

Introduction
The concept that cAMP has restricted access to different pools of intracellular effectors was articulated more than 30 years ago. Observations made by Corbin et al. (1977) demonstrated that the two isoenzymes of the major intracellular cAMP target, PKA I and II, are located in different cell compartments. PKA II was recovered predominantly in the particulate fraction of heart extracts, whereas PKA I was mostly soluble. Epinephrine activates the holoenzyme present in the particulate fraction and causes its translocation to the cytosol. In the same vein, Hayes et al. (1979) and Buxton and Brunton (1983) demonstrated that different hormones increase cAMP in cardiac myocytes, but only the β-adrenergic agonists affect heart contractility, providing functional evidence for the selectivity of cAMP action. These findings led to the enunciation of a set of rules describing this unexpected property of cAMP signaling (Steinberg and Brunton, 2001). Although a small diffusible molecule, Steinberg and Brunton (2001) predicted that cAMP does not have equal access to all PKAs present in a cell, implying an uneven cAMP concentration or the presence of compartments of signaling. The demonstration that local β-adrenergic activation does not elicit Ca2+ currents measured at opposite ends of a frog cardiac myocyte provided experimental confirmation of constraints on the spread of the cAMP signal (Jurevicius and Fischmeister, 1996). At about the same time and using unrelated approaches, a field of cell biology was developed that supports the critical role of PKA targeting to different subcellular structures through A kinase anchoring protein (AKAP) scaffolds (Wong and Scott, 2004; Dodge-Kafka et al., 2006). Disruption of these complexes by several means provided additional compelling evidence that physiological cAMP signaling requires PKA anchoring. All of these findings support the conclusion that, functionally, cAMP does not behave as a freely diffusing molecule and that different cAMP concentrations must be present in certain subdomains of a cell. Whereas localized cAMP function is widely accepted and undisputed, the mechanisms causing these localized effects are still a matter of debate. The most obvious obstacle to cAMP diffusion would be the physical barriers generated by membranes or organelles or the presence of buffering systems. An additional hypothesis that has gained credence is that cyclic nucleotide phosphodiesterases (PDEs) provide a functional barrier to cAMP diffusion (Fig. 1 A). According to this view, cAMP diffusion is hindered because of rapid degradation by myriad PDE isoenzymes expressed in a cell. It is undisputed that PDEs have a critical role in the spatiotemporal dynamics of the cyclic nucleotide signal (Conti and Beavo, 2007; Houslay, 2010); however, their contribution to diffusion and generation of cyclic nucleotide gradients or compartments is less clear. Here, we will review the available data in support of, or inconsistent with, this concept (see Table 1 for a summary).
Figure 1.

Scheme illustrating the different hypothetical roles of PDEs in the generation of cyclic nucleotide compartments. Three models are presented. (A) A PDE functions as a barrier to diffusion. (B) A PDE functions as a sink generating a domain of low cAMP. (C) Different, slowly equilibrating compartments are present in a cell with PDE concentration varying among compartments. In this latter model, a PDE contributes to generation of compartments by regulating cAMP concentration and cAMP fluxes between compartments.
Table 1.
Summary of the data on the contribution of different mechanisms to cAMP compartmentalization
| Cell type | AC (cAMP synthesis) | PDE (cAMP degradation) | Hindered diffusion | Cell shape | cAMP buffers | Notes | References |
| HEK293 cells | ND | Yes: PDE inhibition abolishes differences cytosol/membrane | Yes | ND | Yes | Modified CNG channel | Rich et al. (2000, 2001, 2007) |
| HEK293 cells | ND | Yes: Loss of gradient with PDE4 RNAi; no effect of PDE4 inhibitors | No: Computational analysis | ND | ND | PKA/EPAC reporters | Terrin et al. (2006); Oliveira et al. (2010) |
| HEK293 cells | Yes (targeted AC8) | Yes: Loss of membrane compartment after PDE4 inhibition | ND | ND | ND | Gravin-mediated complex | Willoughby et al. (2006) |
| MEFs | ND | No: PDE4B ablation increases cytosol/membrane gradient | ND | ND | ND | CNGC/EPAC reporters | Blackman et al. (2011) |
| Neonatal cardiac myocytes | ND | Yes: IBMX abolishes local cAMP accumulation | ND | ND | ND | PKA reporter | Zaccolo and Pozzan (2002); Mongillo et al. (2004) |
| Neonatal cardiac myocytes | Yes (receptor-dependent βAR vs. PGE2) | Yes: PDE inhibition decreases the PKA-phosphorylation gradients | Yes | ND | Yes | Membrane/cytosol restricted diff. local cAMP uncaging | Saucerman et al. (2006) |
| Neurons (dendrites) | Yes: Role of negative regulatory loop | No | Yes: Dendrite diameter | Bacskai et al. (1993); Neves et al. (2008) | |||
| Adult cardiac myocytes | Differences β1/β2 responses | No: No PDE effect on diffusion of the β2AR restricted signal | Yes | ND | ND | HCN2-cAMP probe in transgenic mice | Nikolaev et al. (2006) |
| Vascular endothelium | Yes: Membrane vs. cytosol AC | Yes: PDE4D4 displacement affects membrane cAMP; Rolipram disrupts the gradient | Yes | Yes | Yes | Manipulation of site of cAMP synthesis | Sayner et al. (2006); Feinstein et al. (2012) |
Are the structural and kinetic properties of PDEs compatible with a function as a barrier to cAMP diffusion?
The superfamily of PDEs includes 21 genes coding for proteins with distinct properties (Conti and Beavo, 2007). To add to the complexity, multiple splicing variants increase the number of PDE proteins expressed in mammalian cells and tissues to >50. PDE4, PDE7, and PDE8 families hydrolyze cAMP; PDE1, PDE2, PDE3, PDE10, and PDE11 hydrolyze both cAMP and cGMP (Bender and Beavo, 2006); and PDE5, PDE6, and PDE9 are cGMP specific. The affinity of different isoenzymes for cAMP varies widely. PDE3 and PDE8 have affinities for cAMP in the range of 10–100 nM (Manganiello et al., 1995). PDE4s are abundant in most cells and have an affinity in the range of 2–8 µM (Salanova et al., 1998). PDE1C has similar affinity for cAMP and cGMP in the low micromolar range (Yan et al., 1995). Other dual-substrate PDEs have low affinity for cAMP that often exceeds 10 µM, such as PDE1A or PDE2 (Bender and Beavo, 2006). Estimates of cAMP concentrations in different models vary according to the cell, ranging from 10 nM to 10–50 µM (Rich et al., 2001; Iancu et al., 2007; Neves et al., 2008; Blackman et al., 2011; Börner et al., 2011; Feinstein et al., 2012). Given that multiple PDE isoenzymes are expressed, the cell is endowed with enzymes that degrade cAMP over a wide range of concentrations. As a consequence, cAMP degradation is rarely saturated certainly under basal, unstimulated conditions or during hormone and neurotransmitter activation. Mathematical models have demonstrated that the Km of a PDE affects the steepness of the cAMP gradients (Feinstein et al., 2012).
