence of KP10, the 72-kDa KISS1R band remained stable, whereas the smaller 42 and 50 kDa bands decreased in intensity (Figure 5A). Conversely, in the absence of CHX, KP10 stimulation did not alter the intensity of any of the 3 bands (Figure 5B).
Figure 5.

Mature KISS1R is resistant to rapid degradation. HEK-myc-KISS1R cells were pretreated with (A) or without (B) CHX for 10 minutes and then incubated with or without 10−7 M KP10 for the times indicated. The lysates were analyzed by Western blotting using an anti-myc antibody. A representative blot of 3 independent experiments with similar results is shown. C, HEK-myc-KISS1R cells were biotinylated and incubated in the presence or absence of 10−7 M KP10 for 4 or 24 hours. The cells were lysed and immunoprecipitated (IP) with an anti-myc antibody. The immunoprecipitant was analyzed by Western blotting (IB) using HRP-streptavidin (top panel) or HRP-anti-myc antibody (bottom panel). A representative blot of 3 independent experiments with similar results is shown.
We speculated that the 72-kDa band represented mature cell surface KISS1R. To confirm this hypothesis, a biotinylation assay was performed to specifically detect cell surface KISS1R (Figure 5C). In this assay, the biotinylated cell surface proteins were immunoprecipitated with an anti-myc antibody (5 μg/sample; Millipore) followed by Western blot analysis with HRP-streptavidin. The resulting immunocomplex was 72 kDa in size; the 42- and 50-kDa complexes were not detected in this assay (Figure 5C). The results of this biotinylation assay also showed no significant change in the amount of biotinylated KISS1R after 4 hours of incubation after biotinylation. After 24 hours, most of the biotinylated KISS1R was depleted both in the presence and absence of KP10 (Figure 5C). Given the much more rapid degradation of the internalized ligand (Figure 4), the results suggest dissociated processing of the internalized kisspeptin/KISS1R complex, with a slower rate of receptor degradation.
Discussion
Although significant advances have been made in our understanding of the neuroanatomy of kisspeptin neurons and in the physiology of kisspeptin as a potent upstream stimulator of GnRH release to regulate gonadotropin secretion (35), our knowledge regarding KISS1R trafficking and its influence on cellular signaling and responses to ligand stimulation is limited. Increases in intracellular calcium in GnRH neurons after kisspeptin stimulation have been reported previously (10, 36). In these studies using GnRH neurons derived from either prenatal mouse nasal explants or from young adult rats, kisspeptin stimulated a sustained calcium response. Our data show that in CHO-KISS1R cells, KP10 stimulates a biphasic increase in intracellular calcium, with an acute first-phase response followed by a more sustained second-phase response (Figure 1A). A lower concentration of KP10 is correlated with a shorter duration of the sustained second-phase response, which may result from the rapid degradation of internalized KP10, because we have shown that the sustained calcium response is dependent on the continued availability of exogenous KP10 (Figure 1C). The rapid degradation of internalized KP10 is supported by our 125I-KP10 degradation study (Figure 4). We have previously explored the source of the calcium mediating the response by measuring the KP10-stimulated intracellular calcium response in calcium-free medium (37). Only a monophasic calcium response was observed in calcium-free medium, indicating that intracellular calcium stores are the primary source for the acute first-phase calcium response, whereas the sustained second-phase calcium response is primarily dependent on extracellular calcium (37). In human studies, continuous infusion of KP10 resulted in persistent pulsatile LH secretion for up to 22 hours (13). Because intracellular Ca2+ is a key second messenger for triggering neurotransmitter (38) and neuropeptide (39) release, our findings provide a potential underlying molecular mechanism for the sustained in vivo LH response: KISS1R-mediated sustained [Ca2+]i signaling in response to KP10 may contribute to the persistent LH secretory response to kisspeptin.
