

Abstract
AIM
To investigate the genetic findings and phenotypic characteristics of a Chinese family with Norrie disease (ND).
METHODS
Molecular genetic analysis and clinical examinations were performed on a Chinese family with ND. Mutations in the Norrie disease pseudoglioma (NDP) gene were detected by direct sequencing. Haplotypes were constructed and compared with the phenotypes in the family. Evolutionary comparisons and mutant open reading frame (ORF) prediction were also undertaken.
RESULTS
Two family members with ocular manifestations were diagnosed with ND. No signs of sensorineural hearing loss were observed in either patient, while one of them showed signs of mild mental retardation. A novel heterozygous mutation in the NDP gene, c.-1_2delAAT, was detected in both patients. The mutation and the mutation bearing haplotype co-segregated with the ND phenotype in males and was transmitted from their mothers and/or grandmothers (II:2). The male without ND did not harbor the mutation. The mutation occurred at the highly conserved nucleotides. ORF finder predicted that the mutation would lead to the production of a truncated protein that lacks the first 11 N-terminal amino acids.
CONCLUSION
A novel mutation, c.-1_2delAAT in the NDP gene, was identified in a Chinese family with ND. This mutation caused ND without obvious sensorineural hearing loss. Mental disorder was found in one but not the other patients. The clinical heterogeneity in the family indicated that other genetic variants and epigenetic factors may also play a role in the disease presentation.
Keywords: Norrie disease, pseudoglioma, mutation, Chinese
INTRODUCTION
Norrie disease (ND, MIM#310600) is a rare X-linked recessive disorder characterized by bilateral congenital blindness in males due to degenerative and proliferative changes in the neuroretina. Approximately 30%-50% of patients show some forms of progressive mental disorder and/or sensorineural hearing loss[1],[2].
The Norrie disease pseudoglioma gene (NDP, NM_00266) is the causal gene that underlies ND and is located on chromosome Xp11.4[3],[4]. It contains three exons, but only the second and the third exons are translated. This gene encodes a 133-amino-acid secretory protein, norrin, with a cystein-knot motif that plays a critical role in retinal vascular development[5]-[8].
More than 143 NDP mutations (HGMD Professional 2013.2) (http://www.hgmd.cf.ac.uk) have been identified to date in patients with ND or other NDP-related retinopathies, including persistent hyperplastic primary vitreous (PHPV)[9]-[11], X-linked familial exudative vitreoretinopathy (XL-FEVR)[9],[10],[12],[13], retinopathy of prematurity (ROP)[9],[10],[14],[15] and Coats disease[16].
In this study, we identified a novel NDP mutation responsible for ND in a Chinese family with three affected males, two of which were evaluated in this study.
SUBJECTS AND METHODS
Subjects
The family from Guangdong province, China comprised 4 generations with 3 affected patients (Figure 1). Pedigree analysis indicated that the mode of inheritance was most likely to be X-linked recessive. Informed consent was obtained from all recruited subjects or their parents or guardians. The study was approved by the Ethics Committee of the University for Human Study. The principles outlined in the Declaration of Helsinki were followed. Eight subjects from the family were recruited for genetic analysis, among which 2 patients underwent comprehensive clinical examinations. One hundred unrelated healthy individuals from the same province and ethnic background were recruited as controls.
Figure 1. Pedigree and haplotype construction for a family with Norrie disease.

Markers are listed on the upper left of the figure. The arrow indicates the proband. Black bars indicate the affected haplotype. Inferred genotypes are indicated in parentheses, and unknown genotypes in question mark.
Mutation Detection and Haplotype Construction
Genomic DNA was extracted from peripheral blood samples using a standard phenol-chloroform technique. Mutations were detected by sequencing the PCR products flanking all 3 exons and intron-exon splice sites of the NDP gene. The guidelines for describing sequence variations and numbering (http://www.hgvs.org/mutnomen/) were followed[17]. Haplotypes were constructed using the informative intragenic single nucleotide polymorphisms (SNPs).