Adenylyl cyclases are characterized by a relatively slow turnover number (20/s) compared with cAMP diffusion (Sunahara et al., 1996). This kinetic property prevents accumulation of the cAMP product in the vicinity of the enzyme unlike that described, for instance, for the Ca2+ channels (Rich et al., 2000). Yet, sequestration of these enzymes in different membrane subdomains may contribute to compartmentalized signaling (Cooper and Crossthwaite, 2006). If one considers the kcat of the different PDE enzymes, these are in the order of 5–100/s (Rocque et al., 1997; Salanova et al., 1998). Only the retina-specific PDE6 has a kcat of approximately 1,000/s, a property compatible with a role in limiting cGMP diffusion in the rod outer segment. Given this high turnover number and local concentration, PDE6 and guanylyl cyclases play an important role in generating cGMP gradients in this specialized structure (Olson and Pugh, 1993). The 100-fold lower kcat of other PDEs renders it unlikely that the cyclic nucleotide kinetics measured in the retina are possible in other cells.
It should be noted that PDE activity is dynamic and changes during the time course of hormone/neurotransmitter stimulation (Conti and Jin, 1999). For instance, PDE4s are phosphorylated and activated by PKA (Sette and Conti, 1996); thus, as cAMP and PKA activity increase in response to hormone stimulation, PKA serves to terminate the cAMP signal by activating PDE4. In some cases, this feedback occurs in a macromolecular complex, in which both PKA and PDE4 bind to the same scaffold AKAP (Dodge-Kafka et al., 2006). Similar feedback regulation of cGMP signaling via PKG has been described for PDE5 (Francis et al., 2011). Other PDEs are regulated by numerous intracellular signals (Bender and Beavo, 2006). Thus, it is possible that during cell stimulation, cAMP hydrolysis may reach sufficient velocity to impact local cyclic nucleotide concentration and equilibration between compartments. It has been difficult to evaluate this possibility, however, because of the limitations of cell-free measurements of PDE activity (see below).
An additional property of the PDE system potentially important in the genesis of cAMP compartments is that activation of different Gs protein–coupled receptors leads to activation of different PDE isoenzymes. For instance, activation of β1ARs in cardiac myocytes causes preferential activation of the PDE4D variant PDE4D8, whereas stimulation of the related β2AR causes activation of a different variant, PDE4D5 (Richter et al., 2008). More recent data show that β1ARs but not β2ARs activate a splicing variant of PDE4B, confirming that different receptors cause activation of distinct PDEs (Mika et al., 2014). Similarly, differential sensitivity of PKA type I and II to inhibition by different PDEs is consistent with the idea that PKA and PDEs function together in discrete subcellular compartments, likely in macromolecular complexes organized by scaffold proteins (Di Benedetto et al., 2008). Thus, one can envision that selective activation of PKA and PDEs by a given receptor can lead to changes in cAMP in a subcompartment without affecting other regions of a cell.
PDE subcellular localization/anchoring/interaction with scaffold proteins
Nonuniform concentration of PDEs in the cell is consistent with the large body of work published over the last two decades, opening the possibility that high concentrations of PDEs can be achieved in distinct subcellular compartments (Houslay, 2010; Francis et al., 2011). Isoforms of PDE2, PDE3, PDE4, PDE5, PDE6, PDE7, and PDE10 are recovered in the particulate fraction of homogenates from different tissues, indicating direct interaction of the isoenzymes with different organelles or indirect anchoring through other mechanisms (McCahill et al., 2008; Francis et al., 2011). PDE6 isoenzymes are inserted into the membrane through posttranslational prenylation and carboxymethylation (Anant et al., 1992). A splicing variant of PDE2 with a unique N terminus is recovered in the particulate fraction of brain homogenates (Yang et al., 1994), whereas other variants are not. Similarly, different PDE3A forms are detected in heart homogenate (Shakur et al., 2000), with those possessing domains of hydrophobic helices being particulate, whereas shorter forms are recovered in the soluble fraction. A splicing variant of PDE4A, PDE4A1, which shares most of its sequence with all other PDE4A isoforms except for its variant-specific N terminus, is recovered mostly in the particulate fraction, providing a strong indication that the N terminus of different splicing variants may be an important determinant of PDE4 localization (Houslay, 2010). Subcellular enrichment of PDEs in discrete compartments using immunofluorescence has also been widely reported (Juilfs et al., 1997; Jin et al., 1998; Verde et al., 2001; Mongillo et al., 2004; Stefan et al., 2007). Using expression of recombinant proteins and, in many cases, coimmunoprecipitation with endogenous proteins, it has been shown that PDE4s interact with several classes of scaffold proteins. The most notable example is the interaction of PDE4D isoforms with various AKAPs, including AKAP18 (McSorley et al., 2006), mAKAP (Dodge et al., 2001), and AKAP450/Yotiao (Taskén et al., 2001; Terrenoire et al., 2009). Of note, these scaffold proteins also interact with PKA, indicating that the PKA–PDE4 feedback may function within a macromolecular complex. A complete survey of all the PDE interactions with scaffolds can be found in recent reviews (McCahill et al., 2008; Houslay, 2010).
It also should be mentioned that PDE targeting is often dynamic and may be regulated, for instance, by phosphorylation. The phosphorylation of a splicing variant of PDE10, PDE10A2, coincides with translocation of the protein to the cytosolic compartment, again suggesting that this posttranslational modification controls the interaction with the membrane or with anchoring proteins (Charych et al., 2010). Dissociation of PDE4D from an AKAP (Carlisle Michel et al., 2004) or from complexes with the β-adrenergic receptors upon receptor ligation has been reported (Richter et al., 2008, 2013; De Arcangelis et al., 2009). Binding of PDE inhibitors to the catalytic site may also affect localization, as shown for the PDE4 variant PDE4A4 (Day et al., 2011). Thus, PDE subcellular localization is often dynamic, a property which should be taken into consideration when investigating cyclic nucleotide compartments. From the above examples, there is ample evidence that cAMP hydrolysis can be focal and localized in subcellular compartments or microdomains.
PDE concentration in subcellular compartments
Do their properties allow PDEs to reach concentrations sufficient to prevent cAMP diffusion? To answer this question, accurate methods to assess local PDE concentrations in a cell are needed. Measuring PDE activity and calculating its molar concentration would appear to be a simple task, but reliable measurements of the actual concentration of a PDE in a given compartment are still lacking. PDE activity can be measured in a cell extract and the actual concentration in a cell then calculated after correction for cell volume. However, it is not clear whether activities measured in cell-free preparations reflect activities in an intact cell; indeed, data are available showing that this assumption may be incorrect. We have revisited this issue by calculating the rate of decay of cAMP with live cell cAMP sensors under conditions where synthesis should be negligible (Fig. 2). In this experimental paradigm, cells expressing β-adrenergic receptors are stimulated with an agonist such as isoproterenol. When the cAMP concentration has reached a maximum, excess antagonist is added and the rate of decay is followed. This rate of decay should be proportional to the rate of degradation in a compartment and/or the rate of diffusion from the compartment. With these measurements, we estimate PDE activity in situ at the plasma membrane of 2–6 µM/min, which corresponds to a concentration of PDE protein in the low nanomolar range. By repeating measurements in cells derived from PDE4 knockout mice, one can extrapolate the contribution of different PDE4 isoforms in a local environment. Using this strategy, we calculated a concentration of PDE4B in the membrane compartment of cardiac myocytes in the order of 3–4 nM. These measurements suggest PDE4 concentrations 100-fold lower than those used for mathematical simulations of cAMP signaling in the cell (Neves et al., 2008; Oliveira et al., 2010), requiring a reassessment of the contribution of a PDE4 to signal compartments. It should also be mentioned that this method assesses PDE activity after cAMP stimulation and, thus, under conditions in which the PKA–PDE feedback is activated. Therefore, one should expect even lower PDE activity under basal conditions.
