

Abstract
BACKGROUND AND PURPOSE
The cyclin-dependent kinase CDK9 is an important therapeutic target but currently available inhibitors exhibit low specificity and/or narrow therapeutic windows. Here we have used a new highly specific CDK9 inhibitor, LDC000067 to interrogate gene control mechanisms mediated by CDK9.
EXPERIMENTAL APPROACH
The selectivity of LDC000067 was established in functional kinase assays. Functions of CDK9 in gene expression were assessed with in vitro transcription experiments, single gene analyses and genome-wide expression profiling. Cultures of mouse embryonic stem cells, HeLa cells, several cancer cell lines, along with cells from patients with acute myelogenous leukaemia were also used to investigate cellular responses to LDC000067.
KEY RESULTS
The selectivity of LDC000067 for CDK9 over other CDKs exceeded that of the known inhibitors flavopiridol and DRB. LDC000067 inhibited in vitro transcription in an ATP-competitive and dose-dependent manner. Gene expression profiling of cells treated with LDC000067 demonstrated a selective reduction of short-lived mRNAs, including important regulators of proliferation and apoptosis. Analysis of de novo RNA synthesis suggested a wide ranging positive role of CDK9. At the molecular and cellular level, LDC000067 reproduced effects characteristic of CDK9 inhibition such as enhanced pausing of RNA polymerase II on genes and, most importantly, induction of apoptosis in cancer cells.
CONCLUSIONS AND IMPLICATIONS
Our study provides a framework for the mechanistic understanding of cellular responses to CDK9 inhibition. LDC000067 represents a promising lead for the development of clinically useful, highly specific CDK9 inhibitors.
Keywords: cyclin-dependent kinase, CDK9, gene expression, RNA polymerase II, transcription
Introduction
Cyclin-dependent kinases (CDKs) are a family of evolutionarily conserved serine/threonine kinases that form heterodimers with regulatory cyclin partner proteins (Malumbres and Barbacid, 2005). CDKs can generally be classified into two major groups based on whether they control cell cycle progression or regulate gene transcription by RNA polymerase II (RNAPII). The first group includes CDK1 to CDK6, while CDK8, CDK9, CDK12 and CDK19 are linked to regulation of transcription (Loyer et al., 2005). CDK7 and CDK20 act in both cell cycle control and transcription processes (Fisher, 2005; Wohlbold et al., 2006). Several CDKs (such as CDK10, CDK11A, CDK11B, CDK13) are involved in RNA processing (Loyer et al., 2005), while other CDKs have specialized roles in proliferation and other processes such as cellular survival, homeostasis or development (Malumbres and Barbacid, 2005). Taken together, the CDKs present particularly promising drug targets for therapeutic interference in human pathologies where these processes are affected (Shapiro, 2006; Malumbres and Barbacid, 2009), and several CDK inhibitors are currently under evaluation in clinical trials (http://clinicaltrials.gov/). In particular, inhibition of transcriptional CDKs such as CDK9 might present an effective strategy against proliferative diseases like cancer (Shapiro, 2006; Wang and Fischer, 2008). This idea is supported by the observation that cancer cells often rely on the production of short-lived anti-apoptotic regulator proteins in order to resist programmed cell death. Transcriptional down-regulation of such survival factors through pharmacological CDK9 inhibition would result in antitumour activity due to reinstatement of apoptosis. Indeed, down-regulation of the anti-apoptotic MCL1 gene by the known CDK9 inhibitor flavopiridol (alvocidib) appears to be the primary mechanism underlying its antitumor activity in chronic lymphocytic leukaemia (Chen et al., 2005; Byrd et al., 2007). While these results demonstrate the potential of CDK9 as a therapeutic target, flavopiridol has been shown to inhibit multiple CDKs (Liu et al., 2012) as well as other kinases such as Akt (Caracciolo et al., 2012). Thus, inhibitors that exhibit higher specificity for CDK9 are needed to establish final target validation and to investigate the underlying molecular mechanisms of gene expression control by CDK9, in greater detail.
CDK9 and cyclin T form the positive transcription elongation factor b (P-TEFb) complex that was originally identified as an activity controlling the early phase of transcription elongation via release of RNAPII from inherent promoter-proximal pause sites (Marshall and Price, 1995; Li and Gilmour, 2011). This release involves phosphorylation of the inhibitor 5,6-dichloro-1-β-D-ribofuranosyl-benzimidazole (DRB) sensitivity-inducing factor (DSIF), which consists of SPT4 and SPT5 subunits, as well as subunits of negative elongation factor (NELF) (Wada et al., 1998; Isel and Karn, 1999; Yamaguchi et al., 1999; Ivanov et al., 2000; Fujinaga et al., 2004). DSIF and NELF are thought to cooperate to cause promoter-proximal pausing of RNAPII (Garriga and Grana, 2004; Peterlin and Price, 2006). Subsequent phosphorylation of SPT5 and NELF subunits leads to dissociation of NELF and conversion of DSIF into a positive processivity factor (Martinez-Rucobo et al., 2011; Martinez-Rucobo and Cramer, 2013). CDK9 also phosphorylates the carboxy-terminal heptarepeat (consensus sequence Y1S2P3T4S5P6S7) within the carboxy-terminal domain (CTD) of the largest subunit of RNAPII. The CTD is modified at various stages of transcription. RNAPII is recruited into the preinitiation complex with a hypophosphorylated CTD, and the CTD is phosphorylated on Ser5 (Ser5-P) during initiation and then on Ser2 (Ser2-P) during elongation (Fuda et al., 2009). The latter task is performed by CDK9 (Shim et al., 2002). The degree of phosphorylation at Ser2 increases towards the 3'-ends of genes, which correlates to binding of termination and RNA processing factors to the CTD (Bird et al., 2004). Ser2-P at promoter-proximal sites further helps to release RNAPII from initiation and early elongation complexes (Zhou et al., 2012).