Alignment of NDP cDNA Orthologues
CLUSTAL X (1.83) was used to compare human NDP cDNA sequence (NM_000266.3) with the nucleotide sequences of chimpanzee (XM_001139783.2), gorilla (XM_004064036.1), cow (NM_001046090.2), pig (XM_003469362.2), mouse (NM_010883.2), rat (NM_001108814.1) and chicken (NM_001278086.1)[18].
Mutant Open Reading Frame Prediction
Open reading frame (ORF) finder (http://www.ncbi.nlm.nih.gov/gorf/gorf.html) was used to predict the ORF in the mutant DNA.
RESULTS
Clinical Findings
Of the 3 affected subjects, 2 were alive (III:5 and IV-1) and one died at young age from an accident (III:3) (Figure 1). The proband, IV-1, was born in September 2007 after an uncomplicated pregnancy. He was found after birth by his parents for not following moving light stimuli. He was taken to Zhongshan Ophthalmic Center for examination when he was 3-month old. The cornea of both eyes were transparent, while the diameter of left side cornea (9mm) was smaller than that of the right (10.5mm); The lens of both eyes were clear, while the anterior surface of lens attached to the posterior surface of the cornea in the right eye; Gray-whitish opacity was observed behind the lens in both eyes. Ultrasound B-scan examination revealed massive opacity in the vitreous cavity with complete retinal detachment in both eyes. He was diagnosed with ND and pars plana vitrectomy and lensectomy were performed in the left eye. Extensive retinal proliferation was observed during surgery, and retinal reattachment was not achieved. Since the surgical outcome of the left eye was not satisfactory, no surgical intervention was attempted in the right eye. No obvious sensorineural hearing loss was observed. During the five-year follow-up, signs of mild mental retardation were documented for this patient.
Patient III:5 was also found to be blind in both eyes after birth and didn't receive any eye examination until 31 years of age. Both eyes were enophthalmos and both cornea appeared milky white (Figure 2). Ultrasound B-scan revealed calcification of ocular wall, massive vitreous opacity and complete retinal detachment (Figure 3). No obvious signs of mental retardation or sensorineural hearing loss were observed based on clinical observation. No medical intervention was attempted for this patient.
Figure 2. Slit-lamp photograph of patient III:5.
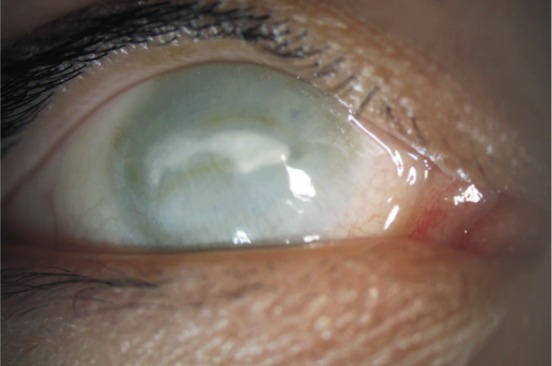
It showed sunken eye and corneal opacification.
Figure 3. Ultrasound B-scan of patient III:5.

It showed calcification of the ocular wall, massive vitreous opacity and complete retinal detechment in both eyes.
Fundus examination found no obvious abnormality in the proband's 30-year-old mother (III:2).
Genetic Findings
Direct sequencing on both DNA strands in individuals with ND (III:5 and IV:1) revealed a novel mutation, c.-1-_2delAAT, in exon 2 of the NDP gene (NM_00266.3) (Figure 4). This mutation was found to be heterozygous in their mothers (II:4 and III:2) and/or grandmothers (II:2), but was absent in the other unaffected family members and normal controls. Three intragenic SNPs were identified in this family: one was rs2004594, the insertion of additional GGCCTCTT repeat at 79 bases upstream of the untranslated exon 1; the other two SNPs were in the intron 1, rs1159223 and a novel SNP c.-208+125delT. Haplotypes were constructed using the mutation and the two informative intragenic SNPs, rs2004594 and c.-208+125delT.
Figure 4. Partial nucleotide sequences of exon 2 of the NDP gene in affected, unaffected and heterozygous family members.