Prolonged GPCR activation has been reported for Gs-coupled receptors such as PTH receptor (15) and TSHR (17). In these studies, removal of the ligand did not terminate the persistent cAMP signaling. Among Gq-coupled GPCRs, the angiotensin AT1 receptor (40) and the protease-activated receptors PAR1 and PAR4 (41) displayed biphasic ligand-stimulated intracellular calcium responses. In addition, in a human neuroblastoma cell line, either carbachol or bradykinin stimulation resulted in sustained biphasic inositol triphosphate accumulation, suggesting that both muscarinic and bradykinin receptors mediate sustained ligand-dependent signaling through Gq-coupled pathways (42). A more recent study suggested that Gq-coupled receptors mediated sustained protein kinase D activation via protein kinase C-dependent and -independent pathways (43).
Neuromedin U (NmU) receptors couple to both Gq/11 and Gi and undergo ligand-dependent rapid internalization (44). Agonist-stimulated sustained Ca2+ signaling was observed for NmU receptors and agonist-free buffer failed to return the response to basal levels. The underlying mechanism was unclear although the authors suggested that high affinities of ligand binding to NmU receptors contributed to the prolonged intracellular signaling (44). Conversely, our data indicated that KISS1R-mediated sustained [Ca2+]i signaling is dependent on the continued presence of exogenous ligand in both GnRH neurons and nonneuronal cells and is thus not likely to be the result of prolonged KISS1R activation or prolonged ligand/receptor binding (Figure 1, C and D). In turn, the nature of the exogenous ligand-dependent sustained [Ca2+]i response suggests that KISS1R trafficking contributes to the maintenance of the sustained signaling, because GPCR internalization and recycling modulate receptor desensitization and resensitization (for review, see Ref. 45). The finding that dynasore, a dynamin inhibitor (30), PAO, an endocytosis inhibitor (46), and BFA, a receptor recycling inhibitor (32), specifically blunted the sustained second-phase [Ca2+]i response in CHO and GT1–7 cells (Figure 2) supports our hypothesis that KISS1R trafficking contributes to the sustained signaling. This hypothesis is further supported by our findings that KISS1R undergoes ligand-dependent dynamic internalization and recycling (Figure 3).
Transient exposure to kisspeptin induces sustained depolarization in GnRH neurons in most studies (10–12). However, this is not consistently the case for the intracellular calcium response. Because membrane depolarization and intracellular calcium responses are distinct events, it is possible to see a dissociation of the duration of these 2 responses. Transient kisspeptin stimulation has been reported to induce persistent [Ca2+]i elevations in both GnRH-like neurons from embryonic mouse nasal explants (10) and the GT1–7 cell line (47). In both cases, the [Ca2+]i response was sustained after removal of the ligand. On the other hand, in a study using 200-μm-thick-cut coronal brain slices of adult mice, transient kisspeptin exposure stimulated sustained membrane depolarization but only a brief intracellular calcium response (7). In our study, we found that KP10 removal abolished the sustained [Ca2+]i response in both CHO-KISS1R cells, a nonneuronal cell line, and GnRH neuronal GT1–7-myc-KISS1R cells. The exact reason for the differences in the duration of the [Ca2+]i response to transient kisspeptin exposure in these different studies is not entirely clear, but different sources of the cells and different techniques for calcium measurement and ligand removal may contribute to the diverse intracellular calcium responses. In our study, to exclude any artifacts induced by washing the cells, we included control groups of cells washed in washing buffer in the presence or absence of KP10. Washing the cells with ligand-free washing buffer abolished the [Ca2+]i response, but washing with the buffer containing KP10 did not affect the sustained [Ca2+]i response (Figure 1, C and D). Furthermore, the cells responded to restimulation with kisspeptin 10 to 30 minutes after ligand removal (Figure 1, C and D), indicating that the abolished [Ca2+]i response was not the result of loss of the cells during the washing step.