Figure 2.
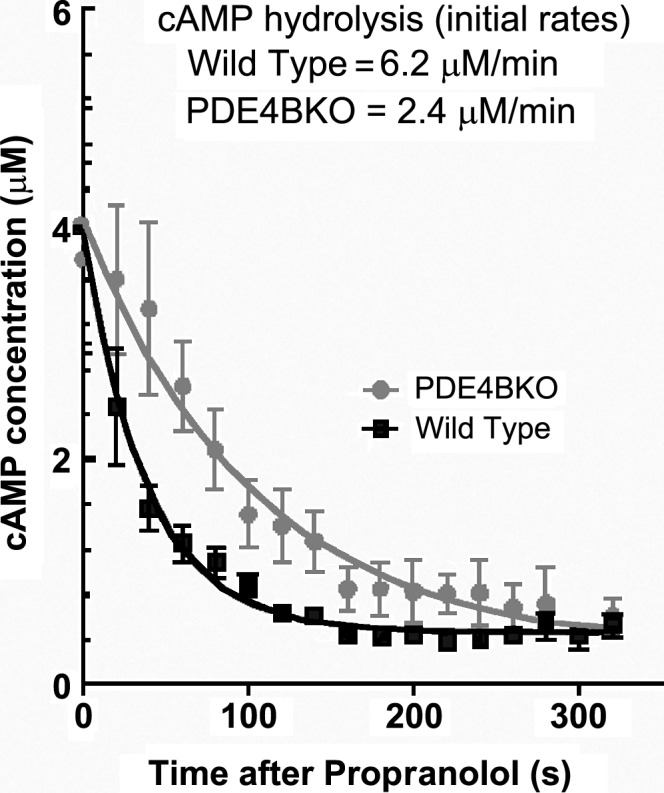
Rate of cAMP decay at the membrane in wild-type and PDE4BKO neonatal cardiac myocytes. Myocytes expressing EPAC2-PM were stimulated with 10 nM isoproterenol, followed by treatment with 1 µM propranolol 80 s later. R/R0 recordings for each cell were transformed into concentrations of cAMP using the equation of Börner et al. (2011). Data were averaged and fitted with an exponential decay equation, and the initial rate of cAMP hydrolysis was calculated from the K constant and initial [cAMP]. Data represent the mean ± SEM of n ≥ 25 cells analyzed.
Pharmacological inhibition of PDEs allows spreading of the cAMP signal
One of the pillars of evidence supporting the theory that PDEs constitute a barrier to cyclic nucleotide diffusion is the ubiquitous finding that PDE inhibition increases the spreading of the cAMP signal. Classical experiments performed in frog ventricular myocytes by Jurevicius and Fischmeister (1996) and Fischmeister et al. (2006) demonstrated that a local β-adrenergic stimulation could be detected at distal L-type Ca2+ channels only after PDE inhibition with IBMX. Similar observations have been reported for activation of the CFTR channel, where stimulation of adenosine receptors could activate the channel at a distance only in the presence of PDE4 inhibitors (Barnes et al., 2005). Together, all these data strongly imply that when PDEs are inhibited, cAMP induces a broader set of actions, implying wide-ranging diffusion.
Direct evidence for extended diffusion of cAMP signals when PDEs are inhibited has been obtained with the development of live cell cAMP/PKA sensors (Zaccolo et al., 2000; Nikolaev et al., 2004). The highly structured and compartmentalized cardiac myocytes have been used to show that PDE inhibition causes broad spreading of an otherwise localized signal. In neonatal cardiomyocytes, β-adrenergic stimulation causes activation of a PKA sensor localized in proximity to the Z bands. When PDEs are inhibited with IBMX or PDE4 inhibitors, sensor activation is no longer localized to the Z bands but occurs diffusely throughout the cell (Zaccolo and Pozzan, 2002; Mongillo et al., 2004). On the basis of this finding, it has been proposed that PDE4s limit the diffusion of the cAMP signal away from these regions, therefore acting as a barrier (Fig. 1 A). Immunolocalization data indicate PDE4 enrichment in these areas (Mongillo et al., 2004; Leroy et al., 2011). When a nonanchored unimolecular HCN2-cAMP sensor was used in adult cardiac myocytes, no striated pattern of cAMP accumulation could be detected (Nikolaev et al., 2006). It has been reasoned that nonanchored sensors diffuse during the measurements, thus preventing detection of local cAMP accumulation in the Z bands. β1- and β2-adrenergic responses were shown to be spatially different, with the β1 signal being far reaching, whereas the β2 signal is restricted to small subcellular compartments. PDE inhibition did not induce spreading of this spatially restricted β2AR response, a finding inconsistent with a PDE role as a diffusional barrier. Spatial restriction of the β2AR signal is lost in rat failing heart (Nikolaev et al., 2010), underscoring the physiological relevance of compartments. In this rat model, the β2AR-restricted response was sensitive to rolipram.
In a slightly different enunciation of the barrier function caused by cAMP hydrolysis, it has been proposed that PDEs, and PDE4 in particular, protect a compartment or a macromolecular complex from cAMP influx. In other words, PDEs function as a sink, generating subcellular domains where cAMP concentration is maintained sufficiently low to prevent PKA activation (see below and Fig. 1 B; Terrin et al., 2006).
Although the above findings are consistent with increased cAMP diffusion when PDEs are inhibited, there are additional effects of PDE inhibition that need to be considered in interpreting the data. By altering cAMP degradation and increasing cAMP concentration, it is possible that important mechanisms of cAMP buffering become saturated (e.g., cAMP binding to the PKA regulatory subunit). Computational models have addressed some of these issues; however, these additional effects need to be more thoroughly assessed.
