

Abstract
The JNKs are master protein kinases that regulate many physiological processes, including inflammatory responses, morphogenesis, cell proliferation, differentiation, survival and death. It is increasingly apparent that persistent activation of JNKs is involved in cancer development and progression. Therefore, JNKs represent attractive targets for therapeutic intervention with small molecule kinase inhibitors. However, evidence supportive of a tumour suppressor role for the JNK proteins has also been documented. Recent studies showed that the two major JNK proteins, JNK1 and JNK2, have distinct or even opposing functions in different types of cancer. As such, close consideration of which JNK proteins are beneficial targets and, more importantly, what effect small molecule inhibitors of JNKs have on physiological processes, are essential. A number of ATP-competitive and ATP-non-competitive JNK inhibitors have been developed, but have several limitations such as a lack of specificity and cellular toxicity. In this review, we summarize the accumulating evidence supporting a role for the JNK proteins in the pathogenesis of different solid and haematological malignancies, and discuss many challenges and scientific opportunities in the targeting of JNKs in cancer.
Keywords: JNK1, JNK2, PARP14, survival, apoptosis, multiple myeloma, hepatocellular carcinoma, cancer targets
Introduction
Protein kinases are the largest family of enzymes encoded by the human genome and have emerged as one of the most popular drug target class in the pharmaceutical industry (Zhang et al., 2009). The role of protein kinases is to catalyse the transfer of the terminal phosphate group of ATP to specific amino acid residues in target proteins (substrates). This modifies the functions of the target proteins by affecting either the activities or by controlling subcellular localization, degradation and association with other binding partners (Manning et al., 2002). Furthermore, protein kinases themselves can be activated by addition of phosphate groups to their specific amino acid residues, either by an autophosphorylation event or phosphorylation by other kinases. Of these kinases, the MAPKs comprise a well-studied family of serine-threonine kinases that play important regulatory roles in the cell (Johnson and Lapadat, 2002). MAPKs are activated via a kinase signalling cascade in which a MAP3K activates a MAP2K, that in turn activates a MAPK. There are three well-characterized subfamilies of MAPKs in mammals: the ERKs, the p38 kinases and the JNKs, and all three MAPKs have been conserved from unicellular organisms to complex organisms including humans (Johnson and Lapadat, 2002).
The JNK kinase family includes three proteins (JNK1, JNK2 and JNK3) that are encoded by three separate genes jnk1 (Mapk8), jnk2 (Mapk9) and jnk3 (Mapk10), and are alternatively spliced to create at least 10 variants of 46 and 55 kDa (Davis, 2000). Whereas JNK1 and JNK2 are expressed in most tissues, the expression of JNK3 is largely restricted to brain, heart and testes (Bogoyevitch and Kobe, 2006). The JNK proteins – also known as stress-activated protein kinases (SAPKs) – similar to other MAPKs, are activated by a series of phosphorylation events (i.e. the MAPK model) in response to multiple stimuli such as cytokines, growth factors, pathogens, stress, toxins and drugs (Bogoyevitch and Kobe, 2006; Seki et al., 2012; Figure 1). MKK4 and MKK7 are the two MAP2K protein kinases that directly phosphorylate JNKs on threonine 183 (Thr183) and tyrosine (Tyr185) residues present in a conserved tripeptide motif (Thr-Pro-Tyr) within their activation loop (Davis, 2000). In turn, MKK4 and MKK7 are activated via dual phosphorylation by MAP3Ks, such as members of MEKK family, the mixed-lineage kinase family, the apoptosis signal-regulating kinase family, TAK1 and TPL2 (Davis, 2000). Moreover, the kinase activity of JNKs is regulated by interaction with scaffold proteins (Whitmarsh et al., 2001), as well as dual-specificity phosphatases (Owens and Keyse, 2007) and NF-κB transcription factors (Papa et al., 2006).
Figure 1.
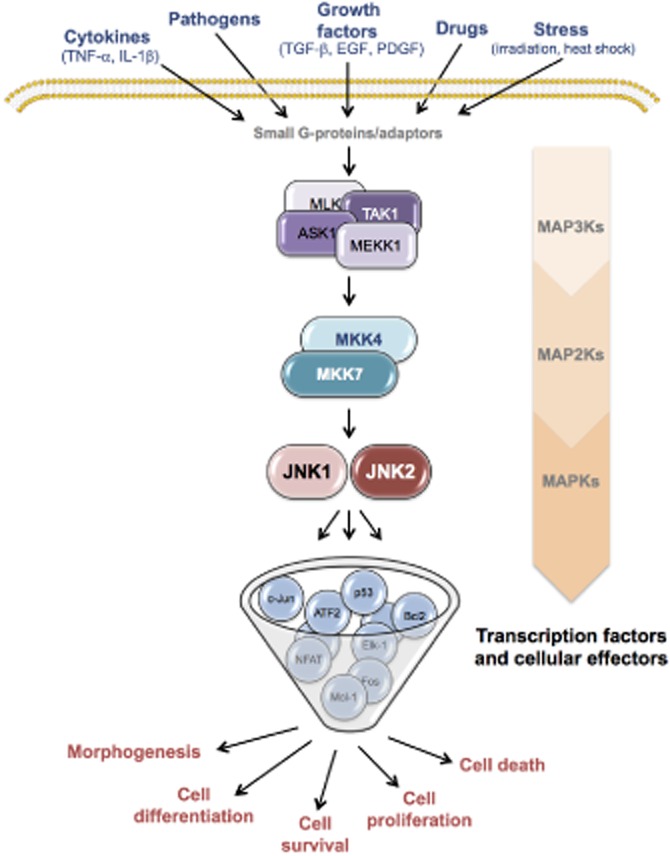
Schematic representation of the JNK signalling cascade. Like all other MAPK, the JNK signalling cascade consists of a three-component module of upstream MAP3Ks that couples the signals from the cell surface to intracellular protein effectors. Different MAP3Ks have been implicated in the JNK cascade, depending on the type of stimulus the cell receives. Once activated, MAP3Ks phosphorylate and activate components of the MAP2K module, such as MKK4 and MKK7, which in turn phosphorylate and stimulate the activity of distinct JNK isoforms through dual phosphorylation on threonine and tyrosine residues within a conserved Thr-X-Tyr motif. Upon activation, each JNK protein itself can phosphorylate serine and threonine residues on specific substrates, delivering different cellular activities.
Upon activation by the upstream MAP2Ks, JNKs phosphorylate and activate a number of nuclear and non-nuclear proteins, including the transcription factor activator protein-1 (AP-1) – which is formed by dimerization of the Jun proteins (c-Jun, JunB, JunD) with the Fos proteins (c-Fos, FosB, Fra-1, Fra-2) – activating transcription factor 2 (ATF-2), c-Myc, p53, Elk1, NFAT, as well as cell death regulators of the Bcl-2 family in the mitochondria (Bogoyevitch and Kobe, 2006). These proteins control a diversity of cellular responses, such as proliferation, differentiation, cell death and survival.
The diversity of cellular functions of JNKs underscores the diversity of disease conditions in which JNKs are implicated, including cancer (Figure 2). Indeed, aberrant expression and activation of JNKs are found in many cancer cell lines as well as in patient samples (Wagner and Nebreda, 2009). Moreover, abnormalities in JNK activity have also been associated with diabetes (Hirosumi et al., 2002), inflammatory and neurodegenerative disorders (Hunot et al., 2004; Roy et al., 2008). Consequently, the signalling mechanisms that underline JNKs expression and activities in these diseases, and the identification of JNK inhibitory molecules, are the subject of intense ongoing research. JNKs are also essential regulators of normal physiological functions, such as immune responses and cell and tissue morphogenesis (Waetzig and Herdegen 2005). Therefore, although targeting JNKs in the above diseases represents a worthwhile therapeutic strategy, inhibition of the JNK proteins could also be deleterious. Here, we particularly review the current knowledge on the role of JNK signalling pathways in cancer pathogenesis, with a focus on selected solid and haematological malignancies, in which the progress in understanding their pathogenesis is proceeding at a rapid pace. We also offer a perspective on the principal challenges that are relevant to the development of drugs targeting JNKs.
Figure 2.
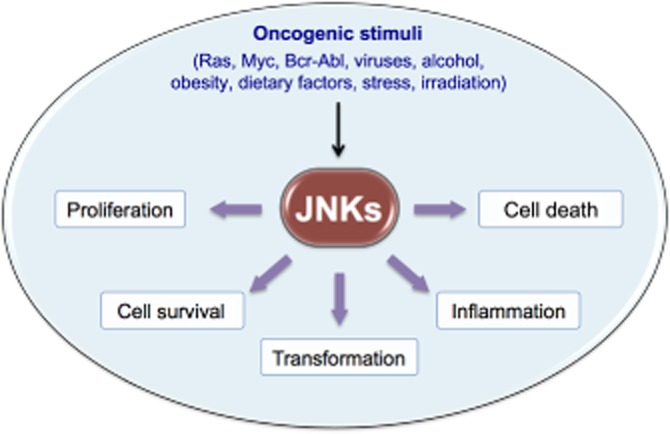
JNKs regulate key cellular activities involved in cancer. JNKs are activated in response to different extracellular and intracellular stimuli, such as oncogenes, dietary agents, obesity, alcohol, infectious agents and irradiation. Persistent activation of JNKs affect tumourigenesis by both transcription-dependent and transcription-independent mechanisms involved in cell proliferation, survival, transformation, inflammation, migration and suppression of cell death.
The role of JNKs in cancer
The early findings that the transforming actions of several oncogenes such as Ras, c-fos, Met and Bcr-Abl could be JNKs dependent (Smeal et al., 1991; Raitano et al., 1995; Rodrigues et al., 1997; Behrens et al., 2000; Manning and Davis, 2003), suggested that the JNK signalling could contribute to the cellular transformation that supports cancer development. Indeed, c-Jun, the prototypical downstream target of the JNK pathway, is required for transformation induced by the oncogene Ras (Lloyd et al., 1991) and for liver cancer development in mice (Eferl and Wagner, 2003; Min et al., 2012). Moreover, studies in a mouse model of intestinal cancer (Apcmin) have shown that the oncogenic function of c-Jun is dependent on phosphorylation by JNK, as mice expressing a non-phosphorylatable mutant form of c-Jun (JunAA/JunAA/Apcmin) developed smaller and fewer polyps (Nateri et al., 2005), thus implying a pro-oncogenic role for JNK/c-Jun axis in this context.
In support of this view, hyperactivation of the JNK proteins themselves has been reported in multiple cancer cell lines and tissue samples (Hui et al., 2008; Chang et al., 2009a; 2009b; Barbarulo et al., 2013), and depletion of the individual JNKs can suppress tumourigenesis depending on the tissue specificity (Wagner and Nebreda, 2009). In primary hepatocellular carcinomas (HCCs), for example, activation of JNK1, but not JNK2, is increased as compared with the non-neoplastic lesions, and absence of JNK1 impaired liver cell proliferation and tumour formation (Hui et al., 2008; Chang et al., 2009a). Additionally, overactivation of JNK1 in HCC tissues has been shown to associate with both a poor disease prognosis and expression of progenitor cell biomarkers (Chang et al., 2009b). JNK1 has also been shown to be an important contributor to the tumour-promoting activity of tobacco smoke in the lungs (Takahashi et al., 2010). In contrast, other studies have described an oncogenic function for JNK2 in various tumour-derived cell lines (Bost et al., 1999; Yang et al., 2003; Cui et al., 2006; Nitta et al., 2011; Yoon et al., 2012) and animal cancer models (Chen et al., 2001; Chromik et al., 2007; Ke et al., 2010). Myeloma cell lines and primary tumours have been shown to exhibit elevated JNK2 activity, which appears to be the key determinant to enhanced cell survival (Barbarulo et al., 2013). Moreover, genetic deletion of JNK2 in mice reduced skin carcinogenesis induced by 7,12-dimethylbenz[α]anthracene (DMBA) and 12-O-tetradecanoylphorbol-13-acetate (TPA) treatment (Chen et al., 2001), further outlining the role of JNK2 in tumourigenesis. In addition, a recent study established that both JNK1 and JNK2 are required for lung tumour formation induced by inactivation of endogenous KRas gene (Cellurale et al., 2011). These findings are consistent with the hypothesis that the JNK proteins may promote tumour development in a tissue- or cell-specific manner, and suggest that selective inhibition of these kinases may be effective in halting specific tumour formation.