Whereas Ozcan et al observed (47) a kisspeptin-stimulated intracellular calcium response in untransfected GT1–7 cells, suggesting the presence of functional KISS1R in their cells, we did not detect a KP10-induced [Ca2+]i response in our GT1–7 cell line. Several factors may contribute to the varied findings between the study of Ozcan et al and our studies. One difference is that Ozcan et al used the reagent Fura 2, whereas we used Fluo-4 to measure the [Ca2+]i response. Second, there may be differences in the phenotypes and features of the GT1–7 cell line subclones in each laboratory. The stable line we generated, GT1–7-myc-KISS1R, responded to KP10 stimulation, indicating that our cells had appropriate postreceptor pathways to mount a response. The presence of exogenously introduced receptors may modulate observed responses, for example, if receptor levels are excessively high. To minimize this concern, we selected a GT1–7-myc-KISS1R clone for further study with moderate levels of KISS1R (based on binding assays) and an intermediate KP10-induced [Ca2+]i response, compared with other clones purified after antibiotic selection. How the KISS1R expression levels in our GT1–7-myc-KISS1R cell line compare with levels in primary GnRH neurons is not known.
The rapid KISS1R internalization rate is consistent with earlier findings using immunofluorescence coupled to confocal imaging, flow cytometry, and indirect receptor radiolabeling to measure KISS1R internalization (48). The discordance in the rate of ligand-dependent KISS1R internalization measured by 125I-KP10 and ELISAs suggested that receptor recycling occurred concomitantly with internalization. First, the internalization of the 125I-KP10/KISS1R complex is very rapid (Figure 3A). Second, there is rapid constitutive KISS1R recycling (Figure 3C). Third, KP10 stimulation resulted in only a modest decrease in cell surface KISS1R (Figure 3B). Without simultaneous receptor recruitment, we would expect to see a more profound loss of cell surface receptors. The 125I-KP10 assay could detect only trafficking of the labeled KP10/KISS1R complex, whereas the ELISA could detect all cell surface receptors, reflecting the original cell surface KISS1R pool, minus internalized receptors, and plus KISS1R that recruited to the cell surface. When the recycling of internalized kisspeptin/KISS1R complex is tracked using 125I-KP10, only a small fraction of internalized 125I-KP10/KISS1R, represented by released intact and surface-bound 125I-KP10, returned to the cell surface (Figure 4). In light of the robust KISS1R recycling that occurred after proteolytic removal of cell surface KISS1R (Figure 3B), it is possible that ligand-stimulated cell surface KISS1R recruitment occurs primarily from an intracellular KISS1R pool. It is not clear whether kisspeptin triggers KISS1R recruitment to replete the cell surface KISS1R pool or whether KISS1R recruitment is ligand independent, because we and others have demonstrated that dynamic and rapid constitutive KISS1R recycling exists (Ref. 48 and Figure 3C). In either case, the homeostasis of cell surface KISS1Rs is maintained by either ligand-dependent and/or -independent receptor recruitment that keeps sufficient cell surface receptors available for ligand binding to provide continued calcium signaling if exogenous kisspeptin is present (see model in Figure 6).
Figure 6.

Proposed model for the contribution of KISS1R trafficking to prolonged Ca2+ signaling. Kisspeptin binds to KISS1R to stimulate a rapid acute-phase increase in [Ca2+]i as well as rapid KISS1R internalization. The internalized ligand undergoes rapid degradation and is released extracellularly, whereas the KISS1R undergoes a much slower degradation. In addition to KISS1R recycling, there is also dynamic recruitment of KISS1R from intracellular pools to the cell surface that maintains sufficient cell surface KISS1R to bind with exogenous kisspeptin and maintain prolonged Ca2+ signaling. ER, endoplasmic reticulum.