The forward (A) and the reverse (B) sequences of the affected family members show the AAT deletion at position c. -1-_2. The forward (C) and the reverse (D) sequences of the other unaffected family members and the general population show no nucleotide changes. The forward (E) and the reverse (F) sequences of the patients' mothers and grandmother show heterozygous AAT deletion.
Haplotypes constructed showed that the mutation-bearing haplotype co-segregated with the ND phenotype in males (III:5 and IV:1) which was transmitted from their mothers (II:4 and III:2) and/or grandmothers (II:2). The male without ND inherited normal haplotype.
Alignment of the gene in different species showed conserved c.-1_2 AAT in the cDNA sequence in chimpanzee, gorilla, cow, pig, mouse, rat and chicken (Figure 5).
Figure 5. Alignment of NDP cDNA orthologues.

It shows that the nucleotides AAT where the mutation occurs (c.-1_2) are conserved in these species (highlighted by square frame).
The c.-1_2delAAT mutation deletes the last nucleotide A of the 5′UTR and the first two nucleotides AT of the initiation codon, changing the start of translation initiation site. ORF finder predicts that the mutant ORF begins 33 bp further downstream which causes the production of a truncated protein lacking the first 11 N-terminal amino acids.
DISCUSSION
Norrie disease (ND) is an X-linked recessive disorder in which retinal degeneration occurs before or very shortly after birth[1],[2]. The ocular abnormalities include retinal dysplasia with early vascular proliferation (pseudoglioma), retinal detachment, corneal opacities, cataract and atrophic irides. In addition to the ocular findings, some affected males have varying degree of mental retardation or show cognitive decline with age. About a third of cases develop progressive sensorineural hearing loss that is usually apparent in the second decade of life. In this study two patients were diagnosed with ND based on their ocular manifestations. No signs of sensorineural hearing loss were observed in either patient, while one of them showed signs of mild mental retardation.
ND is caused by mutations in the NDP gene[3],[4]. The gene encodes norrin, a 133-amino-acid secretory protein with two primary domains: a signal peptide (the first 24 amino acids of the N-terminus) responsible for anchoring the molecule and a cysteine knot that plays important roles in receptor binding and signal transduction activation[5]-[8]. Over 143 NDP mutations have been identified, which have been associated with various phenotypes including ND[9],[10],[12],[13], PHPV[9]-[11], XL-FEVR [9],[10],[12],[13], ROP[9],[10],[14],[15] and Coats disease[16]. It has been observed that mutations disrupting the cysteine-knot motif are associated with severe retinal dysgenesis, whereas noncysteine mutations are present in various abnormality in vascular and retinal development[10],[13]. The 5′UTR contains elements that regulate gene expression, mutations in the region may alter gene regulation[19]. The 5′UTR mutations in NDP have been reported in ROP[9],[10]. In this study, a novel mutation responsible for ND, c.-1_2delAAT, was identified. The mutation deletes the last nucleotide A of the 5′UTR and the first two nucleotides AT of the initiation codon. Since the translation start site and its context sequence play an important role in the control of translation efficiency and the correct translation of eukaryotic mRNA[20], the c.-1_2delAAT mutation is expected to cause the failure of ND gene translation or the production of an aberrant protein. ORF finder predicted that it would eliminate the first 11 N-terminal amino acids of norrin, disrupting its putative signal peptide. Loss of the signal peptide would lead to altered secretory pathway and absence of functional protein. The other initiation codon point mutations, c.1A>G (ATG>GTG, p.Met1Val?) and c.2T>G (ATG>AGG, p.Met1Arg?), have been reported to be responsible for ND[21],[22]. Congenital blindness is common among patients with either of the two mutations or the c.-1_2delAAT mutation in this study. However, three Japanese patients with c.1A>G mutation remained unremarkable in neurological and otological studies, while the patient with c.2T>G mutation had hearing loss and autistic features[22]. The difference in the mutations and the subsequent differences in the translation efficiency and aberrant protein products may contribute to the differences in phenotypes[21]. Patients with c.-1_2delAAT mutation in this study had no obvious sensorineural hearing loss. One patient showed mild mental retardation, but not the other one. The clinical heterogeneity in the same family with the same NDP mutation has also been found in another report and indicates that other genetic and epigenetic factors may also play a role in the disease presentation[9],[22],[23]. Characterization of the mutant protein and the regulation of NDP gene expression would provide further understanding on the mechanism of this disease.