In cardiac myocytes, phosphorylation of PKA substrates has been used to map different subcellular compartments. Phospholamban (PLB) is a critical modulator of Ca2+ reuptake in the sarcoplasmic reticulum, as it inhibits the activity of the sarcoplasmic reticulum Ca2+ transport ATPase (SERCA). PKA phosphorylation of PLB relieves its negative constraint on SERCA and Ca2+ reuptake, a regulation critical for the relaxation of the cardiac muscle (lusitropic effects). Whereas activation of β1-adrenergic receptors causes an increase in phosphorylation of PLB by PKA, other ligands such as prostaglandin E1 or E2 (PGE1/2) do not affect the PLB phosphorylation state (Di Benedetto et al., 2008; Liu et al., 2012). A compartmentalized PGE2-dependent cAMP response was further documented using different fluorescence resonance energy transfer (FRET) probes that monitor PKA-mediated phosphorylations (Liu et al., 2012). Whereas isoproterenol causes a major increase in FRET signal from a sarcoplasmic reticulum–anchored AKAR3 probe, PGE2 has no effect. Some PGE2 responses in this compartment can be restored by treating the cells with the PDE4 inhibitor rolipram. On the basis of these findings, the authors propose that PGE2 responses in cardiomyocytes are limited to the plasma membrane because PDE4D is readily phosphorylated by activation of these receptors. This activation prevents the diffusion of cAMP generated from the membrane to the SR compartment. Of note, Saucerman et al. (2006) reported that PGE2 response in the cytosol of cardiac myocytes is higher than that in the membrane (see below). Nevertheless, PDE4D regulation of the PKA pool that controls PLB phosphorylation is consistent with numerous other observations. For instance, it has been reported that PDE4D, but not PDE4B, affects PLB phosphorylation by PKA (Beca et al., 2011; Leroy et al., 2011). This finding would indicate that PDE4D, but not PDE4B, protects the PKA–PLB complex from cAMP access. This kind of experiment strongly suggests that PDE4D activation may be a mechanism that limits cAMP diffusion.
There is additional experimental evidence indicating that PDEs, and in particular PDE4s, act as a barrier, protecting a macromolecular complex or a subdomain from cAMP intrusion. Using anchored PKA I and PKA II sensors, it has been shown that their activation is dependent on the ligand used and is sensitive to different PDE inhibitors (Di Benedetto et al., 2008). A microdomain where cAMP is lower than the bulk cytosol is also present in the caveolae (Iancu et al., 2007).
A complex formed by the relaxin family peptide receptor 1 (RXFP1), the scaffold protein AKAP79, the adenylyl cyclase AC2, β-arrestin 2, and PDE4D3 has been described in HEK293 cells overexpressing the receptor (Halls and Cooper, 2010). In this model, relaxin causes an increase in cAMP in a biphasic manner, with the most sensitive response observed in the femtomolar range. This response is obliterated in the presence of a PDE4 inhibitor. On the basis of these data and immunoprecipitation experiments, the authors conclude that the PDE4D3–PKA–β-arrestin complex generates a subdomain where cAMP is maintained at low level, necessary for a highly sensitive relaxin signal. These findings support a role for a PDE to act as a sink to create a microdomain of low cAMP (Fig. 1 B). A similar conclusion is indicated by recent data in which the effect of antagonists of β1AR signaling was investigated (Richter et al., 2013). The receptor is in complex with PDE4, and ligation of the receptor with antagonists causes dissociation of the complex. Using different experimental approaches, it was shown that although antagonists decrease bulk cAMP in a cell, they produce a local increase in cAMP in the vicinity of the receptor because of PDE4 displacement (Richter et al., 2013). These findings again support the possibility that PDEs may be able to maintain a subdomain of low cAMP by acting as a sink (Fig. 1 B).
PDEs and equilibration of membrane/cytosol compartments
A complementary experimental paradigm used to explore nonuniform cAMP concentration in a cell and the role of PDEs was developed by comparing cytosolic and membrane cAMP with different sensors. By expressing a modified olfactory CNG channel to monitor cAMP levels near the plasma membrane in cell populations and single cells, Rich et al. (2000, 2001) demonstrated that although cAMP signals after PGE stimulation are transient, measurements of global cAMP in the bulk cytosol showed a sustained response. Further experiments manipulating PKA activity documented that the transient increase in cAMP at the membrane is caused by activation of PDE4s (Rich et al., 2007). Importantly, activation of the channel at the membrane was insensitive to washout of cAMP in the cytosol (Rich et al., 2000). These findings provided a first indication that cAMP concentrations at the membrane and in the bulk cytosol of a cell may be different and the two compartments do not equilibrate in the timeframe of the experiment. Computational modeling of the two compartments suggests that PDE activity by itself would not be sufficient to stabilize the compartmentalization of the cyclic nucleotide (Rich et al., 2001), although it did affect the rate of equilibration between the two compartments modeled. Thus, diffusional restrictions between the membrane and the cytosol, as well as differential regulation of PDE activity, are required to generate temporally distinct patterns of cAMP accumulation (Fig. 1 C).
In HEK293 cells probed with either PKA-FRET reporters or unimolecular exchange protein directly activated by cAMP (EPAC) probes, Terrin et al. (2006) measured the cAMP dynamics after PGE2 stimulation in the bulk cytosol or at the membrane. Differences in FRET emission were consistently found, with the reporter targeted to the membrane giving higher responses than the probe confined to the cytosol. The PKA holoenzyme sensor gave larger differences, a finding interpreted by the authors as an indication that PKA plays a role in generation of these gradients by activating PDE4. These measurements imply that differences in cAMP concentration at the membrane and in the cytosol are present, a conclusion similar to that reached by Rich et al. (2001). The kinetics of activation of the reporters were also different, with the membrane probe being activated faster than the cytosol probe, which is a strong argument in favor of compartmentalization of cAMP. The authors propose that PDE-mediated hydrolysis is the major determinant of these differences. In support of this conclusion, they showed that when PDE4D is ablated with siRNAs, the gradient is no longer detected and cAMP reaches higher concentrations in the cytosol than at the membrane. Knockdown of a different PDE, PDE4B, has no effect on the cytosol/membrane gradient. In contrast to the knockdown experiments, the difference in response between membrane and cytosol was maintained in the presence of rolipram and IBMX. The authors argue that the PDE inhibitors are ineffective in dissipating the gradient because they are competitive and would not be effective at the membrane when cAMP is high. This reasoning is difficult to reconcile with the fact that the concentrations of inhibitor used are >10-fold higher than the cAMP concentration expected at the membrane and that, even if PDEs were not completely inhibited, changes in cytosol/membrane ratios should have been detected. Differences between cytosol and membrane FRET responses were reproduced in a subsequent paper where modeling was used to verify the concept of PDE-dependent limited cAMP diffusion (Oliveira et al., 2010). In this study, changing the rate of diffusion of cAMP modified but did not eliminate the gradient, leading to the conclusion that diffusion plays a limited role in the generation of the cytosol/membrane compartment.
Using a PKA phosphorylation–sensitive probe (AKAR2) in neonatal cardiac myocytes, Saucerman et al. (2006) again found that β-adrenergic agonists stimulate a membrane probe faster than a cytosolic probe, whereas the response to PGE1 is the opposite. They hypothesized that a restricted diffusion was the cause of these divergent time courses with a diffusional constant between the membrane and cytosol of 2 µm2/s. Gradients of phosphorylation were also observed by uncaging a cAMP analogue in the cytosol. Mathematical modeling allowed them to investigate the contribution of different components including cAMP buffering or PDEs. Inhibition of the PDE activity substantially decreased the PKA phosphorylation gradients.