However, despite having a role in tumour development, a substantial body of evidence implicates JNK proteins in tumour suppression (Davis, 2000; Wagner and Nebreda, 2009). In the DMBA/TPA-induced skin cancer model, for example, JNK1-null mice developed a greater number of papillomas compared with the wild-type mice (She et al., 2002), indicating that JNK1 negatively regulates tumourigenesis. The tumour-suppressing function for JNK1 has been further supported by experiments, which demonstrated that JNK1-null mice are highly susceptible to tumour development after inoculation of both melanoma and lymphoma cell lines (Gao et al., 2005). Moreover, JNK1-deficient mice spontaneously develop intestinal tumours (Tong et al., 2007). It has been proposed that the tumour suppressor activity of JNKs results from their well-recognized apoptotic functions involving the mitochondrial pathway (Davis, 2000). Detailed analysis demonstrated that primary murine embryonic fibroblasts (MEFs), isolated from mice that lack expression of both JNK1 and JNK2 proteins, were resistant to the induction of apoptosis and failed to release pro-apoptotic molecules from mitochondria, including cytochrome C in response to UV irradiation (Tournier et al., 2000). This indicates that stress-induced JNK signalling leads to apoptosis via activation of the mitochondrial pathway. In line with these findings, studies in multiple myeloma (MM) cell lines showed that activation of the JNK pathway by 2-methoxyestradiol, a potent antitumour agent, is associated with apoptosis and the release of Smac/DIABLO protein from mitochondria into the cytosol (Chauhan et al., 2003). Moreover, the activation of the JNK pathway is required for microtubule-damaging agent-induced apoptosis of breast cancer cells through its ability to phosphorylate and inactivate the mitochondrial anti-apoptotic protein Bcl2 (Srivastava et al., 1999; Yamamoto et al., 1999). In addition, JNK2 has been shown to phosphorylate the tumour suppressor p53 on Ser6, which is a key site for p53 stabilization and activation (Fuchs et al., 1998; Oleinik et al., 2007). Indeed, knockdown of JNK1 and JNK2 by small interfering RNA impaired the p53-dependent apoptosis in response to anticancer drugs (Oleinik et al., 2007).
In summary, the information outlined above indicate that JNK1 and JNK2 can act as either tumour promoter or as tumour suppressor kinases in different types of cancer, and highlight the importance in understanding fully both the roles of JNKs and the molecular basis for these distinct functions in different tumours, in order to validate the therapeutic potential of JNK inhibition. It has been proposed that differential regulation of specific cellular targets is the underlying mechanism for the distinct functions of JNK1 and JNK2 in cancer, although targets regulated by the individual JNK proteins remain largely unidentified (Figure 3). In the succeeding section, we focus on various cancers, both solid and haematological malignancies in which JNKs are reported to be involved and may be targetable.
Figure 3.
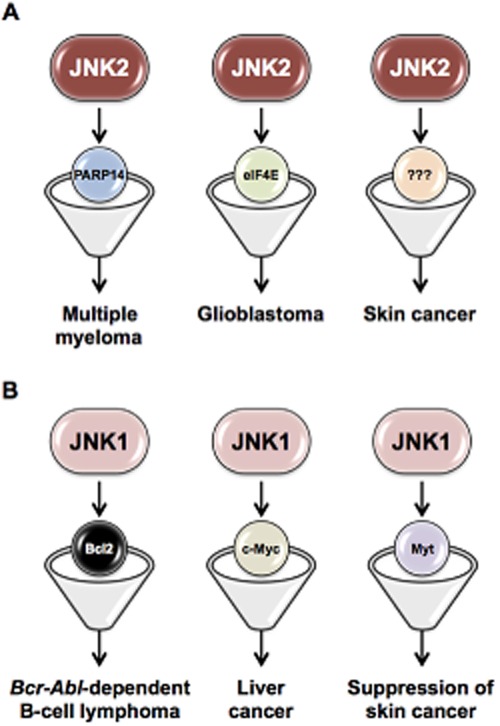
Specific cellular targets of JNK1 and JNK2 in different types of cancer. A diagram showing the different targets regulated by the individual JNK proteins. Distinct functions of JNK1 and JNK2 in various cancers appear through differing regulation of specific substrates and/or downstream effectors. The development of inhibitors that target specific downstream effectors of the individual JNK proteins would provide unique opportunities for therapeutic interventions.
JNKs in solid cancers
Liver cancer
Liver cancer is a compelling example of the involvement of the JNK pathway in the initiation and progression of cancer (see Seki et al., 2012). The most common type of primary liver cancer is HCC, with approximately 750 000 cases per year worldwide and few approved treatment options (Llovet et al., 2003; Jemal et al., 2011). HCC could arise from cirrhosis (scarring of the liver) or severe liver fibrosis, caused by multiple rounds of hepatocyte death, inflammatory responses and compensatory liver regeneration, all of which involve overactivation of the JNK pathway in injured hepatocytes (Farazi and DePinho, 2006; Papa et al., 2009; Seki et al., 2012). Although the exact contribution of the JNK pathway to HCC development remains to be fully elucidated, evidence from studies in mice with diethylnitrosamine (DEN)-induced HCC suggests that the JNK pathway contributes to the development of HCC by inducing hepatocyte cell death that promotes compensatory proliferation (Seki et al., 2012). For example, deletion of the IκB kinase β (IKKβ) in hepatocytes induced activation of JNKs, cell death that triggers compensatory proliferation of surviving hepatocytes, and greatly enhanced the development of HCC in response to DEN treatment (Maeda et al., 2005; reviewed by Papa et al., 2009). Interestingly, deletion of jnk1, but not jnk2, was able to abolish the effect of deletion of IKKβ in hepatocytes on cell death, compensatory proliferation and hepatocarcinogenesis in response to DEN induction, indicating overactivation of JNK1 as a key player in DEN-induced HCC (Sakurai et al., 2006). This was further confirmed by studies using JNK1 deficient mice, which were much less susceptible to DEN-induced HCC development. This impaired formation of HCC was associated with decreased expression of the cell cycle regulator cyclin D and VEGF, suggesting that JNK1 likely acts as a tumour promoter in HCC by enhancing hepatocyte proliferation and neovascularization (Sakurai et al., 2006). The critical role for JNK1 activation in HCC was further supported by experiments which demonstrated that hepatocyte-specific deletion of another IκB kinase (IKKγ; also called NEMO) and activator of NF-κB, caused spontaneous formation of HCC in parallel with increased JNK1 activation, hepatocyte death and compensatory hepatocyte proliferation (Luedde et al., 2007). Additionally, administration of antioxidants to these mice resulted in significantly reduced JNK activation and HCC development, further indicating a role for JNK activation in promoting HCC formation (Luedde et al., 2007). JNK1 has also been shown to regulate the expression of the p53 up-regulated modulator of apoptosis (PUMA), a pro-apoptotic BH3-only protein, during fatty acid-induced hepatocyte apoptosis (see Huang and Strasser, 2000; Cazanave et al., 2009). Indeed, in the DEN model, mice deficient in PUMA exhibit defects in HCC formation due to death and compensatory proliferation reduction of hepatocytes. Such effects were prevented in these mice by treatment with the JNK inhibitor SP600125, indicating that the JNK1/PUMA axis plays a central role in DEN-induced hepatocyte apoptosis and subsequent proliferation (Qiu et al., 2011).
A further proposed mechanism that accounts for the pro-tumourigenic role of JNK1 in HCC development is that JNK1 may regulate hepatocyte proliferation directly. In the DEN model, JNK1 was found to down-regulate the expression of the proliferation inhibitor p21 through transcriptional up-regulation of the growth promoter c-Myc (Hui et al., 2008). Mice that lack JNK1 display decreased tumour cell proliferation following DEN administration, and genetic inactivation of p21 in JNK1-deficient mice has been shown to restore hepatocyte proliferation in these mice. Furthermore, overexpression of c-Myc has been shown to increase proliferation of JNK1-/- liver cells (Hui et al., 2008). JNK1, but not JNK2, was constitutively active in more than 50% of tissue samples from patients with HCC, relative to adjacent normal tissues (Hui et al., 2008; Chang et al., 2009a). The hyperactivation of JNK1 was associated with increased tumour size and an elevated expression of genes controlling cell cycle and proliferation, indicating the relevance of JNK1 for the proliferation of liver cancer cells (Chang et al., 2009a). Indeed, knockdown of JNK1 by short hairpin RNA lentiviruses impaired cell proliferation in a human HCC cell line, and JNK1 has been shown to reduce expression of p21 through up-regulation of c-Myc (Hui et al., 2008).
More recently, an additional aspect has emerged, with relevance to the effects of the JNK signalling on HCC development. Davis and colleagues, using mice with compound deficiency of JNK1 and JNK2 in hepatocytes, demonstrated that JNKs have both tumour promoting and suppressing roles in hepatocarcinogenesis that depend on the cell types of the liver. The authors generated a genetically engineered mouse model, deleted of JNK1 in hepatocytes and JNK2 in the entire organism, and discovered that these mice developed more HCCs than wild-type mice following administration of DEN. The increased development of HCC was associated with increased hepatocyte death, and consequently increased compensatory proliferation as well as increased c-Myc and c-Jun expression. This indicates that JNK activity in hepatocytes functions to reduce tumour development (Das et al., 2011). However, in non-parenchymal cells of liver, such as hepatic immune cells (Kupffer cells), JNK activity is reported to promote HCC development. The authors went further to demonstrate that this was mediated through creation of an inflammatory environment, including the expression of pro-tumourigenic cytokines such as IL6 and TNF-α, that contribute to compensatory hepatocyte proliferation (Das et al., 2011).
To date, JNK1 inhibitors have not been developed to treat patients with HCC. Nevertheless, the functional roles of JNK1 in hepatocyte death and compensatory proliferation make drugs of this type promising anti-HCC therapies. Indeed, an inhibitory peptide directed against the substrate-docking domain of JNK proteins (D-JNKI1) was shown to suppress JNK activity and reduce tumour growth in the DEN-induced HCC model and in a human HCC xenograft model (Hui et al., 2008). In a rat DEN-induced HCC model, the administration of the JNK inhibitor SP600125 reduced the number and size of HCCs (Nagata et al., 2009). Additionally, inhibition of JNK activity has been shown to improve the efficacy of some current chemotherapeutic agents. For example, SP600125, in combination with the chemotherapy drug TNF-related apoptosis-inducing ligand, was shown to increase apoptosis in human HCC cultures (Mucha et al., 2009). Indeed, activated JNK1 was found in tissue samples from patients with HCC (Hui et al., 2008), and correlates directly with poor therapeutic response to sorafenib, a multikinase inhibitor that is currently being tested in clinical trials for HCC (Hagiwara et al., 2012). Taken together, the findings described above illustrate the importance of JNK1 in promoting HCC and the potential of JNK1 to be a target, alone or in combination with other therapeutic agents, for anti-HCC therapies.
Breast cancer
In sharp contrast to the tumour-promoting role of JNK1 described above, in breast cancer models, the JNK pathway has been shown to suppress tumour development. One example came from the work of Cellurale and colleagues (Cellurale et al., 2010). They used mice heterozygous for the p53 tumour suppressor, which spontaneously develop breast cancer in a pattern and frequency similar to that reported for the Li-Fraumeni breast cancer in humans (Kuperwasser et al., 2000). Crossing of p53−/+ mice with JNK1−/− mice and JNK2−/− mice causes significantly shortened tumour-free survival compared with control p53−/+ mice, suggesting that JNK1 and JNK2 may function to reduce breast cancer. A similar tumour-suppressing role for JNK proteins, especially JNK2, was also observed in transgenic mice that express the polyomavirus (PyV) middle T (MT) oncogene in mammary epithelial cells (Webster et al., 1998). PyV MT transgenic mice develop multifocal mammary tumours, which frequently metastasize to the lung, thus emulating both early and late stages of human breast cancer. Systemic deletion of JNK2 in PyV MT transgenic mice was found to shorten latency and increase tumour multiplicity. Interestingly, PyV MT/jnk2−/− mammary tumours showed more genomic instability, aneuploidy and impaired DNA damage response due to a reduction of DNA repair proteins and activation of the cell-cycle inhibitor p21, suggesting that in this case, JNK2 expression prevents breast cancer development by regulating cell cycle progression and DNA repair mechanisms (Chen et al., 2010). Further evidence for the tumour-suppressing role of JNKs in breast cancer has been provided from studies that show compound JNK deficiency in mammary epithelial cells to promote tumour formation in a KRas/p53 mouse model of breast cancer (Cellurale et al., 2012). In line with these findings, inactivating mutations in the JNK activator Mkk4 gene have been identified in human breast cancer (Su et al., 1998; 2002). In addition, deletion of Mkk7 gene (the other upstream activator of JNK) in primary murine epithelial cells facilitates oncogenic transformation in the mammary gland (Schramek et al., 2011), further validating the tumour-suppressive role of the JNK cascade in breast cancer. Activation of the JNK pathway has also been shown to mediate the cytotoxic effects of the chemotherapy drug taxol in MCF7 breast cancer cells (Mamay et al., 2003). While these recent findings implicate the JNK cascade in mammary carcinoma suppression, they also emphasize that JNK1 and JNK2 have redundant, or at least overlapping roles. Inducing both JNK1 and JNK2 would therefore be exploited for therapeutic benefit in patients with breast cancer.