Acknowledgments
The authors thank all the patients and their family members for their participation.
Footnotes
Foundation items: National Natural Science Foundation of China (No.81200402 and No.81200670); the Science and Technology Project of Guangdong Province (No.2011B050400022)
REFERENCES
- 1.Warburg M. Norrie's disease-differential diagnosis and treatment. Acta Ophthalmol (Copenh) 1975;53(2):217–236. doi: 10.1111/j.1755-3768.1975.tb01156.x. [DOI] [PubMed] [Google Scholar]
- 2.Sims KB. NDP-Related Retinopathies. In: Pagon RA, Adam MP, Bird TD, Dolan CR, Fong CT, Stephens K, editors. GeneReviews™ [Internet] Seattle: University of Washington, Seattle; 1999. [updated 2009]:1993-2013 http://www.ncbi.nlm.nih.gov/books/NBK1331/ [PubMed] [Google Scholar]
- 3.Chen ZY, Hendriks RW, Jobling MA, Breakefield XO, Sims KB, Craig IW. Isolation and characterization of a candidate gene for Norrie disease. Nat Genet. 1992;1(3):204–208. doi: 10.1038/ng0692-204. [DOI] [PubMed] [Google Scholar]
- 4.Berger W, Meindl A, van de Pol TJ, Ropers HH, Döerner C, Monaco A, Bergen AAB, Lebo R, Warburg M, Zergollern L, Lorenz B, Gal A, Bleeker-Wagemakers EM, Meitinge T. Isolation of a candidate gene for Norrie disease by positional cloning. Nat Genet. 1992;1(3):199–203. doi: 10.1038/ng0692-199. [DOI] [PubMed] [Google Scholar]
- 5.Chen ZY, Battinelli EM, Hendriks RW, Powell JF, Middleton-Price H, Sims KB, Breakefield XO, Craig IW. Norrie disease gene: characterization of deletions and possible function. Genomics. 1993;16(2):533–535. doi: 10.1006/geno.1993.1224. [DOI] [PubMed] [Google Scholar]
- 6.eitinger T, Meindl A, Bork P, Rost B, Sander C, Haasemann M, Murken J. Molecular modelling of the Norrie disease protein predicts a cystine knot growth factor tertiary structure. Nat Genet. 1993;5(4):376–380. doi: 10.1038/ng1293-376. [DOI] [PubMed] [Google Scholar]
- 7.Ohlmann A, Tamm ER. Norrin: molecular and functional properties of an angiogenic and neuroprotective growth factor. Prog Retin Eye Res. 2012;31(3):243–257. doi: 10.1016/j.preteyeres.2012.02.002. [DOI] [PubMed] [Google Scholar]
- 8.Wang Y, Rattner A, Zhou Y, Williams J, Smallwood PM, Nathans J. Norrin/Frizzled4 signaling in retinal vascular development and blood brain barrier plasticity. Cell. 2012;151(6):1332–1344. doi: 10.1016/j.cell.2012.10.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Dickinson JL, Sale MM, Passmore A, FitzGerald LM, Wheatley CM, Burdon KP, Craig JE, Tengtrisorn S, Carden SM, Maclean H, Mackey DA. Mutations in the NDP gene: contribution to Norrie disease, familial exudative vitreoretinopathy and retinopathy of prematurity. Clin Experiment Ophthalmol. 2006;34(7):682–688. doi: 10.1111/j.1442-9071.2006.01314.x. [DOI] [PubMed] [Google Scholar]
- 10.Wu WC, Drenser K, Trese M, Capone A, Jr, Dailey W. Retinal phenotype-genotype correlation of pediatric patients expressing mutations in the Norrie disease gene. Arch Ophthalmol. 2007;125(2):225–30. doi: 10.1001/archopht.125.2.225. [DOI] [PubMed] [Google Scholar]
- 11.Aponte EP, Pulido JS, Ellison JW, Quiram PA, Mohney BG. A novel NDP mutation in an infant with unilateral persistent fetal vasculature and retinal vasculopathy. Ophthalmic Genet. 2009;30(2):99–102. doi: 10.1080/13816810802705755. [DOI] [PubMed] [Google Scholar]