Using mouse embryonic fibroblasts (MEFs) deficient in different PDE4s derived from the respective PDE4KO mice, a somewhat different conclusion was reached regarding the function of a specific PDE4 in a compartment (Blackman et al., 2011). The EPAC-cAMP probes targeted to the cytosol (EPAC-cyto) and to the plasma membrane (EPAC-PM) detected changes in cAMP induced by both β-adrenergic agonists and PGE2. Moreover, PDE4D ablation caused a marked increase in cAMP both in the bulk cytosol as well as in a putative membrane compartment, a finding different from that reported for HEK293 cells (Terrin et al., 2006). When PDE4B was ablated, an effect on cAMP decay was observed at the membrane but not the cytosol, consistent with the concept of a sequestered cAMP membrane compartment. However, PDE4B ablation accentuates rather than dissipates differences between cytosol and membrane compartments. This latter finding indicates that PDE4B does not function as a barrier to cAMP diffusion and is not necessary to stabilize this compartment. Phosphorylation of membrane and cytoplasmic substrates independently confirmed the divergent effects of PDE4B ablation in the two compartments. Of note, Terrin et al. (2006) did not detect any effect of PDE4B down-regulation on membrane cAMP. These differences may be caused by the cells used (MEFs vs. HEK293 cells) or by quantitative differences in PDE down-regulation in knockdown versus knockout. A possibility yet to be explored is that PDEs other than PDE4B control cAMP diffusion out of this particular membrane compartment.
Endothelial cells play a critical function in controlling vascular permeability, and development of lung edema associated with vascular leakage indicates how critical this regulation may be (Sayner, 2011). Cyclic nucleotide signaling through PKA and EPAC plays a critical role in the control of endothelial cell permeability via regulation of the cytoskeleton (Sayner, 2011). Differences in biological effects of membrane and cytosolic cAMP on endothelial cell function are an additional example of two cAMP compartments with distinct functions that do not appear to equilibrate readily. Cyclic AMP production at the plasma membrane reinforces pulmonary microvascular endothelial barrier integrity, whereas cAMP production in the cytosol disrupts this barrier (Sayner et al., 2006). This conclusion is supported by experiments manipulating cAMP at the membrane via the adenylyl cyclase activator forskolin, or in the cytosol either by expression of an engineered soluble cyclase (sACI/II), by using the exotoxin cyclase produced by Pseudomonas aeruginosa (exoY gene product), or by manipulation of the endogenous soluble adenylyl cyclase apparently expressed in these cells (Sayner et al., 2006). The Tau protein phosphorylation by PKA in the cytosol alters endothelial barrier integrity, whereas PKA phosphorylation of filamin at the membrane reinforces the barrier (Zhu et al., 2010). PDE4D has been implicated in vascular permeability and in the control of cAMP equilibration between these two compartments (Table 1).
Conclusions
A large body of biochemical and functional data has established that the cAMP concentration in a cell is not uniform. A localized membrane signal activates a restricted pool of effector molecules such as PKA, EPAC, or CNGs, often without perturbing other compartments. This restricted activation of a subpopulation of effectors is associated with distinct biological responses, thus enforcing signaling specificity. This property of cAMP signaling is clearly dependent on the many PDE isoenzymes expressed in a cell. However, the contribution of different PDEs to signaling is complex (Table 1). We favor a scenario in which PDEs are located in distinct, slowly equilibrating compartments (Fig. 1 C). Any given PDE plays a critical role in controlling cAMP concentration in each of these compartments because of its enzymatic properties, regulation, and participation in macromolecular complexes. Indirectly, and by controlling cAMP concentration, PDEs regulate cAMP fluxes and the equilibration time between different compartments. Aside from the extreme case of the retina outer segment where PDE concentration and kinetic properties generate compartmentalization, the activity of PDEs in other cells alone is not sufficient to generate stable pools of cAMP. PDEs cooperate with other mechanisms that limit diffusion to stabilize and isolate a compartment from the surrounding cellular environment (Fig. 1 C). These additional components likely include buffering systems, physicochemical properties of the intracellular environment, cell shape, and ratio surface/volume, as well other physical barriers (Fig. 1, A and B).
Future studies should be devoted to dissecting the contribution of each of these mechanisms to cyclic nucleotide compartmentalization. Computational models will certainly aid in understanding how the diffusion of the cAMP signal is controlled. For realistic modeling, more accurate parameters for the concentration of PDEs in each compartment should be generated. We have proposed a strategy based on in situ measurements of the rate of decay that should aid in this task. Using cAMP sensors tethered to different macromolecular complexes, one should be able to obtain more accurate information on PDE concentration within restricted compartments and, possibly, macromolecular complexes. Understanding the boundaries, size, and location of a compartment in which a PDE functions should also help in refining the different models. The kinetic properties of signaling components in isolation or when assembled in macromolecular complexes have not been systematically investigated, but they should provide additional important information on how local cAMP signaling develops. Novel strategies to probe the physicochemical properties of the different subcellular compartments and their contributions to diffusion should also be developed. All these strategies will provide a better understanding of local signaling, its role during disease development and, ultimately, more accurate predictions of the consequences of a pharmacological intervention affecting cAMP signaling.
This Perspectives series includes articles by Karpen, Kapiloff et al., Rich et al., and Saucerman et al.
Acknowledgments
We are indebted to Dr. Thomas C. Rich (University of South Alabama, Mobile, AL) for the extensive discussions of this topic.
Work performed in our laboratory is supported by National Institutes of Health grants HL0927088 and HL107960. D. Mika is supported by a fellowship from the American Heart Association.
Olaf S. Andersen served as editor.