Skin cancer
Persistent exposure to UV radiation is the major cause for the development of skin cancer, which comprises several, histologically distinct, skin neoplasms. These include squamous cell carcinoma (SCC), basal cell carcinoma and melanoma, named after the types of skin cell from which they arise. Among these, SCC is the most common and invasive type of non-melanoma skin cancer, with limited treatment options at present, especially for the late stage (see Zhang and Selim, 2012). As JNK proteins, also termed SAPKs, are highly responsive to UV radiation (Dérijard et al., 1994), it is not surprising that hyperactivation of JNKs is frequently observed in SCC. Early studies on this topic revealed opposing functions for JNK1 and JNK2 in regulating skin carcinogenesis (see Zhang and Selim, 2012). Using a well-characterized multistage model of mouse skin carcinogenesis, in which tumours are initiated with DMBA and promoted with TPA, Dong and colleagues found that in JNK2-deficient mice, the multiplicity of papillomas induced by TPA was lower than that in wild-type mice, indicating that JNK2 is a crucial tumour promoter of skin cancer development (Chen et al., 2001). Moreover, Mkk4 deficiency prevented DMBA/TPA-induced skin cancer by suppressing JNK2 expression, providing the first clear evidence that the MKK4-JNK2 axis is essential for tumour formation in the skin (Finegan and Tournier, 2010). In contrast, JNK1 appears to be a crucial suppressor of skin cancer, as JNK1-deficient mice exhibited a significantly higher papilloma incidence compared with the wild-type mice (She et al., 2002). This was further confirmed by studies using the regenerated human SCC model, in which primary keratinocytes were transduced for expression of constitutively active JNK1 or JNK2, along with the oncogenic Ras mutant and then used for human skin regeneration on immunodeficient mice (Ke et al., 2010). While tissue-expressing JNK2 were hyperproliferative and displayed clinical features of human SCC, tissue expressing JNK1 exhibited normal histological features of human skin. This indicates that JNK2, but not JNK1, cooperates with Ras to transform normal epidermal cells to malignancy with SCC features (Ke et al., 2010). In line with these findings, analyses of both biopsies and keratinocytes from patients with SCC reported increased levels of JNK2 activity, as compared with healthy controls (Zhang et al., 2007; Ke et al., 2010). The exact mechanism for the distinct functions of JNK1 and JNK2 in skin cancer development is not fully clear, but has been proposed to include ERKs and Akt signalling and the AP-1 DNA-binding activity (She et al., 2002).
More recently, JNK1 has been shown to prevent skin cancer via a novel mechanism. After UV irradiation, JNK1 associates with and phosphorylates the cell cycle inhibitory kinase myelin transcription factor 1 (Myt1), which induces caspase-3 cleavage and DNA fragmentation, resulting in apoptosis. Knockdown of Myt1 suppresses UV-induced apoptosis in skin cancer cells, and Myt1 has been shown to phosphorylate the mitotic kinase cdc2 on Tyr15, which is a key site for kinase inactivation (Choi et al., 2009). The opposing functions of JNK1 and JNK2 in skin cancer suggest that therapeutic agents discriminating between JNK1 and JNK2 proteins may offer a promising therapeutic opportunity.
Brain tumours
The most frequent primary brain tumours in adults are gliomas, which begin in the glial tissue. Indeed, gliomas account for about 70% of adult malignant primary brain tumours. According to the World Health Organization classification system, gliomas are further distinguished into three subtypes: grade 2 (low-grade astrocytoma, oligodendroglioma and oligoastrocytoma), grade 3 (anaplastic gliomas) and grade 4 (glioblastoma; Li et al., 2008). Approximately half of all gliomas are glioblastoma, by far the most severe and aggressive subtype (Ricard et al., 2012). A number of cellular and xenograft studies revealed a role for JNKs in the development and progression of gliomas. For example, elevated levels of JNKs activity were observed in a panel of glioblastoma cell lines and in human glioblastoma tumour samples (Antonyak et al., 2002). Similarly, phosphorylation of JNKs and c-Jun has been shown to strongly correlate with the histological grade of glioblastoma (Li et al., 2008). Compared with JNK1 and JNK3, JNK2 has a dominant pro-tumourigenic role in glioblastoma. Tsuiki et al. (2003) reported that JNK2 possesses autophosphorylation activity and constitutive substrate kinase activity. This is in line with data showing that JNK2 is the major JNK protein that is constitutively activated in glioblastoma. JNK2 was also shown to support tumourigenesis in vivo by activating Akt and up-regulating the expression of eukaryotic translation initiation factor 4 (Cui et al., 2006), which is frequently overexpressed in many human cancers (Graff et al., 2008). More recent evidence indicates a role for JNKs in maintaining glioma stem cell properties, which are responsible for the poor prognosis of glioblastoma patients. Elevated levels of the phosphorylated (active) form of JNKs were observed in sphere-cultured U87 and U373 glioma cells and in stem cells isolated from patient-derived glioma cells. Pharmacological inhibition of JNKs with SP600125 or knockdown of JNK1 or JNK2 significantly suppressed sphere formation and soft-agar colony formation, which are assay methods to evaluate “stemness” of stem-like cancer cells in vitro, and reduced the expression of stem cell markers in sphere-cultured glioma cells. Moreover, knockdown of JNK2 in glioma stem cells suppressed tumour formation in vivo (Yoon et al., 2012). The same group reported that JNK2 promotes stemness of glioma stem-like cells through up-regulation of Notch-2 expression (Yoon et al., 2012). This study reveals a new tumourigenic function for JNKs, in particular, JNK2. However, there is also evidence supporting a role for JNK3, expression of which is largely restricted to the brain as a tumour suppressor. Indeed, loss of expression of the jnk3 gene was identified in 10 of the 19 human brain tumour cell lines examined (Yoshida et al., 2001).
JNKs in haematological malignancies
While the roles of JNK proteins in solid tumours have been described in recent studies, the specific functions of JNKs in haematological malignancies remain limited. This stems in part from a lack of genetic animal models of these conditions, which makes the task even more difficult.
Leukaemia
Leukaemia is a broad term used to describe numerous cancers of the blood and bone marrow (Murati et al., 2012). These haematological malignancies are characterized by an increase in white blood cell proliferation, coupled with the failure of normal pro-apoptotic responses to neoplastic cells. Leukaemia is clinically and pathologically classified as ‘acute’ and ‘chronic’. Additionally, these two classes are further subdivided according to which kind of blood cell is affected, such as lymphoblastic leukaemia and myeloid leukaemia. Acute myeloid leukaemia (AML) is a heterogeneous clonal disorder of haematopoietic progenitor cells (‘blasts’), which lose the ability to differentiate normally and to respond to normal regulators of proliferation (see Estey and Döhner, 2006). Chronic myeloid leukaemia conversely involves the unregulated proliferation of more differentiated mature haematopoietic cells (reviewed by Deininger et al., 2000).
One proposed therapeutic strategy for treatment of leukaemia is to induce differentiation of circulating leukaemia cells. This is a first step to induce apoptosis of these neoplastic cells. The precise mechanism that triggers the activation of apoptosis in mature differentiated cells has not been elucidated. JNK has been shown to enhance monocytic differentiation of human leukaemia cells induced by 1,25-dihydroxyvitamin D3, a derivative of vitamin D3 (Wang et al., 2005). However, the precise molecular mechanism of how JNK promotes differentiation is not completely understood. Recent studies have highlighted the distinct role of JNK1 and JNK2 in this differentiation process and described for the first time a negative interplay between JNK2 and JNK1. While JNK2 is a negative regulator of monocytic differentiation, JNK1 is a positive regulator. Strikingly, silencing of JNK2 increased the activity of JNK1p46, which is associated with increased cellular differentiation after treatment with 1,25D. In contrast, silencing of JNK1 reduced the intensity of differentiation induced by 1,25-dihydroxyvitamin D3, and partially blocked activation of c-Jun/AP-1 transcription factors (Chen-Deutsch et al., 2009). Accordingly, specific inhibitors of JNK2, and not JNK1, may complement the differentiation therapy induced by the vitamin D3 derivatives, and thus, may be used as an anti-leukaemia drug.
Constitutive activation of JNK has been also found in T-cell acute lymphoblastic leukaemia (T-ALL). Suppression of JNK activity in T-ALL cells by using general (pan)-JNK chemical inhibitors promoted cell cycle arrest and apoptosis, and increased sensitivity to Fas-mediated apoptosis, whereas constitutive activation of JNK in T-ALL cells resulted in accelerated cell cycle progression and resistance to Fas-mediated apoptosis. Silencing of JNK1, but not JNK2, exhibited similar effects to JNK inhibitors, suggesting that JNK1 is the predominant form of JNK proteins and, in this case, the identification of JNK1-specific inhibitors may have some important therapeutic implications in the treatment of T-ALL (Cui et al., 2009).
Other studies have demonstrated that JNK activity has been also associated with treatment failure in AML. Biochemical analyses of primary AML bone marrow samples have shown that JNK activity is associated with increased efflux activity of the multidrug transporter protein (MRP), whose function promotes the efflux of chemotherapeutic drugs from the treated cells, with consequential resistance to chemotherapy and hyperproliferation of AML cells. A central cellular programme involving the signalling pathway JNK→c-Jun/AP-1 might coordinate the expression of MRP, thus affecting the multidrug-resistant state of AML cells (Cripe et al., 2002). Notably, the involvement of JNK in multidrug resistance has been also characterized in other human cancers (Ronaldson et al., 2010; Yan et al., 2010). However, there is no clear understanding of how JNK regulates the functioning of MRP, or any other member of the ATP-binding cassette family of membrane efflux pumps.
In summary, studies to precisely identify the specific function of JNK in certain type of leukaemia cells may provide valuable information for the design of new inhibitors with clinical translational potential.
Multiple myeloma
MM is a malignant tumour of terminally differentiated B-cells, characterized by accumulation of clonal, long-lived plasma cells in the bone marrow and extramedullary sites. Notable characteristics of myeloma cells include a low proliferative capacity of plasma cells and the failure of normal apoptotic index in vivo. Accordingly, pro-survival mechanisms play central roles in the accumulation of the malignant clones in MM. Although the role of JNK signalling in the pathogenesis of MM has been extensively addressed, the function of JNK in promoting or suppressing MM is still a debate. Initial studies from Anderson and colleagues described the biological effect of JNK inhibition in MM cell lines. The JNK-specific inhibitor SP600125 induced growth arrest of MM cell lines. Importantly, SP600125 induced activation of the transcription factor NF-κB and its upstream molecules IKKα, suggesting that JNK may play a role in the survival of MM cells by suppressing constitutive activation of NF-κB in MM cells (Hideshima et al., 2003a). However, these results conflict with the fact that NF-κB is important for the survival of these cancer cells (see Gilmore, 2007). Constitutive activation of NF-κB was indeed highly scored in CD138+ cells from the bone marrow of 22 MM patients. In contrast to MM patients, activated forms of NF-κB were absent in cells from healthy individuals (Bharti et al., 2004). Diverse genetic aberrations accumulated in many myeloma samples activate both the canonical and alternative pathways of NF-κB activation (Annunziata et al., 2007; Keats et al., 2007; Hideshima et al., 2009). A possible explanation for this apparent discrepancy is that the JNK inhibitor SP600125 might overstimulate NF-κB signalling independently of JNK activation. Whichever is the case, the molecular mechanisms elucidating how JNK promotes survival of MM cells are not clear (discussed below).