Neuropeptides are secreted from the neurons, but unlike classic neurotransmitters, they convey actions with less spatial and temporal specificity (49). In addition, it appears that mechanisms for reuptake of neuropeptides to terminate the ligand action do not exist (49). Instead, extracellular enzymatic degradation plays an important role in elimination of neuropeptides. Our findings of rapid processing and degradation of internalized kisspeptin suggests an alternative mechanism for termination of neuropeptide action. The much slower degradation of cell surface KISS1R (Figure 5B) suggests dissociated processing of the internalized kisspeptin/KISS1R complex that may contribute to the exogenous kisspeptin-dependent sustained signaling. Although 3 bands of myc-KISS1R were visualized by Western blot (Figure 5A), only the receptors represented by the heaviest band at 72 kDa were detected on the cell surface (Figure 5C), suggesting that the other 2 bands represent immature, intracellular KISS1R. In an earlier study, Western blot analysis of KISS1R-GFP revealed a dominant band close to 75 kDa (50). Given that the molecular weight of GFP is about 27 kDa, the apparent size of KISS1R in this study is closer to that observed in a previous study from our group, in which the predominant band of myc-KISS1R was at 42 kDa (18). In the present study, a stable cell line expressing KISS1R was used for the Western blots, whereas in the previous studies, transient expression systems were used (18, 50). In transient expression systems, modified (mature) receptors may not be as readily formed and detected. We do not have evidence that the 72-kDa band is glycosylated, although glycosylation is a common modification for GPCRs. Inhibition of new protein synthesis by CHX had little effect on mature KISS1R turnover for as long as 2 hours, further supporting the dissociated processing of kisspeptin ligand and receptor (Figure 5B). Intriguingly, although the mature KISS1R is relatively stable, the 2 lower molecular weight forms of the receptor, probably reflecting immature intracellular KISS1R, undergo more rapid turnover (Figure 5A). The more rapid turnover of the lower molecular weight KISS1R forms may reflect dynamic posttranslational processing and maturation of these immature forms to contribute to the mature KISS1R pool.
In conclusion, we have demonstrated that KP10 stimulates a biphasic intracellular calcium response, with an acute rapid increase in intracellular calcium followed by a more sustained second-phase calcium response. This second-phase calcium response is dependent on the continued presence of exogenous ligand. Our findings are consistent with human studies showing that continuous KP10 infusion stimulates persistent pulsatile LH secretion (13). We have provided evidence that KISS1R undergoes kisspeptin-dependent and -independent dynamic internalization and recycling to contribute to the sustained ligand-dependent second-phase [Ca2+]i response. Based on our findings of ligand- and internalization-dependent sustained Ca2+ signaling, ligand-dependent dynamic KISS1R trafficking (internalization and recycling), and dissociated processing of the kisspeptin/KISS1R complex, we propose that dynamic KISS1R trafficking is important for maintaining stable levels of sensitive, ligand-responsive cell surface KISS1R to allow the prolonged intracellular calcium signaling responses.
Acknowledgments
This work was supported by the Eunice Kennedy Shriver National Institute of Child Health and Human Development (NICHD), National Institutes of Health (NIH), through cooperative agreement U54 HD28138 as part of the Specialized Cooperative Centers Program in Reproduction and Infertility Research (to U.B.K.), by National Institute of Diabetes and Digestive and Kidney Diseases (NIDDK)/NIH T32 DK007529–22 (to L.M.), and by NICHD/NIH K08 HD070957 (to L.M.).
Disclosure Summary: The authors have nothing to disclose.
Footnotes
- BFA
- brefeldin A
- CHX
- cycloheximide
- CHO
- Chinese hamster ovary
- GPCR
- G protein–coupled receptor
- HEK
- human embryonic kidney
- HRP
- horseradish peroxidase
- NmU
- neuromedin U
- PAO
- phenylarsine oxide
- RFU
- relative fluorescence units
- TCA
- trichloroacetic acid
- TRHR
- TRH receptor
- TSHR
- TSH receptor.