Footnotes
Abbreviations used in this paper:
- AKAP
- A kinase anchoring protein
- EPAC
- exchange protein directly activated by cAMP
- FRET
- fluorescence resonance energy transfer
- MEF
- mouse embryonic fibroblast
- PDE
- phosphodiesterase
- PLB
- phospholamban
References
- Anant J.S., Ong O.C., Xie H.Y., Clarke S., O’Brien P.J., Fung B.K. 1992. In vivo differential prenylation of retinal cyclic GMP phosphodiesterase catalytic subunits. J. Biol. Chem. 267:687–690 [PubMed] [Google Scholar]
- Bacskai B.J., Hochner B., Mahaut-Smith M., Adams S.R., Kaang B.K., Kandel E.R., Tsien R.Y. 1993. Spatially resolved dynamics of cAMP and protein kinase A subunits in Aplysia sensory neurons. Science. 260:222–226 10.1126/science.7682336 [DOI] [PubMed] [Google Scholar]
- Barnes A.P., Livera G., Huang P., Sun C., O’Neal W.K., Conti M., Stutts M.J., Milgram S.L. 2005. Phosphodiesterase 4D forms a cAMP diffusion barrier at the apical membrane of the airway epithelium. J. Biol. Chem. 280:7997–8003 10.1074/jbc.M407521200 [DOI] [PubMed] [Google Scholar]
- Beca S., Helli P.B., Simpson J.A., Zhao D., Farman G.P., Jones P.P., Tian X., Wilson L.S., Ahmad F., Chen S.R., et al. 2011. Phosphodiesterase 4D regulates baseline sarcoplasmic reticulum Ca2+ release and cardiac contractility, independently of L-type Ca2+ current. Circ. Res. 109:1024–1030 10.1161/CIRCRESAHA.111.250464 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bender A.T., Beavo J.A. 2006. Cyclic nucleotide phosphodiesterases: molecular regulation to clinical use. Pharmacol. Rev. 58:488–520 10.1124/pr.58.3.5 [DOI] [PubMed] [Google Scholar]
- Blackman B.E., Horner K., Heidmann J., Wang D., Richter W., Rich T.C., Conti M. 2011. PDE4D and PDE4B function in distinct subcellular compartments in mouse embryonic fibroblasts. J. Biol. Chem. 286:12590–12601 10.1074/jbc.M110.203604 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Börner S., Schwede F., Schlipp A., Berisha F., Calebiro D., Lohse M.J., Nikolaev V.O. 2011. FRET measurements of intracellular cAMP concentrations and cAMP analog permeability in intact cells. Nat. Protoc. 6:427–438 10.1038/nprot.2010.198 [DOI] [PubMed] [Google Scholar]
- Buxton I.L., Brunton L.L. 1983. Compartments of cyclic AMP and protein kinase in mammalian cardiomyocytes. J. Biol. Chem. 258:10233–10239 [PubMed] [Google Scholar]
- Carlisle Michel J.J., Dodge K.L., Wong W., Mayer N.C., Langeberg L.K., Scott J.D. 2004. PKA-phosphorylation of PDE4D3 facilitates recruitment of the mAKAP signalling complex. Biochem. J. 381:587–592 10.1042/BJ20040846 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Charych E.I., Jiang L.X., Lo F., Sullivan K., Brandon N.J. 2010. Interplay of palmitoylation and phosphorylation in the trafficking and localization of phosphodiesterase 10A: implications for the treatment of schizophrenia. J. Neurosci. 30:9027–9037 10.1523/JNEUROSCI.1635-10.2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conti M., Beavo J. 2007. Biochemistry and physiology of cyclic nucleotide phosphodiesterases: essential components in cyclic nucleotide signaling. Annu. Rev. Biochem. 76:481–511 10.1146/annurev.biochem.76.060305.150444 [DOI] [PubMed] [Google Scholar]
- Conti M., Jin S.L. 1999. The molecular biology of cyclic nucleotide phosphodiesterases. Prog. Nucleic Acid Res. Mol. Biol. 63:1–38 10.1016/S0079-6603(08)60718-7 [DOI] [PubMed] [Google Scholar]
- Cooper D.M., Crossthwaite A.J. 2006. Higher-order organization and regulation of adenylyl cyclases. Trends Pharmacol. Sci. 27:426–431 10.1016/j.tips.2006.06.002 [DOI] [PubMed] [Google Scholar]
- Corbin J.D., Sugden P.H., Lincoln T.M., Keely S.L. 1977. Compartmentalization of adenosine 3′:5′-monophosphate and adenosine 3′:5′-monophosphate-dependent protein kinase in heart tissue. J. Biol. Chem. 252:3854–3861 [PubMed] [Google Scholar]
- Day J.P., Lindsay B., Riddell T., Jiang Z., Allcock R.W., Abraham A., Sookup S., Christian F., Bogum J., Martin E.K., et al. 2011. Elucidation of a structural basis for the inhibitor-driven, p62 (SQSTM1)-dependent intracellular redistribution of cAMP phosphodiesterase-4A4 (PDE4A4). J. Med. Chem. 54:3331–3347 10.1021/jm200070e [DOI] [PubMed] [Google Scholar]
- De Arcangelis V., Liu R., Soto D., Xiang Y. 2009. Differential association of phosphodiesterase 4D isoforms with beta2-adrenoceptor in cardiac myocytes. J. Biol. Chem. 284:33824–33832 10.1074/jbc.M109.020388 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Benedetto G., Zoccarato A., Lissandron V., Terrin A., Li X., Houslay M.D., Baillie G.S., Zaccolo M. 2008. Protein kinase A type I and type II define distinct intracellular signaling compartments. Circ. Res. 103:836–844 10.1161/CIRCRESAHA.108.174813 [DOI] [PubMed] [Google Scholar]
- Dodge K.L., Khouangsathiene S., Kapiloff M.S., Mouton R., Hill E.V., Houslay M.D., Langeberg L.K., Scott J.D. 2001. mAKAP assembles a protein kinase A/PDE4 phosphodiesterase cAMP signaling module. EMBO J. 20:1921–1930 10.1093/emboj/20.8.1921 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dodge-Kafka K.L., Langeberg L., Scott J.D. 2006. Compartmentation of cyclic nucleotide signaling in the heart: the role of A-kinase anchoring proteins. Circ. Res. 98:993–1001 10.1161/01.RES.0000218273.91741.30 [DOI] [PubMed] [Google Scholar]
- Feinstein W.P., Zhu B., Leavesley S.J., Sayner S.L., Rich T.C. 2012. Assessment of cellular mechanisms contributing to cAMP compartmentalization in pulmonary microvascular endothelial cells. Am. J. Physiol. Cell Physiol. 302:C839–C852 10.1152/ajpcell.00361.2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fischmeister R., Castro L.R., Abi-Gerges A., Rochais F., Jurevicius J., Leroy J., Vandecasteele G. 2006. Compartmentation of cyclic nucleotide signaling in the heart: the role of cyclic nucleotide phosphodiesterases. Circ. Res. 99:816–828 10.1161/01.RES.0000246118.98832.04 [DOI] [PubMed] [Google Scholar]
- Francis S.H., Houslay M.D., Conti M. 2011. Phosphodiesterase inhibitors: factors that influence potency, selectivity, and action. Handbook Exp. Pharmacol. 204:47–84 10.1007/978-3-642-17969-3_2 [DOI] [PubMed] [Google Scholar]