Although the function of JNK has been linked to MM cell survival, JNK activation is also required for apoptotic cell death of MM cells induced by chemotherapeutic drugs. The proteasome inhibitor bortezomib (originally named PS-341) has proven successful for the treatment of relapsed and newly diagnosed MM. Notably, bortezomib mediates anti-MM activity by inducing expression of p53 and MDM2 protein and also induces the phosphorylation of p53 protein and activation of JNK activity, which is essential for the induction of caspase-3 activation, and therefore, apoptosis of MM cells. In addition, SP600125 blocked bortezomib-induced apoptosis in MM cells (Hideshima et al., 2003b).
How can one signal pathway mediate two very different cellular responses? One plausible answer is that the different JNK proteins, JNK1 and JNK2, may serve different functions in MM by regulating specific downstream effectors. Recently, we systematically assessed the functional roles of JNK1 and JNK2 in MM by silencing the expression of either JNK1 or JNK2 in MM cell lines and examined their activity in MM primary tumours. Interestingly, we evaluated the level of phospho-active JNK in myeloma plasma cells from trephine biopsies of bone marrow of newly diagnosed MM patients, compared with normal plasma cells from tonsillar samples, and found that JNK was constitutively active in MM plasma. In addition, we observed that phosphorylation of JNK2 was significantly higher than JNK1 in a panel of MM cell lines and primary CD138+ MM cells, suggesting a pathogenic role for JNK2 in this disease. Silencing JNK2 enhanced MM apoptotic cell death. On the other hand, ablation of JNK1 did not alter the proliferation rate of MM cells. More fascinating was the observation that silencing of JNK2 in MM cells was associated with increased JNK1 activity, and that suppression of JNK1 activity in JNK2-depleted cells restored the regular growth rate of these malignant cells, suggesting that JNK2 controls basal JNK1-mediated pro-apoptotic responses to abnormal signals (Barbarulo et al., 2013). It is important to point out that the JNK2-mediated control of JNK1 activity is not a novel concept in tumourigenesis. In a range of human cell lines, JNK1 was actively pro-apoptotic, but was constitutively inhibited by the presence of active JNK2 (Ahmed and Milner, 2009). Moreover, JNK2 antagonized the JNK1-mediated differentiation signal in human myeloid leukaemia cells resistant to vitamin D (Chen-Deutsch et al., 2009). This regulation of JNK1 by JNK2, however, was not observed in non-malignant cells (Ahmed and Milner, 2009), suggesting that the opposing roles of JNK1 and JNK2 may exist only in cancerous cells. Indeed, ablation of the Jnk2 gene in JNK1+/+ MEFs caused increased expression of c-Jun and cellular proliferation (Tournier et al., 2000; Sabapathy et al., 2004), whereas selective inhibition of JNK2 activity by a chemical genetic approach in JNK1+/+ MEFs caused no changes in c-Jun expression or proliferation, suggesting compensatory increases in JNK1 function (Jaeschke et al., 2006). Interestingly, it has also been proposed that increased JNK1 function detected in JNK2-/- MEFs are primarily due to a competition between JNK1 and JNK2 for activation by the upstream MAPKK (Jaeschke et al., 2006).
How does JNK2 suppress the JNK1-mediated pro-apoptotic signal in MM? By investigating the mechanisms for the distinct role of JNK1 and JNK2 in MM, we identified and characterized a novel signalling pathway that involves JNK2-PARP14-JNK1 (Barbarulo et al., 2013). Previous studies have established that PARP14 is an ADP ribosyl-transferase that transduces survival signals in murine primary B-cells by regulating expression of B-cell survival factors, as well as by repressing an apoptotic programme involving caspases (Goenka and Boothby, 2006). In the mouse, PARP14 has been shown to facilitate B-lymphoid tumourigenesis driven by oncogenic c-Myc (Cho et al., 2011). Our recent study has investigated whether expression of PARP14 is under control of JNK2 in promoting survival of malignant MM cells. Silencing of JNK2 reduced the levels of PARP14 protein and promoted apoptosis in a panel of MM cell lines. Notably, ectopic expression of PARP14 in JNK2-depleted MM cells restored the levels of PARP14 similar to those of the endogenous protein expressed in MM cells, and rescued cells from undergoing apoptosis induced by the permanent deficiency of JNK2 expression. Moreover, the rescue of JNK2-depleted cells by ectopic expression of PARP14 is associated with the suppression of JNK1 activity in MM cells. Clearly, these data indicate that JNK2 regulates expression of PARP14, which is important for the suppression of JNK1 activity. However, how this regulation occurs is not determined yet. It is plausible that JNK2 cooperates with certain transcription factors. To date, however, the identity of the transcription factors regulating Parp14 gene expression is not known. Moreover, regulation of PARP14 by JNK2 does not explain the constitutive inhibition of JNK1-mediated apoptosis in MM cells. In an attempt to evaluate how JNK2-PARP14 suppresses JNK1 activity, we have examined whether PARP14 protein is able to interact with either JNK1 or JNK2 in cells. Remarkably, we showed that PARP14 specifically interacts with JNK1, through its C-terminal portion, thus inhibiting JNK1 kinase activity and ultimately apoptosis. Although at the present, it is not directly proved whether JNK1 kinase inactivation by PARP14 involves the enzymatic activity of PARP14, our data suggest that this might be the case. The JNK1 kinase activity is, indeed, enhanced after treatment of MM cells with PJ-34, a pan-inhibitor of PARP enzymic activity (see Jagtap and Szabó, 2005; Barbarulo et al., 2013). However, whether this overactivation of JNK1 is due to specific inhibition of PARP14 enzymic activity remains to be elucidated. In summary, we have identified a novel pathway whereby constitutively active JNK2 promotes survival of myeloma cells via PARP14, which in turn associates with JNK1 and suppresses its kinase activity, suggesting that JNK2-PARP14 is a potential therapeutic target in myeloma.
Lymphoma
Bcr-Abl is a chimeric oncogene generated by the translocation of sequences from the c-Abl protein-TK gene on chromosome 9 into the Bcr gene on chromosome 22. Bcr-Abl is an activator of several signal transduction pathways that mediates malignant transformation and entry of the cell cycle, and is also a potent inhibitor of cell death (Cilloni and Saglio, 2012). Previous studies have demonstrated that survival signalling of Bcr-Abl-transformed B-lymphoblasts is mediated by JNK1, as knockdown of Jnk1 gene in mice causes defective transformation of pre-B-cells by Bcr-Abl. The JNK1-mediated survival is supported via regulation of Bcl2 expression. Notably, ectopic expression of Bcl2 rescued the defective phenotype of Jnk1-/- B-cells (Hess et al., 2002). These data are consistent with a model, whereby JNK1-Bcl2 mediates the pathogenesis of transformed pre-B-cells. In line with this observation, JNK signalling via Egr-1 and c-Myc activation was required for survival and proliferation of B-lymphoma cells (Gururajan et al., 2005). Suppression of JNK activity by SP600125 strongly inhibited cell growth in a panel of murine and human B-lymphoma cells, indicating that JNK is essential for suppression of basal apoptosis (Gururajan et al., 2005). Recent observations by Sabapathy and colleagues, however, underscored the redundant function of JNK1 and JNK2 in B-cell lymphomas (Anbalagan and Sabapathy, 2012). These authors used the murine Eμ-Myc-induced B-cell lymphoma model to induce lymphomagenesis and investigate the role of JNK1 and JNK2 in the development of B-cell lymphoma. The Eμ-Myc transgenic mice model resemble the MYC/Ig t(8:14) translocation in human Burkitt's lymphoma, where c-Myc is overexpressed in B-lymphocytes by the immunoglobulin heavy chain enhancer (Eμ; Adams et al., 1985). In this model, Myc-induced lymphoma is preceded by a prolonged precancerous phase, in which the high proliferative rates of Eμ-Myc B-cells are counterbalanced by a high apoptotic index (Maclean et al., 2003). Surprisingly, the overall survival of Myc-transgenic mice deficient for Jnk1 is not significantly different from wild-type mice, and there is also no significant difference between the wild-type and Jnk2-/- group. Moreover, loss of either Jnk1 or Jnk2 in Myc-transgenic mice neither revealed any statistical difference in the survival of these two groups of animals nor appreciably affected the maturation status of B220+ B-cells. Therefore, JNK1 or JNK2 alone is not dispensable for Myc-induced lymphomagenesis (Anbalagan and Sabapathy, 2012). Although in our study we have characterized only one B-lymphoma cell line, our observations are in line with the latter findings, suggesting that JNK1 and JNK2 play redundant roles in B-lymphoma cells and the absence of either JNK1 or JNK2 is compensated for by the other remaining JNK isoforms for the proliferation of B-lymphoma cells (Barbarulo et al., 2013). These observations, therefore, suggest that JNK plays an important role in B-cell lymphoma pathogenesis, and targeting the catalytic activities of both JNK1 and JNK2 would be beneficial for the treatment of these conditions.
Strategies to target the JNK proteins
Clearly, the JNKs play key roles in the pathobiology of many cancer types. This information has stimulated both the academic research community and the pharmaceutical industry to design, synthesize and screen chemical libraries of compounds that can act as selective inhibitors of the JNKs. Based on their mode of action, the majority of these compounds can be classified into two categories, ATP-competitive inhibitors and ATP-non-competitive inhibitors (see Siddiqui and Reddy, 2010).
Well recognized and thoroughly studied, the ATP-competitive JNK inhibitors include small molecules from a variety of scaffolds such as indazoles, aminopyrazoles, aminopyridines, pyridine carboxamides, benzothien-2-ylamides and benzothiazol-2-yl acetonitriles, quinoline derivatives and aminopyrimidines (Szczepankiewicz et al., 2006; LoGrasso and Kamenecka, 2008; Gaillard et al., 2010; He et al., 2011; Kamenecka et al., 2010; Plantevin Krenitsky et al., 2012). Although, to date, no clinical use of these inhibitors has been reported, efficacy of a subset of these molecules has been explored in cell-based experiments and animal models of disease. For example, the anthrapyrazolone SP600125, one of the earliest and most studied ATP-competitive JNK inhibitor (Bennett et al., 2001), was effective in a mouse model of Parkinson's disease (Wang et al., 2004). Another compound, the benzothiazolone AS601245, provided neuroprotective effects in rats after focal cerebral ischaemia (Carboni et al., 2004), and was effective in ischaemia-reperfusion injury (Ferrandi et al., 2004). These inhibitors, however, display poor kinase specificity as they target the highly conserved ATP-binding site. For example, SP600125 displays action against all three JNK proteins, and at higher concentrations, also displays ability to inhibit the closely related ERKs and p38 MAPKs (Bennett et al., 2001; Bain et al., 2003). Likewise, AS601245 inhibits phosphorylation of c-Jun at high doses, which is due to a moderate specificity for JNKs (Gaillard et al., 2010). To overcome the specificity problem, Zhang et al. (2012) have very recently developed and characterized a potent and irreversible inhibitor of all three JNK proteins, JNK-IN-8, that forms a covalent bond with a conserved cysteine in the ATP site. This has been achieved through the use of structure-based drug design. The major advantage of cysteine-directed covalent inhibitors is the ability to control kinase selectivity and prolong pharmacodynamics despite competition with high endogenous levels of ATP. It will be of interest to see how this novel inhibitor of JNK activity is broadly applied in the treatment of a range of JNK-associated diseases, including cancer, and whether desired therapeutic outcomes would be achieved without unwanted side effects.
The alternative strategy for the inhibition of JNKs is the development of peptide or small molecule ATP-non-competitive inhibitors that prevent the protein–protein interactions of JNKs with the JNK-interacting protein-1 (JIP1), or with the upstream kinases or the downstream substrates (Bogoyevitch et al., 2010). JIP1 is a ‘scaffolding’ protein that enhances JNK signalling specificity and efficiency by bringing JNKs into proximity with their upstream activators and substrates (Whitmarsh, 2006). The site of interaction between JNK and JIP1 contains a conserved consensus sequence, which is called the d-domain (Stebbins et al., 2008). A peptide, pepJIP1, consisting of 11 amino acids (153–163) derived from the d-domain of JIP1, inhibits JNK activity potently in vitro and exhibits noteworthy selectivity for JNKs, with little inhibition of other MAPKs (Barr et al., 2002; Heo et al., 2004). The inhibitory activity of pepJIP1 relies either on the competition of this peptide with the d-domains of substrates or upstream kinases, or on allosteric effects (Barr et al., 2002; Ho et al., 2003; Heo et al., 2004). Moreover, Ngoei et al. (2011) have recently reported the characterization of a novel JNK inhibitory peptide PYC71N, which prevents the interaction of JNK with c-Jun, and represents a novel tool to explore the JNK-mediated substrate interactions and phosphorylation.