References
- 1. Seminara SB, Messager S, Chatzidaki EE, et al. The GPR54 gene as a regulator of puberty. N Engl J Med. 2003;349:1614–1627 [DOI] [PubMed] [Google Scholar]
- 2. Ohtaki T, Shintani Y, Honda S, et al. Metastasis suppressor gene KiSS-1 encodes peptide ligand of a G-protein-coupled receptor. Nature. 2001;411:613–617 [DOI] [PubMed] [Google Scholar]
- 3. Kotani M, Detheux M, Vandenbogaerde A, et al. The metastasis suppressor gene KiSS-1 encodes kisspeptins, the natural ligands of the orphan G protein-coupled receptor GPR54. J Biol Chem. 2001;276:34631–34636 [DOI] [PubMed] [Google Scholar]
- 4. Muir AI, Chamberlain L, Elshourbagy NA, et al. AXOR12, a novel human G protein-coupled receptor, activated by the peptide KiSS-1. J Biol Chem. 2001;276:28969–28975 [DOI] [PubMed] [Google Scholar]
- 5. Stafford LJ, Xia C, Ma W, Cai Y, Liu M. Identification and characterization of mouse metastasis-suppressor KiSS1 and its G-protein-coupled receptor. Cancer Res. 2002;62:5399–5404 [PubMed] [Google Scholar]
- 6. Pampillo M, Camuso N, Taylor JE, et al. Regulation of GPR54 signaling by GRK2 and β-arrestin. Mol Endocrinol. 2009;23:2060–2074 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Liu X, Lee K, Herbison AE. Kisspeptin excites gonadotropin-releasing hormone neurons through a phospholipase C/calcium-dependent pathway regulating multiple ion channels. Endocrinology. 2008;149:4605–4614 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Pielecka-Fortuna J, Chu Z, Moenter SM. Kisspeptin acts directly and indirectly to increase gonadotropin-releasing hormone neuron activity and its effects are modulated by estradiol. Endocrinology. 2008;149:1979–1986 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Krsmanovi LZ, Stojilkovi SS, Merelli F, Dufour SM, Virmani MA, Catt KJ. Calcium signaling and episodic secretion of gonadotropin-releasing hormone in hypothalamic neurons. Proc Natl Acad Sci USA. 1992;89:8462–8466 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Constantin S, Caligioni CS, Stojilkovic S, Wray S. Kisspeptin-10 facilitates a plasma membrane-driven calcium oscillator in gonadotropin-releasing hormone-1 neurons. Endocrinology. 2009;150:1400–1412 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Han SK, Gottsch ML, Lee KJ, et al. Activation of gonadotropin-releasing hormone neurons by kisspeptin as a neuroendocrine switch for the onset of puberty. J Neurosci. 2005;25:11349–11356 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Zhang C, Roepke TA, Kelly MJ, Rønnekleiv OK. Kisspeptin depolarizes gonadotropin-releasing hormone neurons through activation of TRPC-like cationic channels. J Neurosci. 2008;28:4423–4434 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. George JT, Veldhuis JD, Roseweir AK, et al. Kisspeptin-10 is a potent stimulator of LH and increases pulse frequency in men. J Clin Endocrinol Metab. 2011;96:E1228–E1236 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Chan YM, Butler JP, Pinnell NE, et al. Kisspeptin resets the hypothalamic GnRH clock in men. J Clin Endocrinol Metab. 2011;96:E908–E915 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Ferrandon S, Feinstein TN, Castro M, et al. Sustained cyclic AMP production by parathyroid hormone receptor endocytosis. Nat Chem Biol. 2009;5:734–742 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Calebiro D, Nikolaev VO, Gagliani MC, et al. Persistent cAMP-signals triggered by internalized G-protein-coupled receptors. PLoS Biol. 2009;7:e1000172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Neumann S, Geras-Raaka E, Marcus-Samuels B, Gershengorn MC. Persistent cAMP signaling by thyrotropin (TSH) receptors is not dependent on internalization. FASEB J. 2010;24:3992–3999 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Bianco SD, Vandepas L, Correa-Medina M, et al. KISS1R intracellular trafficking and degradation: effect of the Arg386Pro disease-associated mutation. Endocrinology. 