- Halls M.L., Cooper D.M. 2010. Sub-picomolar relaxin signalling by a pre-assembled RXFP1, AKAP79, AC2, beta-arrestin 2, PDE4D3 complex. EMBO J. 29:2772–2787 10.1038/emboj.2010.168 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayes J.S., Brunton L.L., Brown J.H., Reese J.B., Mayer S.E. 1979. Hormonally specific expression of cardiac protein kinase activity. Proc. Natl. Acad. Sci. USA. 76:1570–1574 10.1073/pnas.76.4.1570 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Houslay M.D. 2010. Underpinning compartmentalised cAMP signalling through targeted cAMP breakdown. Trends Biochem. Sci. 35:91–100 10.1016/j.tibs.2009.09.007 [DOI] [PubMed] [Google Scholar]
- Iancu R.V., Jones S.W., Harvey R.D. 2007. Compartmentation of cAMP signaling in cardiac myocytes: a computational study. Biophys. J. 92:3317–3331 10.1529/biophysj.106.095356 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin S.L., Bushnik T., Lan L., Conti M. 1998. Subcellular localization of rolipram-sensitive, cAMP-specific phosphodiesterases. Differential targeting and activation of the splicing variants derived from the PDE4D gene. J. Biol. Chem. 273:19672–19678 10.1074/jbc.273.31.19672 [DOI] [PubMed] [Google Scholar]
- Juilfs D.M., Fülle H.J., Zhao A.Z., Houslay M.D., Garbers D.L., Beavo J.A. 1997. A subset of olfactory neurons that selectively express cGMP-stimulated phosphodiesterase (PDE2) and guanylyl cyclase-D define a unique olfactory signal transduction pathway. Proc. Natl. Acad. Sci. USA. 94:3388–3395 10.1073/pnas.94.7.3388 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jurevicius J., Fischmeister R. 1996. cAMP compartmentation is responsible for a local activation of cardiac Ca2+ channels by beta-adrenergic agonists. Proc. Natl. Acad. Sci. USA. 93:295–299 10.1073/pnas.93.1.295 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leroy J., Richter W., Mika D., Castro L.R., Abi-Gerges A., Xie M., Scheitrum C., Lefebvre F., Schittl J., Mateo P., et al. 2011. Phosphodiesterase 4B in the cardiac L-type Ca2+ channel complex regulates Ca2+ current and protects against ventricular arrhythmias in mice. J. Clin. Invest. 121:2651–2661 10.1172/JCI44747 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu S., Li Y., Kim S., Fu Q., Parikh D., Sridhar B., Shi Q., Zhang X., Guan Y., Chen X., Xiang Y.K. 2012. Phosphodiesterases coordinate cAMP propagation induced by two stimulatory G protein-coupled receptors in hearts. Proc. Natl. Acad. Sci. USA. 109:6578–6583 10.1073/pnas.1117862109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manganiello V.C., Taira M., Degerman E., Belfrage P. 1995. Type III cGMP-inhibited cyclic nucleotide phosphodiesterases (PDE3 gene family). Cell. Signal. 7:445–455 10.1016/0898-6568(95)00017-J [DOI] [PubMed] [Google Scholar]
- McCahill A.C., Huston E., Li X., Houslay M.D. 2008. PDE4 associates with different scaffolding proteins: modulating interactions as treatment for certain diseases. Handbook Exp. Pharmacol. 186:125–166 10.1007/978-3-540-72843-6_6 [DOI] [PubMed] [Google Scholar]
- McSorley T., Stefan E., Henn V., Wiesner B., Baillie G.S., Houslay M.D., Rosenthal W., Klussmann E. 2006. Spatial organisation of AKAP18 and PDE4 isoforms in renal collecting duct principal cells. Eur. J. Cell Biol. 85:673–678 10.1016/j.ejcb.2006.01.005 [DOI] [PubMed] [Google Scholar]
- Mika D., Richter W., Westenbroek R.E., Catterall W.A., Conti M. 2014. PDE4B mediates local feedback regulation of β1-adrenergic cAMP signaling in a sarcolemmal compartment of cardiac myocytes. J. Cell Sci. In press [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mongillo M., McSorley T., Evellin S., Sood A., Lissandron V., Terrin A., Huston E., Hannawacker A., Lohse M.J., Pozzan T., et al. 2004. Fluorescence resonance energy transfer-based analysis of cAMP dynamics in live neonatal rat cardiac myocytes reveals distinct functions of compartmentalized phosphodiesterases. Circ. Res. 95:67–75 10.1161/01.RES.0000134629.84732.11 [DOI] [PubMed] [Google Scholar]
- Neves S.R., Tsokas P., Sarkar A., Grace E.A., Rangamani P., Taubenfeld S.M., Alberini C.M., Schaff J.C., Blitzer R.D., Moraru I.I., Iyengar R. 2008. Cell shape and negative links in regulatory motifs together control spatial information flow in signaling networks. Cell. 133:666–680 10.1016/j.cell.2008.04.025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nikolaev V.O., Bünemann M., Hein L., Hannawacker A., Lohse M.J. 2004. Novel single chain cAMP sensors for receptor-induced signal propagation. J. Biol. Chem. 279:37215–37218 10.1074/jbc.C400302200 [DOI] [PubMed] [Google Scholar]
- Nikolaev V.O., Bünemann M., Schmitteckert E., Lohse M.J., Engelhardt S. 2006. Cyclic AMP imaging in adult cardiac myocytes reveals far-reaching beta1-adrenergic but locally confined beta2-adrenergic receptor-mediated signaling. Circ. Res. 99:1084–1091 10.1161/01.RES.0000250046.69918.d5 [DOI] [PubMed] [Google Scholar]
- Nikolaev V.O., Moshkov A., Lyon A.R., Miragoli M., Novak P., Paur H., Lohse M.J., Korchev Y.E., Harding S.E., Gorelik J. 2010. Beta2-adrenergic receptor redistribution in heart failure changes cAMP compartmentation. Science. 327:1653–1657 10.1126/science.1185988 [DOI] [PubMed] [Google Scholar]
- Oliveira R.F., Terrin A., Di Benedetto G., Cannon R.C., Koh W., Kim M., Zaccolo M., Blackwell K.T. 2010. The role of type 4 phosphodiesterases in generating microdomains of cAMP: large scale stochastic simulations. PLoS ONE. 5:e11725 10.1371/journal.pone.0011725 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olson A., Pugh E.N., Jr 1993. Diffusion coefficient of cyclic GMP in salamander rod outer segments estimated with two fluorescent probes. Biophys. J. 65:1335–1352 10.1016/S0006-3495(93)81177-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rich T.C., Fagan K.A., Nakata H., Schaack J., Cooper D.M., Karpen J.W. 2000. Cyclic nucleotide-gated channels colocalize with adenylyl cyclase in regions of restricted cAMP diffusion. J. Gen. Physiol. 116:147–162 10.1085/jgp.116.2.147 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rich T.C., Fagan K.A., Tse T.E., Schaack J., Cooper D.M., Karpen J.W. 2001. A uniform extracellular stimulus triggers distinct cAMP signals in different compartments of a simple cell. Proc. Natl. Acad. Sci. USA. 98:13049–13054 10.1073/pnas.221381398 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rich T.C., Xin W., Mehats C., Hassell K.A., Piggott L.A., Le X., Karpen J.W., Conti M. 2007. Cellular mechanisms underlying prostaglandin-induced transient cAMP signals near the plasma membrane of HEK-293 cells. Am. J. Physiol. Cell Physiol. 292:C319–C331 10.1152/ajpcell.00121.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richter W., Day P., Agrawal R., Bruss M.D., Granier S., Wang Y.L., Rasmussen S.G., Horner K., Wang P., Lei T., et al. 2008. Signaling from beta1- and beta2-adrenergic receptors is defined by differential interactions with PDE4. EMBO J. 27:384–393 10.1038/sj.emboj.7601968 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richter W., Mika D., Blanchard E., Day P., Conti M. 2013. β1-adrenergic receptor antagonists signal via PDE4 translocation. EMBO Rep. 14:276–283 10.1038/embor.2013.4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rocque W.J., Holmes W.D., Patel I.R., Dougherty R.W., Ittoop O., Overton L., Hoffman C.R., Wisely G.B., Willard D.H., Luther M.A. 1997. Detailed characterization of a purified type 4 phosphodiesterase, HSPDE4B2B: differentiation of high- and low-affinity (R)-rolipram binding. Protein Expr. Purif. 9:191–202 10.1006/prep.1996.0683 [DOI] [PubMed] [Google Scholar]