Studies in vivo using cell-permeable JIP1-based peptides have also shown effective inhibition of JNK signalling. For example, the i.p. administration of pepJIP1 fused to human immunodeficiency virus trans-acting activator of transcription sequence in diabetic mice models restores normoglycaemia without causing hypoglycaemia in lean mice (Kaneto et al., 2004). Other example of the use of JIP1-based peptides for JNKs inhibition includes a cell-permeable and protease-resistant peptide, D-JNKI-1, which was initially found to protect against cerebral ischaemia in two models of middle cerebral artery occlusion (Borsello et al., 2003). Systemic i.p. administration of D-JNKI-1 in mice attenuated the growth of B16-Fluc murine melanoma xenografts and cancer pain (Gao et al., 2009). The same inhibitor suppressed tumour growth of subcutaneously implanted HCC cells in nude mice and in mice with DEN-induced liver cancer. Immunohistochemical analysis of excised tumours confirmed that the anti-growth efficacy resulted from inhibiting levels of activated JNK (p-JNK) in the treatment group which remained inhibited 3 months after D-JNKI-1 injection, whereas tumours from the control group continuously expressed high levels of p-JNK (Hui et al., 2008). Most recently, two separate studies have shown that the inhibition of the JNK activity by D-JNKI-1 in vivo is an effective approach to the treatment of inflammatory bowel diseases (Reinecke et al., 2012; Kersting et al., 2013).
Thus, cell-permeable JIP1-based peptides have beneficial effects to improve a number of JNK-dependent diseases, although there are several challenges that have limited the development of peptide-based inhibitors. Other chemical entities that have the ability to disrupt the interaction between JNKs and JIP1, both in vitro and in vivo, have also recently been identified (Bogoyevitch et al., 2010). In screening 30 000 small molecules in a dissociation-enhanced lanthanide fluoro-immuno assay platform, combined with a time-resolved fluorescence energy transfer-based kinase assay, Stebbins et al. (2008) have identified the inhibitor BI-78D3, which was found to compete with pepJIP1 for binding to JNKs and inhibit the JNK kinase activity. The authors have also shown that BI-78D3 in vivo blocks concanavalin A-induced-liver damage and restores insulin sensitivity in a mouse model of type 2 diabetes. Moreover, through an extensive structure-based virtual screening, Ngoei et al. (2013) have recently identified a thiazole-2,2' diamine compound, JD123, which binds to both the ATP-binding site and the protein substrate site, and so functions as a dual inhibitor of the JNK proteins. JD123 has also been shown to inhibit sorbitol-induced c-Jun phosphorylation in MEFs in a dose-dependent manner. Further structural-based studies will be necessary to improve membrane permeability for its use in cell and in vivo studies, as well as to determine the precise docking modes and interactions with JNKs, and to provide further understanding of its inhibitory mechanisms (Ngoei et al., 2013).
In the cancer setting, however, the strategy of using JNK inhibitors that do not display selectivity across JNK proteins may be flawed, as JNK1 and JNK2, which are more ubiquitous than JNK3 in their tissue distribution, appear to have opposite functions based on tissue specificity (Chen et al., 2001; She et al., 2002; Chen-Deutsch et al., 2009; Yao et al., 2009; Barbarulo et al., 2013). JNK2 is constitutively activated in myeloid leukaemia cells, and has been shown to antagonize differentiation mediated by JNK1 (Chen-Deutsch et al., 2009). Yet, JNK2 appears to be the main JNK protein involved in MM pathobiology via inhibition of the pro-apoptotic activity of JNK1 (Barbarulo et al., 2013). In contrast, JNK1 activity appears to be essential for proliferation of human HCC (Hui et al., 2008). Thus, taken together, the results from these studies and others (Bost et al., 1999; Chen et al., 2001; Yang et al., 2003; Cui et al., 2006; Chromik et al., 2007; Ke et al., 2010; Nitta et al., 2011; Yoon et al., 2012) suggest that highly selective JNK1 or JNK2 inhibitors are necessary for treating cancer. However, to date, selective inhibitors for a specific JNK protein are not yet available commercially. Interestingly, Kaoud et al. (2011) have recently reported the discovery of two potent and selective JNK2-inhibitor peptides that inhibit migration of murine mammary carcinoma cells. A selective inhibitor of JNK1, AV-7, has also been developed and tested in in vitro studies (Yao et al., 2009). Further and more extensive studies, including in vivo pharmacokinetics of these compounds, will be essential to validate them for drug development.
In addition, JNK1 and JNK2 appear to differentially influence tumourigenesis through regulating distinct cellular targets or binding partners (Figure 3). Examples include the interaction of JNK1, but not JNK2, with the membrane-associated kinase Myt1, and the observation that JNK1 phosphorylates Myt1. Phosphorylated Myt1 then inhibits UVA-induced skin cancer development (Choi et al., 2009). In MM, we recently showed that JNK2 inhibits the pro-apoptotic activity of JNK1 through effect on PARP14 expression (Barbarulo et al., 2013). In sharp contrast, JNK1 deficiency impaired liver cancer formation via modulating expression of MYC and the CDK inhibitor p21 (Hui et al., 2008). Few studies, apart from these, have addressed the identification of targets regulated by the individual JNK proteins, and none have yet evaluated the therapeutic potential of their inhibition in cancer. Indeed, so far, most efforts to target JNK pathways in cancer focused on direct inhibition of JNKs themselves. In this respect, it is important to note that selective inhibition of the individual JNK proteins could cause adverse cellular effects and unwanted toxicity because of their relevance to normal cellular functions, such as adaptive immunity and brain development (Waetzig and Herdegen, 2005). Therefore, effective treatment of JNK-associated cancers will require maintaining a delicate balance between suppressing deleterious functions of individual JNK proteins and interfering with cellular physiology. Treatments aimed at inhibiting downstream cellular targets of a specific JNK protein in a tissue-specific manner may have better therapeutic efficacy and reduce adverse side effects.
Concluding remarks
Over the past two decades, JNKs have increasingly been recognized as an attractive molecule target for the treatment of human malignancies because they regulate a number of cellular processes whose deregulation has been associated with cancer (Wagner and Nebreda, 2009). This has triggered extensive drug discovery efforts. To date, the majority of developed JNK inhibitors target the highly conserved ATP-binding site, although multiple steps in the JNK pathway could be targeted for inhibition (see Bogoyevitch et al., 2010; Siddiqui and Reddy, 2010). While a number of these inhibitors have been proven to work in vivo in animal models of disease ranging from inflammatory to neurodegenerative disorders, they have not been translated into clinical use thus far. One of the main concerns associated with ATP-competitive inhibitors is the lack of specificity, as they can also target other kinases, thus, generating off-target effects. Moreover, the high concentrations of intracellular ATP significantly reduce the efficacy of these compounds (Bogoyevitch et al., 2010; Siddiqui and Reddy, 2010). Small molecule and peptide-based inhibitors that target the docking site of downstream substrates or the scaffold proteins are a promising alternative to the traditional ATP-competitive inhibitors with the potential for improved efficacy and specificity towards the JNKs (Chen et al., 2009). Cell-permeable versions of these inhibitors have been successfully used in studies for JNK-associated disorders, including cerebral ischaemia (Borsello et al., 2003), stroke, Parkinson's disease (Kuan and Burke, 2005) and diabetes (Kaneto et al., 2004), and are indeed of increasing interest to the research and drug discovery communities. A considerable additional work, however, will be clearly needed to validate these compounds with in vivo studies.
As the diverse functions of the JNK proteins in cancer are emerging, greater efforts are now being directed towards the development of selective inhibitors (ATP competitive or otherwise) that discriminate between JNK1 and JNK2 (Figure 3). However, because JNKs play a pivotal role in normal biological functions, direct inhibition of the JNKs themselves could also have undesired side effects. Indeed, one of the major challenges facing researchers is to develop selective inhibitors of JNKs aimed at treating different cancers based on their ability to target specific JNK-mediated cellular events. As research is likely to concentrate on new downstream substrates or binding partners, further studies should also focus on developing substrate-specific inhibitors, which could make an important contribution to the alleviation of different types of cancer.
Acknowledgments
The authors would like to thank the members of their laboratories for the constructive discussions and acknowledge the research funding from the Kay Kendall Leukemia Fund, Italian Association for Cancer Research and Foundation for Liver Research.
Abbreviations
- AP
activator protein
- DEN
diethylnitrosamine
- DMBA
7,12-dimethylbenz[α]anthracene
- HCC
hepatocellular carcinoma
- IKK
IκB kinase
- JIP
JNK-interacting protein
- MEFs
murine embryonic fibroblasts
- Myt
myelin transcription factor 1
- MM
multiple myeloma
- PUMA
p53 up-regulated mediator of apoptosis
- SCC
squamous carcinoma cells
- TPA
12-O-tetradecanoylphorbol-13-acetate
Conflict of interests
The authors declare no conflict of interests.