2011;152:1616–1626 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Hislop JN, von Zastrow M. Role of ubiquitination in endocytic trafficking of G-protein-coupled receptors. Traffic. 2011;12:137–148 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Kuohung W, Burnett M, Mukhtyar D, et al. A high-throughput small-molecule ligand screen targeted to agonists and antagonists of the G-protein-coupled receptor GPR54. J Biomol Screen. 2010;15:508–517 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Cook LB, Zhu CC, Hinkle PM. Thyrotropin-releasing hormone receptor processing: role of ubiquitination and proteasomal degradation. Mol Endocrinol. 2003;17:1777–1791 [DOI] [PubMed] [Google Scholar]
- 22. Min L, Galet C, Ascoli M. The association of arrestin-3 with the human lutropin/choriogonadotropin receptor depends mostly on receptor activation rather than on receptor phosphorylation. J Biol Chem. 2002;277:702–710 [DOI] [PubMed] [Google Scholar]
- 23. Nakamura K, Liu X, Ascoli M. Seven non-contiguous intracellular residues of the lutropin/choriogonadotropin receptor dictate the rate of agonist-induced internalization and its sensitivity to non-visual arrestins. J Biol Chem. 2000;275:241–247 [DOI] [PubMed] [Google Scholar]
- 24. Nakamura K, Ascoli M. A dileucine-based motif in the C-terminal tail of the lutropin/choriogonadotropin receptor inhibits endocytosis of the agonist-receptor complex. Mol Pharmacol. 1999;56:728–736 [PubMed] [Google Scholar]
- 25. Wiley HS, Cunningham DD. The endocytotic rate constant. A cellular parameter for quantitating receptor-mediated endocytosis. J Biol Chem. 1982;257:4222–4229 [PubMed] [Google Scholar]
- 26. Gehret AU, Jones BW, Tran PN, Cook LB, Greuber EK, Hinkle PM. Role of helix 8 of the thyrotropin-releasing hormone receptor in phosphorylation by G protein-coupled receptor kinase. Mol Pharmacol. 2010;77:288–297 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Lloyd CE, Ascoli M. On the mechanisms involved in the regulation of the cell-surface receptors for human choriogonadotropin and mouse epidermal growth factor in cultured Leydig tumor cells. J Cell Biol. 1983;96:521–526 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Ascoli M. Internalization and degradation of receptor-bound human choriogonadotropin in Leydig tumor cells. Fate of the hormone subunits. J Biol Chem. 1982;257:13306–13311 [PubMed] [Google Scholar]
- 29. Min L, Ascoli M. Effect of activating and inactivating mutations on the phosphorylation and trafficking of the human lutropin/choriogonadotropin receptor. Mol Endocrinol. 2000;14:1797–1810 [DOI] [PubMed] [Google Scholar]
- 30. Macia E, Ehrlich M, Massol R, Boucrot E, Brunner C, Kirchhausen T. Dynasore, a cell-permeable inhibitor of dynamin. Dev Cell. 2006;10:839–850 [DOI] [PubMed] [Google Scholar]
- 31. Thompson HM, McNiven MA. Discovery of a new 'dynasore.' Nat Chem Biol. 2006;2:355–356 [DOI] [PubMed] [Google Scholar]
- 32. Roy S, Roy SJ, Pinard S, et al. Mechanisms of melanocortin-2 receptor (MC2R) internalization and recycling in human embryonic kidney (HEK) cells: identification of Key Ser/Thr (S/T) amino acids. Mol Endocrinol. 2011;25:1961–1977 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Teles MG, Trarbach EB, Noel SD, et al. A novel homozygous splice acceptor site mutation of KISS1R in two siblings with normosmic isolated hypogonadotropic hypogonadism. Eur J Endocrinol. 2010;163:29–34 [DOI] [PubMed] [Google Scholar]