- Salanova M., Jin S.C., Conti M. 1998. Heterologous expression and purification of recombinant rolipram-sensitive cyclic AMP-specific phosphodiesterases. Methods. 14:55–64 10.1006/meth.1997.0565 [DOI] [PubMed] [Google Scholar]
- Saucerman J.J., Zhang J., Martin J.C., Peng L.X., Stenbit A.E., Tsien R.Y., McCulloch A.D. 2006. Systems analysis of PKA-mediated phosphorylation gradients in live cardiac myocytes. Proc. Natl. Acad. Sci. USA. 103:12923–12928 10.1073/pnas.0600137103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sayner S.L. 2011. Emerging themes of cAMP regulation of the pulmonary endothelial barrier. Am. J. Physiol. Lung Cell. Mol. Physiol. 300:L667–L678 10.1152/ajplung.00433.2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sayner S.L., Alexeyev M., Dessauer C.W., Stevens T. 2006. Soluble adenylyl cyclase reveals the significance of cAMP compartmentation on pulmonary microvascular endothelial cell barrier. Circ. Res. 98:675–681 10.1161/01.RES.0000209516.84815.3e [DOI] [PubMed] [Google Scholar]
- Sette C., Conti M. 1996. Phosphorylation and activation of a cAMP-specific phosphodiesterase by the cAMP-dependent protein kinase. Involvement of serine 54 in the enzyme activation. J. Biol. Chem. 271:16526–16534 10.1074/jbc.271.28.16526 [DOI] [PubMed] [Google Scholar]
- Shakur Y., Takeda K., Kenan Y., Yu Z.X., Rena G., Brandt D., Houslay M.D., Degerman E., Ferrans V.J., Manganiello V.C. 2000. Membrane localization of cyclic nucleotide phosphodiesterase 3 (PDE3). Two N-terminal domains are required for the efficient targeting to, and association of, PDE3 with endoplasmic reticulum. J. Biol. Chem. 275:38749–38761 10.1074/jbc.M001734200 [DOI] [PubMed] [Google Scholar]
- Stefan E., Wiesner B., Baillie G.S., Mollajew R., Henn V., Lorenz D., Furkert J., Santamaria K., Nedvetsky P., Hundsrucker C., et al. 2007. Compartmentalization of cAMP-dependent signaling by phosphodiesterase-4D is involved in the regulation of vasopressin-mediated water reabsorption in renal principal cells. J. Am. Soc. Nephrol. 18:199–212 10.1681/ASN.2006020132 [DOI] [PubMed] [Google Scholar]
- Steinberg S.F., Brunton L.L. 2001. Compartmentation of G protein-coupled signaling pathways in cardiac myocytes. Annu. Rev. Pharmacol. Toxicol. 41:751–773 10.1146/annurev.pharmtox.41.1.751 [DOI] [PubMed] [Google Scholar]
- Sunahara R.K., Dessauer C.W., Gilman A.G. 1996. Complexity and diversity of mammalian adenylyl cyclases. Annu. Rev. Pharmacol. Toxicol. 36:461–480 10.1146/annurev.pa.36.040196.002333 [DOI] [PubMed] [Google Scholar]
- Taskén K.A., Collas P., Kemmner W.A., Witczak O., Conti M., Taskén K. 2001. Phosphodiesterase 4D and protein kinase a type II constitute a signaling unit in the centrosomal area. J. Biol. Chem. 276:21999–22002 10.1074/jbc.C000911200 [DOI] [PubMed] [Google Scholar]
- Terrenoire C., Houslay M.D., Baillie G.S., Kass R.S. 2009. The cardiac IKs potassium channel macromolecular complex includes the phosphodiesterase PDE4D3. J. Biol. Chem. 284:9140–9146 10.1074/jbc.M805366200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Terrin A., Di Benedetto G., Pertegato V., Cheung Y.-F., Baillie G., Lynch M.J., Elvassore N., Prinz A., Herberg F.W., Houslay M.D., Zaccolo M. 2006. PGE(1) stimulation of HEK293 cells generates multiple contiguous domains with different [cAMP]: role of compartmentalized phosphodiesterases. J. Cell Biol. 175:441–451 10.1083/jcb.200605050 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verde I., Pahlke G., Salanova M., Zhang G., Wang S., Coletti D., Onuffer J., Jin S.L., Conti M. 2001. Myomegalin is a novel protein of the golgi/centrosome that interacts with a cyclic nucleotide phosphodiesterase. J. Biol. Chem. 276:11189–11198 10.1074/jbc.M006546200 [DOI] [PubMed] [Google Scholar]
- Willoughby D., Wong W., Schaack J., Scott J.D., Cooper D.M. 2006. An anchored PKA and PDE4 complex regulates subplasmalemmal cAMP dynamics. EMBO J. 25:2051–2061 10.1038/sj.emboj.7601113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong W., Scott J.D. 2004. AKAP signalling complexes: focal points in space and time. Nat. Rev. Mol. Cell Biol. 5:959–970 10.1038/nrm1527 [DOI] [PubMed] [Google Scholar]
- Yan C., Zhao A.Z., Bentley J.K., Loughney K., Ferguson K., Beavo J.A. 1995. Molecular cloning and characterization of a calmodulin-dependent phosphodiesterase enriched in olfactory sensory neurons. Proc. Natl. Acad. Sci. USA. 92:9677–9681 10.1073/pnas.92.21.9677 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Q., Paskind M., Bolger G., Thompson W.J., Repaske D.R., Cutler L.S., Epstein P.M. 1994. A novel cyclic GMP stimulated phosphodiesterase from rat brain. Biochem. Biophys. Res. Commun. 205:1850–1858 10.1006/bbrc.1994.2886 [DOI] [PubMed] [Google Scholar]
- Zaccolo M., Pozzan T. 2002. Discrete microdomains with high concentration of cAMP in stimulated rat neonatal cardiac myocytes. Science. 295:1711–1715 10.1126/science.1069982 [DOI] [PubMed] [Google Scholar]
- Zaccolo M., De Giorgi F., Cho C.Y., Feng L., Knapp T., Negulescu P.A., Taylor S.S., Tsien R.Y., Pozzan T. 2000. A genetically encoded, fluorescent indicator for cyclic AMP in living cells. Nat. Cell Biol. 2:25–29 10.1038/71345 [DOI] [PubMed] [Google Scholar]
- Zhu B., Zhang L., Creighton J., Alexeyev M., Strada S.J., Stevens T. 2010. Protein kinase A phosphorylation of tau-serine 214 reorganizes microtubules and disrupts the endothelial cell barrier. Am. J. Physiol. Lung Cell. Mol. Physiol. 299:L493–L501 10.1152/ajplung.00431.2009 [DOI] [PMC free article] [PubMed] [Google Scholar]