- 12.Pelcastre EL, Villanueva-Mendoza C, Zenteno JC. Novel and recurrent NDP gene mutations in familial cases of Norrie disease and X-linked exudative vitreoretinopathy. Clin Experiment Ophthalmol. 2010;38(4):367–74. doi: 10.1111/j.1442-9071.2010.02245.x. [DOI] [PubMed] [Google Scholar]
- 13.Yang H, Li S, Xiao X, Guo X, Zhang Q. Screening for NDP mutations in 44 unrelated patients with familial exudative vitreoretinopathy or Norrie disease. Curr Eye Res. 2012;37(8):726–729. doi: 10.3109/02713683.2012.675615. [DOI] [PubMed] [Google Scholar]
- 14.Hiraoka M, Takahashi H, Orimo H, Hiraoka M, Ogata T, Azuma N. Genetic screening of Wnt signaling factors in advanced retinopathy of prematurity. Mol Vis. 2010;16:2572–2577. [PMC free article] [PubMed] [Google Scholar]
- 15.Shastry BS. Genetic susceptibility to advanced retinopathy of prematurity (ROP) J Biomed Sci. 2010;17:69. doi: 10.1186/1423-0127-17-69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Black GC, Perveen R, Bonshek R, Cahill M, Clayton-Smith J, Lloyd IC, McLeod D. Coats' disease of the retina (unilateral retinal telangiectasis) caused by somatic mutation in the NDP gene: a role for norrin in retinal angiogenesis. Hum Mol Genet. 1999;8(11):2031–2035. doi: 10.1093/hmg/8.11.2031. [DOI] [PubMed] [Google Scholar]
- 17.den Dunnen JT, Antonarakis SE. Mutation nomenclature extensions and suggestions to describe complex mutations: A discussion. Hum Mutat. 2000;15(1):7–12. doi: 10.1002/(SICI)1098-1004(200001)15:1<7::AID-HUMU4>3.0.CO;2-N. [DOI] [PubMed] [Google Scholar]
- 18.Thompson JD, Gibson TJ, Plewniak F, Jeanmougin F, Higgins DG. The CLUSTAL_X windows interface: flexible strategies for multiple sequence alignment aided by quality analysis tools. Nucleic Acids Res. 1997;25(24):4876–4882. doi: 10.1093/nar/25.24.4876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kenyon JR, Craig IW. Analysis of the 5′ regulatory region of the human Norrie's disease gene: evidence that a non-translated CT dinucleotide repeat in exon one has a role in controlling expression. Gene. 1999;227(2):181–188. doi: 10.1016/s0378-1119(98)00611-8. [DOI] [PubMed] [Google Scholar]
- 20.Kolitz SE, Takacs JE, Lorsch JR. Kinetic and thermodynamic analysis of the role of start codon/anticodon base pairing during eukaryotic translation initiation. RNA. 2009;15(1):138–52. doi: 10.1261/rna.1318509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Isashiki Y, Ohba N, Yanagita T, Hokita N, Doi N, Nakagawa M, Ozawa M, Kuroda N. Novel mutation at the initiation codon in the Norrie disease gene in two Japanese families. Hum Genet. 1995;95(1):105–108. doi: 10.1007/BF00225085. [DOI] [PubMed] [Google Scholar]
- 22.Schuback DE, Chen ZY, Craig IW, Breakefield XO, Sims KB. Mutations in the Norrie disease gene. Hum Mutat. 1995;5(4):285–292. doi: 10.1002/humu.1380050403. [DOI] [PubMed] [Google Scholar]
- 23.Riveiro-Alvarez R, Trujillo-Tiebas MJ, Gimenez-Pardo A, Garcia-Hoyos M, Cantalapiedra D, Lorda-Sanchez I, Rodriguez de Alba M, Ramos C, Ayuso C. Genotype-phenotype variations in five Spanish families with Norrie disease or X-linked FEVR. Mol Vis. 2005;11:705–712. [PubMed] [Google Scholar]