- 34. Schneider-Poetsch T, Ju J, Eyler DE, et al. Inhibition of eukaryotic translation elongation by cycloheximide and lactimidomycin. Nat Chem Biol. 2010;6:209–217 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Roa J, Navarro VM, Tena-Sempere M. Kisspeptins in reproductive biology: consensus knowledge and recent developments. Biol Reprod. 2011;85:650–660 [DOI] [PubMed] [Google Scholar]
- 36. Kroll H, Bolsover S, Hsu J, Kim SH, Bouloux PM. Kisspeptin-evoked calcium signals in isolated primary rat gonadotropin- releasing hormone neurones. Neuroendocrinology. 2011;93:114–120 [DOI] [PubMed] [Google Scholar]
- 37. Babwah AV, Pampillo M, Min L, Kaiser UB, Bhattacharya M. Single-cell analyses reveal that KISS1R-expressing cells undergo sustained kisspeptin-induced signaling that is dependent upon an influx of extracellular Ca2+. Endocrinology. 2012;153:5875–5887 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Wilson M. Synaptic physiology: plenty of models to choose from. Curr Biol. 2004;14:R666–R667 [DOI] [PubMed] [Google Scholar]
- 39. Verhage M, McMahon HT, Ghijsen WE, et al. Differential release of amino acids, neuropeptides, and catecholamines from isolated nerve terminals. Neuron. 1991;6:517–524 [DOI] [PubMed] [Google Scholar]
- 40. Suárez C, Tornadú IG, Cristina C, et al. Angiotensin and calcium signaling in the pituitary and hypothalamus. Cell Mol Neurobiol. 2002;22:315–333 [DOI] [PubMed] [Google Scholar]
- 41. Vaidyula VR, Rao AK. Role of Gαq and phospholipase C-β2 in human platelets activation by thrombin receptors PAR1 and PAR4: studies in human platelets deficient in Gαq and phospholipase C-β2. Br J Haematol. 2003;121:491–496 [DOI] [PubMed] [Google Scholar]
- 42. Willars GB, Nahorski SR. Quantitative comparisons of muscarinic and bradykinin receptor-mediated Ins (1,4,5)P3 accumulation and Ca2+ signalling in human neuroblastoma cells. Br J Pharmacol. 1995;114:1133–1142 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Sinnett-Smith J, Jacamo R, Kui R, et al. Protein kinase D mediates mitogenic signaling by Gq-coupled receptors through protein kinase C-independent regulation of activation loop Ser744 and Ser748 phosphorylation. J Biol Chem. 2009;284:13434–13445 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Brighton PJ, Szekeres PG, Wise A, Willars GB. Signaling and ligand binding by recombinant neuromedin U receptors: evidence for dual coupling to Gαq/11 and Gαi and an irreversible ligand-receptor interaction. Mol Pharmacol. 2004;66:1544–1556 [DOI] [PubMed] [Google Scholar]
- 45. Hanyaloglu AC, von Zastrow M. Regulation of GPCRs by endocytic membrane trafficking and its potential implications. Annu Rev Pharmacol Toxicol. 2008;48:537–568 [DOI] [PubMed] [Google Scholar]
- 46. Brismar H, Hua X, Adachi S, Holtbäck U. The role of endocytosis in renal dopamine D1 receptor signaling. Pflugers Arch. 2006;451:793–802 [DOI] [PubMed] [Google Scholar]
- 47. Ozcan M, Alcin E, Ayar A, Yilmaz B, Sandal S, Kelestimur H. Kisspeptin-10 elicits triphasic cytosolic calcium responses in immortalized GT1–7 GnRH neurones. Neurosci Lett. 2011;492:55–58 [DOI] [PubMed] [Google Scholar]
- 48. Pampillo M, Babwah AV. Assessment of constitutive activity and internalization of GPR54 (KISS1-R). Methods Enzymol. 2010;484:75–93 [DOI] [PubMed] [Google Scholar]
- 49. Parker JA, Bloom SR. Hypothalamic neuropeptides and the regulation of appetite. Neuropharmacology. 2012;63:18–30 [DOI] [PubMed] [Google Scholar]
- 50. Wacker JL, Feller DB, Tang XB, et al. Disease-causing mutation in GPR54 reveals the importance of the second intracellular loop for class A G-protein-coupled receptor function. J Biol Chem. 2008;283:31068–31078 [DOI] [PMC free article] [PubMed] [Google Scholar]