References
- Adams JM, Harris AW, Pinkert CA, Corcoran LM, Alexander WS, Cory S, et al. The c-myc oncogene driven by immunoglobulin enhancers induces lymphoid malignancy in transgenic mice. Nature. 1985;318:533–538. doi: 10.1038/318533a0. [DOI] [PubMed] [Google Scholar]
- Ahmed SU, Milner J. Basal cancer cell survival involves JNK2 suppression of a novel JNK1/c-Jun/Bcl-3 apoptotic network. Plos ONE. 2009;4:e7305. doi: 10.1371/journal.pone.0007305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anbalagan M, Sabapathy K. JNK1 and JNK2 play redundant functions in Myc-induced B cell lymphoma formation. Int J Cancer. 2012;130:1967–1969. doi: 10.1002/ijc.26207. [DOI] [PubMed] [Google Scholar]
- Annunziata CM, Davis RE, Demchenko Y, Bellamy W, Gabrea A, Zhan F, et al. Frequent engagement of the classical and alternative NF-κB pathways by diverse genetic abnormalities in multiple myeloma. Cancer Cell. 2007;12:115–130. doi: 10.1016/j.ccr.2007.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Antonyak MA, Kenyon LC, Godwin AK, James DC, Emlet DR, Okamoto I, et al. Elevated JNK activation contributes to the pathogenesis of human brain tumors. Oncogene. 2002;21:5038–5046. doi: 10.1038/sj.onc.1205593. [DOI] [PubMed] [Google Scholar]
- Bain J, McLauchlan H, Elliott M, Cohen P. The specificities of protein kinase inhibitors: an update. Biochem J. 2003;371:199–204. doi: 10.1042/BJ20021535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barbarulo A, Iansante V, Chaidos A, Naresh K, Rahemtulla A, Franzoso G, et al. Poly(ADP-ribose) polymerase family member 14 (PARP14) is a novel effector of the JNK2-dependent pro-survival signal in multiple myeloma. Oncogene. 2013;32:4231–4242. doi: 10.1038/onc.2012.448. [DOI] [PubMed] [Google Scholar]
- Barr RK, Kendrick TS, Bogoyevitch MA. Identification of the critical features of a small peptide inhibitor of JNK activity. J Biol Chem. 2002;277:10987–10997. doi: 10.1074/jbc.M107565200. [DOI] [PubMed] [Google Scholar]
- Behrens A, Jochum W, Sibilia M, Wagner EF. Oncogenic transformation by ras and fos is mediated by c-Jun N-terminal phosphorylation. Oncogene. 2000;19:2657–2663. doi: 10.1038/sj.onc.1203603. [DOI] [PubMed] [Google Scholar]
- Bennett BL, Sasaki DT, Murray BW, O'Leary EC, Sakata ST, Xu W, et al. SP600125, an anthrapyrazolone inhibitor of Jun N-terminal kinase. Proc Natl Acad Sci U S A. 2001;98:13681–13686. doi: 10.1073/pnas.251194298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bharti AC, Shishodia S, Reuben JM, Weber D, Alexanian R, Raj-Vadhan S, et al. Nuclear factor-kappaB and STAT3 are constitutively active in CD138+ cells derived from multiple myeloma patients, and suppression of these transcription factors leads to apoptosis. Blood. 2004;103:3175–3184. doi: 10.1182/blood-2003-06-2151. [DOI] [PubMed] [Google Scholar]
- Bogoyevitch MA, Kobe B. Uses for JNK: the many and varied substrates of the c-Jun N-terminal kinases. Microbiol Mol Biol Rev. 2006;70:1061–1095. doi: 10.1128/MMBR.00025-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bogoyevitch MA, Ngoei KR, Zhao TT, Yeap YY, Ng DC. c-Jun N-terminal kinase (JNK) signaling: recent advances and challenges. Biochim Biophys Acta. 2010;1804:463–475. doi: 10.1016/j.bbapap.2009.11.002. [DOI] [PubMed] [Google Scholar]
- Borsello T, Clarke PG, Hirt L, Vercelli A, Repici M, Schorderet DF, et al. A peptide inhibitor of c-Jun N-terminal kinase protects against excitotoxicity and cerebral ischemia. Nat Med. 2003;9:1180–1186. doi: 10.1038/nm911. [DOI] [PubMed] [Google Scholar]
- Bost F, McKay R, Bost M, Potapova O, Dean NM, Mercola D. The Jun kinase 2 isoform is preferentially required for epidermal growth factor-induced transformation of human A549 lung carcinoma cells. Mol Cell Biol. 1999;19:1938–1949. doi: 10.1128/mcb.19.3.1938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carboni S, Hiver A, Szyndralewiez C, Gaillard P, Gotteland JP, Vitte PA. AS601245 (1,3-benzothiazol-2-yl (2-[[2-(3-pyridinyl) ethyl] amino]-4 pyrimidinyl) acetonitrile): a c-Jun NH2-terminal protein kinase inhibitor with neuroprotective properties. J Pharmacol Exp Ther. 2004;310:25–32. doi: 10.1124/jpet.103.064246. [DOI] [PubMed] [Google Scholar]
- Cazanave SC, Mott JL, Elmi NA, Bronk SF, Werneburg NW, Akazawa Y, et al. JNK1-dependent PUMA expression contributes to hepatocyte lipoapoptosis. J Biol Chem. 2009;284:26591–26602. doi: 10.1074/jbc.M109.022491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cellurale C, Weston CR, Reilly J, Garlick DS, Jerry DJ, Sluss HK, et al. Role of JNK in a Trp53-dependent mouse model of breast cancer. Plos ONE. 2010;5:e12469. doi: 10.1371/journal.pone.0012469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cellurale C, Sabio G, Kennedy NJ, Das M, Barlow M, Sandy P, et al. Requirement of c-Jun NH(2)-terminal kinase for Ras-initiated tumor formation. Mol Cell Biol. 2011;31:1565–1576. doi: 10.1128/MCB.01122-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cellurale C, Girnius N, Jiang F, Cavanagh-Kyros J, Lu S, Garlick DS, et al. Role of JNK in mammary gland development and breast cancer. Cancer Res. 2012;72:472–481. doi: 10.1158/0008-5472.CAN-11-1628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang Q, Zhang Y, Beezhold KJ, Bhatia D, Zhao H, Chen J, et al. Sustained JNK1 activation is associated with altered histone H3 methylations in human liver cancer. J Hepatol. 2009a;50:323–333. doi: 10.1016/j.jhep.2008.07.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang Q, Chen J, Beezhold KJ, Castranova V, Shi X, Chen F. JNK1 activation predicts the prognostic outcome of the human hepatocellular carcinoma. Mol Cancer. 2009b;8:64. doi: 10.1186/1476-4598-8-64. doi: 10.1186/1476-4598-8-64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chauhan D, Li G, Hideshima T, Podar K, Mitsiades C, Mitsiades N, et al. JNK-dependent release of mitochondrial protein, Smac, during apoptosis in multiple myeloma (MM) cells. J Biol Chem. 2003;278:17593–17596. doi: 10.1074/jbc.C300076200. [DOI] [PubMed] [Google Scholar]
- Chen N, Nomura M, She QB, Ma WY, Bode AM, Wang L, et al. Suppression of skin tumorigenesis in c-Jun NH(2)-terminal kinase-2-deficient mice. Cancer Res. 2001;61:3908–3912. [PubMed] [Google Scholar]
- Chen P, O'Neal JF, Ebelt ND, Cantrell MA, Mitra S, Nasrazadani A, et al. Jnk2 effects on tumor development, genetic instability and replicative stress in an oncogene-driven mouse mammary tumor model. Plos ONE. 2010;5:e10443. doi: 10.1371/journal.pone.0010443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen T, Kablaoui N, Little J, Timofeevski S, Tschantz WR, Chen P, et al. Identification of small-molecule inhibitors of the JIP-JNK interaction. Biochem J. 2009;420:283–294. doi: 10.1042/BJ20081899. [DOI] [PubMed] [Google Scholar]
- Chen-Deutsch X, Garay E, Zhang J, Harrison JS, Studzinski GP. c-Jun N-terminal kinase 2 (JNK2) antagonizes the signaling of differentiation by JNK1 in human myeloid leukemia cells resistant to vitamin D. Leuk Res. 2009;33:1372–1378. doi: 10.1016/j.leukres.2009.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho SH, Ahn AK, Bhargava P, Lee CH, Eischen CM, McGuinness O, et al. Glycolytic rate and lymphomagenesis depend on PARP14, an ADP ribosyltransferase of the B aggressive lymphoma (BAL) family. Proc Natl Acad Sci U S A. 2011;108:15972–15977. doi: 10.1073/pnas.1017082108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi HS, Bode AM, Shim JH, Lee SY, Dong Z. c-Jun N-terminal kinase 1 phosphorylates Myt1 to prevent UVA-induced skin cancer. Mol Cell Biol. 2009;29:2168–2180. doi: 10.1128/MCB.01508-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chromik AM, Müller AM, Körner J, Belyaev O, Holland-Letz T, Schmitz F, et al. Genetic deletion of JNK1 and JNK2 aggravates the DSS-induced colitis in mice. J Invest Surg. 2007;20:23–33. doi: 10.1080/08941930601126140. [DOI] [PubMed] [Google Scholar]
- Cilloni D, Saglio G. Molecular pathways: BCR-ABL. Clin Cancer Res. 2012;18:930–937. doi: 10.1158/1078-0432.CCR-10-1613. [DOI] [PubMed] [Google Scholar]
- Cripe LD, Gelfanov VM, Smith EA, Spigel DR, Phillips CA, Gabig TG, et al. Role for c-jun N-terminal kinase in treatment-refractory acute myeloid leukemia (AML): signaling to multidrug-efflux and hyperproliferation. Leukemia. 2002;16:799–812. doi: 10.1038/sj.leu.2402457. [DOI] [PubMed] [Google Scholar]
- Cui J, Han SY, Wang C, Su W, Harshyne L, Holgado-Madruga M, et al. c-Jun NH(2)-terminal kinase 2alpha2 promotes the tumorigenicity of human glioblastoma cells. Cancer Res. 2006;66:10024–10031. doi: 10.1158/0008-5472.CAN-06-0136. [DOI] [PubMed] [Google Scholar]
- Cui J, Wang Q, Wang J, Lv M, Zhu N, Li Y, et al. Basal c-Jun NH2-terminal protein kinase activity is essential for survival and proliferation of T-cell acute lymphoblastic leukemia cells. Mol Cancer Ther. 2009;8:3214–3222. doi: 10.1158/1535-7163.MCT-09-0408. [DOI] [PubMed] [Google Scholar]
- Das M, Garlick DS, Greiner DL, Davis RJ. The role of JNK in the development of hepatocellular carcinoma. Genes Dev. 2011;25:634–645. doi: 10.1101/gad.1989311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis RJ. Signal transduction by the JNK group of MAP kinases. Cell. 2000;103:239–252. doi: 10.1016/s0092-8674(00)00116-1. [DOI] [PubMed] [Google Scholar]
- Deininger MW, Goldman JM, Melo JV. The molecular biology of chronic myeloid leukemia. Blood. 2000;96:3343–3356. [PubMed] [Google Scholar]
- Dérijard B, Hibi M, Wu IH, Barrett T, Su B, Deng T, et al. JNK1: a protein kinase stimulated by UV light and Ha-Ras that binds and phosphorylates the c-Jun activation domain. Cell. 1994;76:1025–1037. doi: 10.1016/0092-8674(94)90380-8. [DOI] [PubMed] [Google Scholar]
- Eferl R, Wagner EF. AP-1: a double-edged sword in tumorigenesis. Nat Rev Cancer. 2003;3:859–868. doi: 10.1038/nrc1209. [DOI] [PubMed] [Google Scholar]
- Estey E, Döhner H. Acute myeloid leukaemia. Lancet. 2006;368:1894–1907. doi: 10.1016/S0140-6736(06)69780-8. [DOI] [PubMed] [Google Scholar]
- Farazi PA, DePinho RA. Hepatocellular carcinoma pathogenesis: from genes to environment. Nat Rev Cancer. 2006;6:674–687. doi: 10.1038/nrc1934. [DOI] [PubMed] [Google Scholar]
- Ferrandi C, Ballerio R, Gaillard P, Giachetti C, Carboni S, Vitte PA, et al. Inhibition of c-Jun N-terminal kinase decreases cardiomyocyte apoptosis and infarct size after myocardial ischemia and reperfusion in anaesthetized rats. Br J Pharmacol. 2004;142:953–960. doi: 10.1038/sj.bjp.0705873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finegan KG, Tournier C. The mitogen-activated protein kinase kinase 4 has a pro-oncogenic role in skin cancer. Cancer Res. 2010;70:5797–5806. doi: 10.1158/0008-5472.CAN-09-3669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuchs SY, Adler V, Pincus MR, Ronai Z. MEKK1/JNK signaling stabilizes and activates p53. Proc Natl Acad Sci U S A. 1998;95:10541–10546. doi: 10.1073/pnas.95.18.10541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaillard P, Jeanclaude-Etter I, Ardissone V, Arkinstall S, Cambet Y, Camps M, et al. Design and synthesis of the first generation of novel potent, selective, and in vivo active (benzothiazol-2-yl)acetonitrile inhibitors of the c-Jun N-terminal kinase. J Med Chem. 2010;48:4596–4607. doi: 10.1021/jm0310986. [DOI] [PubMed] [Google Scholar]
- Gao Y, Tao J, Li MO, Zhang D, Chi H, Henegariu O, et al. JNK1 is essential for CD8+ T cell-mediated tumor immune surveillance. J Immunol. 2005;175:5783–5789. doi: 10.4049/jimmunol.175.9.5783. [DOI] [PubMed] [Google Scholar]
- Gao YJ, Cheng JK, Zeng Q, Xu ZZ, Decosterd I, Xu X, et al. Selective inhibition of JNK with a peptide inhibitor attenuates pain hypersensitivity and tumor growth in a mouse skin cancer pain model. Exp Neurol. 2009;219:146–155. doi: 10.1016/j.expneurol.2009.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilmore TD. Multiple myeloma: lusting for NF-kappaB. Cancer Cell. 2007;12:95–97. doi: 10.1016/j.ccr.2007.07.010. [DOI] [PubMed] [Google Scholar]
- Goenka S, Boothby M. Selective potentiation of Stat-dependent gene expression by collaborator of Stat6 (CoaSt6), a transcriptional cofactor. Proc Natl Acad Sci U S A. 2006;103:4210–4215. doi: 10.1073/pnas.0506981103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graff JR, Konicek BW, Carter JH, Marcusson EG. Targeting the eukaryotic translation initiation factor 4E for cancer therapy. Cancer Res. 2008;68:631–634. doi: 10.1158/0008-5472.CAN-07-5635. [DOI] [PubMed] [Google Scholar]
- Gururajan M, Chui R, Karuppannan AK, Ke J, Jennings CD, Bondada S. c-Jun N-terminal kinase (JNK) is required for survival and proliferation of B-lymphoma cells. Blood. 2005;106:1382–1391. doi: 10.1182/blood-2004-10-3819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hagiwara S, Kudo M, Nagai T, Inoue T, Ueshima K, Nishida N, et al. Activation of JNK and high expression level of CD133 predict a poor response to sorafenib in hepatocellular carcinoma. Br J Cancer. 2012;106:1997–2003. doi: 10.1038/bjc.2012.145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He Y, Kamenecka TM, Shin Y, Song X, Jiang R, Noel R, et al. Synthesis and SAR of novel quinazolines as potent and brain-penetrant c-jun N-terminal kinase (JNK) inhibitors. Bioorg Med Chem Lett. 2011;21:1719–1723. doi: 10.1016/j.bmcl.2011.01.079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heo YS, Kim SK, Seo CI, Kim YK, Sung BJ, Lee HS, et al. Structural basis for the selective inhibition of JNK1 by the scaffolding protein JIP1 and SP600125. EMBO J. 2004;23:2185–2195. doi: 10.1038/sj.emboj.7600212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hess P, Pihan G, Sawyers CL, Flavell RA, Davis RJ. Survival signaling mediated by c-Jun NH(2)-terminal kinase in transformed B lymphoblasts. Nat Genet. 2002;32:201–205. doi: 10.1038/ng946. [DOI] [PubMed] [Google Scholar]
- Hideshima T, Hayashi T, Chauhan D, Akiyama M, Richardson P, Anderson K. Biologic sequelae of c-Jun NH(2)-terminal kinase (JNK) activation in multiple myeloma cell lines. Oncogene. 2003a;22:8797–8801. doi: 10.1038/sj.onc.1206919. [DOI] [PubMed] [Google Scholar]
- Hideshima T, Mitsiades C, Akiyama M, Hayashi T, Chauhan D, Richardson P, et al. Molecular mechanisms mediating antimyeloma activity of proteasome inhibitor PS-341. Blood. 2003b;101:1530–1534. doi: 10.1182/blood-2002-08-2543. [DOI] [PubMed] [Google Scholar]
- Hideshima T, Chauhan D, Kiziltepe T, Ikeda H, Okawa Y, Podar K, et al. Biologic sequelae of I{kappa}B kinase (IKK) inhibition in multiple myeloma: therapeutic implications. Blood. 2009;113:5228–5236. doi: 10.1182/blood-2008-06-161505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirosumi J, Tuncman G, Chang L, Görgün CZ, Uysal KT, Maeda K, et al. A central role for JNK in obesity and insulin resistance. Nature. 2002;420:333–336. doi: 10.1038/nature01137. [DOI] [PubMed] [Google Scholar]
- Ho DT, Bardwell AJ, Abdollahi M, Bardwell L. A docking site in MKK4 mediates high affinity binding to JNK MAPKs and competes with similar docking sites in JNK substrates. J Biol Chem. 2003;278:32662–32672. doi: 10.1074/jbc.M304229200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang DC, Strasser A. BH3-Only proteins-essential initiators of apoptotic cell death. Cell. 2000;103:839–842. doi: 10.1016/s0092-8674(00)00187-2. [DOI] [PubMed] [Google Scholar]
- Hui L, Zatloukal K, Scheuch H, Stepniak E, Wagner EF. Proliferation of human HCC cells and chemically induced mouse liver cancers requires JNK1-dependent p21 downregulation. J Clin Invest. 2008;118:3943–3953. doi: 10.1172/JCI37156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hunot S, Vila M, Teismann P, Davis RJ, Hirsch EC, Przedborski S, et al. JNK-mediated induction of cyclooxygenase 2 is required for neurodegeneration in a mouse model of Parkinson's disease. Proc Natl Acad Sci U S A. 2004;101:665–670. doi: 10.1073/pnas.0307453101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaeschke A, Karasarides M, Ventura JJ, Ehrhardt A, Zhang C, Flavell RA, et al. JNK2 is a positive regulator of the cJun transcription factor. Mol Cell. 2006;23:899–911. doi: 10.1016/j.molcel.2006.07.028. [DOI] [PubMed] [Google Scholar]
- Jagtap P, Szabó C. Poly(ADP-ribose) polymerase and the therapeutic effects of its inhibitors. Nat Rev Drug Discov. 2005;4:421–440. doi: 10.1038/nrd1718. [DOI] [PubMed] [Google Scholar]
- Jemal A, Bray F, Center MM, Ferlay J, Ward E, Forman D. Global cancer statistics. CA Cancer J Clin. 2011;61:69–90. doi: 10.3322/caac.20107. [DOI] [PubMed] [Google Scholar]
- Johnson GL, Lapadat R. Mitogen-activated protein kinase pathways mediated by ERK, JNK, and p38 protein kinases. Science. 2002;298:1911–1912. doi: 10.1126/science.1072682. [DOI] [PubMed] [Google Scholar]
- Kamenecka T, Jiang R, Song X, Duckett D, Chen W, Ling YY, et al. Synthesis, biological evaluation, X-ray structure, and pharmacokinetics of aminopyrimidine c-jun-N-terminal kinase (JNK) inhibitors. J Med Chem. 2010;53:419–431. doi: 10.1021/jm901351f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaneto H, Nakatani Y, Miyatsuka T, Kawamori D, Matsuoka TA, Matsuhisa M, et al. Possible novel therapy for diabetes with cell-permeable JNK-inhibitory peptide. Nat Med. 2004;10:1128–1132. doi: 10.1038/nm1111. [DOI] [PubMed] [Google Scholar]
- Kaoud TS, Mitra S, Lee S, Taliaferro J, Cantrell M, Linse KD, et al. Development of JNK2-selective peptide inhibitors that inhibit breast cancer cell migration. ACS Chem Biol. 2011;6:658–666. doi: 10.1021/cb200017n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ke H, Harris R, Coloff JL, Jin JY, Leshin B, Miliani de Marval P, et al. The c-Jun NH2-terminal kinase 2 plays a dominant role in human epidermal neoplasia. Cancer Res. 2010;70:3080–3088. doi: 10.1158/0008-5472.CAN-09-2923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keats JJ, Fonseca R, Chesi M, Schop R, Baker A, Chng WJ, et al. Promiscuous mutations activate the noncanonical NF-kappaB pathway in multiple myeloma. Cancer Cell. 2007;12:131–144. doi: 10.1016/j.ccr.2007.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kersting S, Behrendt V, Kersting J, Reinecke K, Hilgert C, Stricker I, et al. The impact of JNK inhibitor D-JNKI-1 in a murine model of chronic colitis induced by dextran sulfate sodium. J Inflamm Res. 2013;6:71–81. doi: 10.2147/JIR.S40092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuan CY, Burke RE. Targeting the JNK signaling pathway for stroke and Parkinson's diseases therapy. Curr Drug Targets CNS Neurol Disord. 2005;4:63–67. doi: 10.2174/1568007053005145. [DOI] [PubMed] [Google Scholar]
- Kuperwasser C, Hurlbut GD, Kittrell FS, Dickinson ES, Laucirica R, Medina D, et al. Development of spontaneous mammary tumors in BALB/c p53 heterozygous mice. A model for Li-Fraumeni syndrome. Am J Pathol. 2000;157:2151–2159. doi: 10.1016/S0002-9440(10)64853-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li JY, Wang H, May S, Song X, Fueyo J, Fuller GN, et al. Constitutive activation of c-Jun N-terminal kinase correlates with histologic grade and EGFR expression in diffuse gliomas. J Neurooncol. 2008;88:11–17. doi: 10.1007/s11060-008-9529-1. [DOI] [PubMed] [Google Scholar]
- Llovet JM, Burroughs A, Bruix J. Hepatocellular carcinoma. Lancet. 2003;362:1907–1917. doi: 10.1016/S0140-6736(03)14964-1. [DOI] [PubMed] [Google Scholar]
- Lloyd A, Yancheva N, Wasylyk B. Transformation suppressor activity of a Jun transcription factor lacking its activation domain. Nature. 1991;352:635–638. doi: 10.1038/352635a0. [DOI] [PubMed] [Google Scholar]
- LoGrasso P, Kamenecka T. Inhibitors of c-jun-N-terminal kinase (JNK) Mini Rev Med Chem. 2008;8:755–766. doi: 10.2174/138955708784912120. [DOI] [PubMed] [Google Scholar]
- Luedde T, Beraza N, Kotsikoris V, van Loo G, Nenci A, De Vos R, et al. Deletion of NEMO/IKKgamma in liver parenchymal cells causes steatohepatitis and hepatocellular carcinoma. Cancer Cell. 2007;11:119–132. doi: 10.1016/j.ccr.2006.12.016. [DOI] [PubMed] [Google Scholar]
- Maclean KH, Keller UB, Rodriguez-Galindo C, Nilsson JA, Cleveland JL. c-Myc augments gamma irradiation-induced apoptosis by suppressing Bcl-XL. Mol Cell Biol. 2003;23:7256–7270. doi: 10.1128/MCB.23.20.7256-7270.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maeda S, Kamata H, Luo JL, Leffert H, Karin M. IKKbeta couples hepatocyte death to cytokine-driven compensatory proliferation that promotes chemical hepatocarcinogenesis. Cell. 2005;121:977–990. doi: 10.1016/j.cell.2005.04.014. [DOI] [PubMed] [Google Scholar]
- Mamay CL, Mingo-Sion AM, Wolf DM, Molina MD, Van Den Berg CL. An inhibitory function for JNK in the regulation of IGF-I signaling in breast cancer. Oncogene. 2003;22:602–614. doi: 10.1038/sj.onc.1206186. [DOI] [PubMed] [Google Scholar]
- Manning AM, Davis RJ. Targeting JNK for therapeutic benefit: from junk to gold? Nat Rev Drug Discov. 2003;2:554–565. doi: 10.1038/nrd1132. [DOI] [PubMed] [Google Scholar]
- Manning G, Whyte DB, Martinez R, Hunter T, Sudarsanam S. The protein kinase complement of the human genome. Science. 2002;298:1912–1934. doi: 10.1126/science.1075762. [DOI] [PubMed] [Google Scholar]
- Min L, Ji Y, Bakiri L, Qiu Z, Cen J, Chen X, et al. Liver cancer initiation is controlled by AP-1 through SIRT6-dependent inhibition of survivin. Nat Cell Biol. 2012;14:1203–1211. doi: 10.1038/ncb2590. [DOI] [PubMed] [Google Scholar]
- Mucha SR, Rizzani A, Gerbes AL, Camaj P, Thasler WE, Bruns CJ, et al. JNK inhibition sensitises hepatocellular carcinoma cells but not normal hepatocytes to the TNF-related apoptosis-inducing ligand. Gut. 2009;58:688–698. doi: 10.1136/gut.2008.154625. [DOI] [PubMed] [Google Scholar]
- Murati A, Brecqueville M, Devillier R, Mozziconacci MJ, Gelsi-Boyer V, Birnbaum D. Myeloid malignancies: mutations, models and management. BMC Cancer. 2012;12:304. doi: 10.1186/1471-2407-12-304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagata H, Hatano E, Tada M, Murata M, Kitamura K, Asechi H, et al. Inhibition of c-Jun NH2-terminal kinase switches Smad3 signaling from oncogenesis to tumor-suppression in rat hepatocellular carcinoma. Hepatology. 2009;49:1944–1953. doi: 10.1002/hep.22860. [DOI] [PubMed] [Google Scholar]
- Nateri AS, Spencer-Dene B, Behrens A. Interaction of phosphorylated c-Jun with TCF4 regulates intestinal cancer development. Nature. 2005;437:281–285. doi: 10.1038/nature03914. [DOI] [PubMed] [Google Scholar]
- Ngoei KR, Catimel B, Church N, Lio DS, Dogovski C, Perugini MA, et al. Characterization of a novel JNK (c-Jun N-terminal kinase) inhibitory peptide. Biochem J. 2011;434:399–413. doi: 10.1042/BJ20101244. [DOI] [PubMed] [Google Scholar]
- Ngoei KR, Ng DC, Gooley PR, Fairlie DP, Stoermer MJ, Bogoyevitch MA. Identification and characterization of bi-thiazole-2,2'-diamines as kinase inhibitory scaffolds. Biochim Biophys Acta. 2013;1834:1077–1088. doi: 10.1016/j.bbapap.2013.02.001. [DOI] [PubMed] [Google Scholar]
- Nitta RT, Del Vecchio CA, Chu AH, Mitra SS, Godwin AK, Wong AJ. The role of the c-Jun N-terminal kinase 2-a-isoform in non-small cell lung carcinoma tumorigenesis. Oncogene. 2011;30:234–244. doi: 10.1038/onc.2010.414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oleinik NV, Krupenko NI, Krupenko SA. Cooperation between JNK1 and JNK2 in activation of p53 apoptotic pathway. Oncogene. 2007;26:7222–7230. doi: 10.1038/sj.onc.1210526. [DOI] [PubMed] [Google Scholar]
- Owens DM, Keyse SM. Differential regulation of MAP kinase signalling by dual-specificity protein phosphatases. Oncogene. 2007;26:3203–3213. doi: 10.1038/sj.onc.1210412. [DOI] [PubMed] [Google Scholar]
- Papa S, Bubici C, Zazzeroni F, Pham CG, Kuntzen C, Knabb JR, et al. The NF-kappaB-mediated control of the JNK cascade in the antagonism of programmed cell death in health and disease. Cell Death Differ. 2006;13:712–729. doi: 10.1038/sj.cdd.4401865. [DOI] [PubMed] [Google Scholar]
- Papa S, Bubici C, Zazzeroni F, Franzoso G. Mechanisms of liver disease: cross-talk between the NF-kappaB and JNK pathways. Biol Chem. 2009;390:965–976. doi: 10.1515/BC.2009.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plantevin Krenitsky V, Nadolny L, Delgado M, Ayala L, Clareen SS, Hilgraf R, et al. Discovery of CC-930, an orally active anti-fibrotic JNK inhibitor. Bioorg Med Chem Lett. 2012;22:1433–1438. doi: 10.1016/j.bmcl.2011.12.027. [DOI] [PubMed] [Google Scholar]
- Qiu W, Wang X, Leibowitz B, Yang W, Zhang L, Yu J. PUMA-mediated apoptosis drives chemical hepatocarcinogenesis in mice. Hepatology. 2011;54:1249–1258. doi: 10.1002/hep.24516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raitano AB, Halpern JR, Hambuch TM, Sawyers CL. The Bcr-Abl leukemia oncogene activates Jun kinase and requires Jun for transformation. Proc Natl Acad Sci U S A. 1995;92:11746–11750. doi: 10.1073/pnas.92.25.11746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reinecke K, Eminel S, Dierck F, Roessner W, Kersting S, Chromik AM, et al. The JNK inhibitor XG-102 protects against TNBS-induced colitis. PLoS ONE. 2012;7:e30985. doi: 10.1371/journal.pone.0030985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ricard D, Idbaih A, Ducray F, Lahutte M, Hoang-Xuan K, Delattre JY. Primary brain tumours in adults. Lancet. 2012;379:1984–1996. doi: 10.1016/S0140-6736(11)61346-9. [DOI] [PubMed] [Google Scholar]
- Rodrigues GA, Park M, Schlessinger J. Activation of the JNK pathway is essential for transformation by the Met oncogene. EMBO J. 1997;16:2634–2645. doi: 10.1093/emboj/16.10.2634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ronaldson PT, Ashraf T, Bendayan R. Regulation of multidrug resistance protein 1 by tumor necrosis factor alpha in cultured glial cells: involvement of nuclear factor-kappaB and c-Jun N-terminal kinase signaling pathways. Mol Pharmacol. 2010;77:644–659. doi: 10.1124/mol.109.059410. [DOI] [PubMed] [Google Scholar]
- Roy PK, Rashid F, Bragg J, Ibdah JA. Role of the JNK signal transduction pathway in inflammatory bowel disease. World J Gastroenterol. 2008;14:200–202. doi: 10.3748/wjg.14.200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sabapathy K, Hochedlinger K, Nam SY, Bauer A, Karin M, Wagner EF. Distinct roles for JNK1 and JNK2 in regulating JNK activity and c-Jun-dependent cell proliferation. Mol Cell. 2004;15:713–725. doi: 10.1016/j.molcel.2004.08.028. [DOI] [PubMed] [Google Scholar]
- Sakurai T, Maeda S, Chang L, Karin M. Loss of hepatic NF-kappa B activity enhances chemical hepatocarcinogenesis through sustained c-Jun N-terminal kinase 1 activation. Proc Natl Acad Sci U S A. 2006;103:10544–10551. doi: 10.1073/pnas.0603499103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schramek D, Kotsinas A, Meixner A, Wada T, Elling U, Pospisilik JA, et al. The stress kinase MKK7 couples oncogenic stress to p53 stability and tumor suppression. Nat Genet. 2011;43:212–219. doi: 10.1038/ng.767. [DOI] [PubMed] [Google Scholar]
- Seki E, Brenner DA, Karin M. A liver full of JNK: signaling in regulation of cell function and disease pathogenesis, and clinical approaches. Gastroenterology. 2012;143:307–320. doi: 10.1053/j.gastro.2012.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- She QB, Chen N, Bode AM, Flavell RA, Dong Z. Deficiency of c-Jun-NH(2)-terminal kinase-1 in mice enhances skin tumor development by 12-O-tetradecanoylphorbol-13-acetate. Cancer Res. 2002;62:1343–1348. [PubMed] [Google Scholar]
- Siddiqui MA, Reddy PA. Small molecule JNK (c-Jun N-terminal kinase) inhibitors. J Med Chem. 2010;53:3005–3012. doi: 10.1021/jm9003279. [DOI] [PubMed] [Google Scholar]
- Smeal T, Binetruy B, Mercola DA, Birrer M, Karin M. Oncogenic and transcriptional cooperation with Ha-Ras requires phosphorylation of c-Jun on serines 63 and 73. Nature. 1991;354:494–496. doi: 10.1038/354494a0. [DOI] [PubMed] [Google Scholar]
- Srivastava RK, Mi QS, Hardwick JM, Longo DL. Deletion of the loop region of Bcl-2 completely blocks paclitaxel-induced apoptosis. Proc Natl Acad Sci U S A. 1999;96:3775–3780. doi: 10.1073/pnas.96.7.3775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stebbins JL, De SK, Machleidt T, Becattini B, Vazquez J, Kuntzen C, et al. Identification of a new JNK inhibitor targeting the JNK-JIP interaction site. Proc Natl Acad Sci U S A. 2008;105:16809–16813. doi: 10.1073/pnas.0805677105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su GH, Hilgers W, Shekher MC, Tang DJ, Yeo CJ, Hruban RH, et al. Alterations in pancreatic, biliary, and breast carcinomas support MKK4 as a genetically targeted tumor suppressor gene. Cancer Res. 1998;58:2339–2342. [PubMed] [Google Scholar]
- Su GH, Song JJ, Repasky EA, Schutte M, Kern SE. Mutation rate of MAP2K4/MKK4 in breast carcinoma. Hum Mutat. 2002;19:81. doi: 10.1002/humu.9002. [DOI] [PubMed] [Google Scholar]
- Szczepankiewicz BG, Kosogof C, Nelson LT, Liu G, Liu B, Zhao H, et al. Aminopyridine-based c-Jun N-terminal kinase inhibitors with cellular activity and minimal cross-kinase activity. J Med Chem. 2006;49:3563–3580. doi: 10.1021/jm060199b. [DOI] [PubMed] [Google Scholar]
- Takahashi H, Ogata H, Nishigaki R, Broide DH, Karin M. Tobacco smoke promotes lung tumorigenesis by triggering IKKβ- and JNK1-dependent inflammation. Cancer Cell. 2010;17:89–97. doi: 10.1016/j.ccr.2009.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tong C, Yin Z, Song Z, Dockendorff A, Huang C, Mariadason J, et al. c-Jun NH2-terminal kinase 1 plays a critical role in intestinal homeostasis and tumor suppression. Am J Pathol. 2007;171:297–303. doi: 10.2353/ajpath.2007.061036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tournier C, Hess P, Yang DD, Xu J, Turner TK, Nimnual A, et al. Requirement of JNK for stress-induced activation of the cytochrome c-mediated death pathway. Science. 2000;288:870–874. doi: 10.1126/science.288.5467.870. [DOI] [PubMed] [Google Scholar]
- Tsuiki H, Tnani M, Okamoto I, Kenyon LC, Emlet DR, Holgado-Madruga M, et al. Constitutively active forms of c-Jun NH2-terminal kinase are expressed in primary glial tumors. Cancer Res. 2003;63:250–255. [PubMed] [Google Scholar]
- Waetzig V, Herdegen T. Context-specific inhibition of JNKs: overcoming the dilemma of protection and damage. Trends Pharmacol Sci. 2005;26:455–461. doi: 10.1016/j.tips.2005.07.006. [DOI] [PubMed] [Google Scholar]
- Wagner EF, Nebreda AR. Signal integration by JNK and p38 MAPK pathways in cancer development. Nat Rev Cancer. 2009;9:537–549. doi: 10.1038/nrc2694. [DOI] [PubMed] [Google Scholar]
- Wang Q, Salman H, Danilenko M, Studzinski GP. Cooperation between antioxidants and 1,25-dihydroxyvitamin D3 in induction of leukemia HL60 cell differentiation through the JNK/AP-1/Egr-1 pathway. J Cell Physiol. 2005;204:964–974. doi: 10.1002/jcp.20355. [DOI] [PubMed] [Google Scholar]
- Wang W, Shi L, Xie Y, Ma C, Li W, Su X, et al. SP600125, a new JNK inhibitor, protects dopaminergic neurons in the MPTP model of Parkinson's disease. Neurosci Res. 2004;48:195–202. doi: 10.1016/j.neures.2003.10.012. [DOI] [PubMed] [Google Scholar]
- Webster MA, Hutchinson JN, Rauh MJ, Muthuswamy SK, Anton M, Tortorice CG, et al. Requirement for both Shc and phosphatidylinositol 3' kinase signaling pathways in polyomavirus middle T-mediated mammary tumorigenesis. Mol Cell Biol. 1998;18:2344–2359. doi: 10.1128/mcb.18.4.2344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whitmarsh AJ. The JIP family of MAPK scaffold proteins. Biochem Soc Trans. 2006;34:828–832. doi: 10.1042/BST0340828. [DOI] [PubMed] [Google Scholar]
- Whitmarsh AJ, Kuan CY, Kennedy NJ, Kelkar N, Haydar TF, Mordes JP, et al. Requirement of the JIP1 scaffold protein for stress-induced JNK activation. Genes Dev. 2001;15:2421–2432. doi: 10.1101/gad.922801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamamoto K, Ichijo H, Korsmeyer SJ. BCL-2 is phosphorylated and inactivated by an ASK1/Jun N-terminal protein kinase pathway normally activated at G(2)/M. Mol Cell Biol. 1999;19:8469–8478. doi: 10.1128/mcb.19.12.8469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan F, Wang XM, Liu ZC, Pan C, Yuan SB, Ma QM. JNK1, JNK2, and JNK3 are involved in P-glycoprotein-mediated multidrug resistance of hepatocellular carcinoma cells. Hepatobiliary Pancreat Dis Int. 2010;9:287–295. [PubMed] [Google Scholar]
- Yang YM, Bost F, Charbono W, Dean N, McKay R, Rhim JS, et al. c-Jun NH(2)-terminal kinase mediates proliferation and tumor growth of human prostate carcinoma. Clin Cancer Res. 2003;9:391–401. [PubMed] [Google Scholar]
- Yao K, Cho YY, Bode AM, Vummenthala A, Park JG, Liu K, et al. A selective small-molecule inhibitor of c-Jun N-terminal kinase 1. FEBS Lett. 2009;583:2208–2212. doi: 10.1016/j.febslet.2009.06.017. [DOI] [PubMed] [Google Scholar]
- Yoon CH, Kim MJ, Kim RK, Lim EJ, Choi KS, An S, et al. c-Jun N-terminal kinase has a pivotal role in the maintenance of self-renewal and tumorigenicity in glioma stem-like cells. Oncogene. 2012;31:4655–4666. doi: 10.1038/onc.2011.634. [DOI] [PubMed] [Google Scholar]
- Yoshida S, Fukino K, Harada H, Nagai H, Imoto I, Inazawa J, et al. The c-Jun NH2-terminal kinase3 (JNK3) gene: genomic structure, chromosomal assignment, and loss of expression in brain tumors. J Hum Genet. 2001;46:182–187. doi: 10.1007/s100380170086. [DOI] [PubMed] [Google Scholar]
- Zhang J, Yang PL, Gray NS. Targeting cancer with small molecule kinase inhibitors. Nat Rev Cancer. 2009;9:28–39. doi: 10.1038/nrc2559. [DOI] [PubMed] [Google Scholar]
- Zhang JY, Selim MA. The role of the c-Jun N-terminal Kinase signaling pathway in skin cancer. Am J Cancer Res. 2012;2:691–698. [PMC free article] [PubMed] [Google Scholar]
- Zhang JY, Adams AE, Ridky TW, Tao S, Khavari PA. Tumor necrosis factor receptor 1/c-Jun-NH2-kinase signaling promotes human neoplasia. Cancer Res. 2007;67:3827–3834. doi: 10.1158/0008-5472.CAN-06-4017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang T, Inesta-Vaquera F, Niepel M, Zhang J, Ficarro SB, Machleidt T, et al. Discovery of potent and selective covalent inhibitors of JNK. Chem Biol. 2012;19:140–154. doi: 10.1016/j.chembiol.2011.11.010. [DOI] [PMC free article] [PubMed] [Google Scholar]