Proteomic studies identified two differently sized forms of the elongation factor P-TEFb, the larger one representing an inactive form of P-TEFb in complex with HEXIM proteins and 7SK RNA (Nguyen et al., 2001; Yang et al., 2001). Targeting CDK9 with pharmacological inhibitors causes its release from this pool, allowing it to bind to genes either on its own or in combination with other proteins. Investigations of individual cellular and viral genes indicate a widespread positive role for CDK9 in gene control (Zhu et al., 1997; Kanazawa et al., 2000; Lis et al., 2000; Chao and Price, 2001). The requirement for CDK9 may quantitatively differ from one gene to another and/or from one activator to another (Hou et al., 2007; Bottardi et al., 2011; Wang et al., 2013).
Here, we have evaluated a new CDK9 inhibitor LDC000067 (abbreviated to LDC067; patent number WO 2008/129080) and shown in Figure 1A. This compound was based on a 2,4-aminopyrimidine scaffold that originated from a small molecule screen of a chemical library that was designed to bind to the ATP binding pockets in kinases. Functional kinase assays confirmed the selectivity for CDKs over other kinases, with a preference for CDK9-cyclin T1. LDC067 inhibited CDK9 in vitro with an IC50 of 44 ± 10 nM. Its selectivity for CDK9 over other CDKs was in the range of 55-fold (vs. CDK2) to over 230-fold (vs. CDK6 and CDK7) and exceeded that of the known and widely used inhibitors DRB and flavopiridol. This higher selectivity of LDC067 was confirmed in an ATP-competitive kinase binding assay. LDC067 also inhibited in vitro transcription in an ATP-competitive and dose-dependent manner. Furthermore, LDC067 decreased phosphorylation of the Ser2 residue within the CTD of RNAPII, both in cells and nuclear extracts as well as in kinase assays using recombinant GST-CTD as substrate. Effects of LDC067 in whole cells included induction of the tumour suppressor protein p53 and apoptosis. Gene expression profiling of cells treated with LDC067 revealed selective reduction of short-lived mRNAs, including those that encode regulators of proliferation and apoptosis such as MCL1 and MYC. Analysis of de novo RNA synthesis demonstrated a broad positive role of CDK9. Finally, after treatment with LDC067, the previously proposed forcing of pausing of RNAPII on MYC and other genes was observed, all of which was consistent with specific inhibition of CDK9. In view of these particular properties, LDC067 may be a valuable tool for the further study of the mechanisms of action CDK9 in vitro and in vivo as well as a potential drug to target CDK9 in disease.
Figure 1.

Inhibition of in vitro transcription by LDC067. (A) Molecular structure of LDC000067. (B) Inhibition of kinase catalytic activity by 10 μM LDC067. A radiometric in vitro kinase assay was used to determine residual kinase activity (expressed as percentage of remaining substrate phosphorylation compared to the DMSO control reaction). Each kinase was measured in duplicate and data shown are means and range of the two measurements. The stippled line indicates residual P-TEFb activity (6.2%) in this assay. (C and D) Influence of inhibitors on in vitro transcription using HEK293T nuclear extract. Transcripts originate from a 380 bp G-free cassette in pGal-ML. Transcript (Tx) levels were determined by phosphorimaging. (E) Comparison of the effects of LDC067 in different nuclear extracts of the indicated cell lines under low (60 μM) and high (500 μM) ATP concentrations. Final inhibitor concentration was 10 μM. (F) Influence of magnesium on transcription inhibition by LDC067 (10 μM) with low ATP (60 μM) in HEK293T nuclear extracts.
Methods
Synthesis of LDC067 (3-((6-(2-methoxyphenyl)pyrimidin-4-yl)amino)phenyl) methanesulfonamide
Step 1
To a solution of 4,6-dichloropyrimidine (3.38 g; 22.7 mmol) in a mixture of dimethoxyethane (30 mL) and water (6 mL) were successively added 2-methoxyphenylboronic acid (3.45 g; 22.7 mmol), PdCl2(PPh3)2 (175 mg; 0.25 mmol) and potassium carbonate (1.69 g; 12.2 mmol). The mixture was stirred for 3 h at 80°C and at room temperature overnight. It was concentrated under reduced pressure. The residue was dissolved in dichloromethane (100 mL), the solution washed with water, dried over MgSO4 and concentrated in vacuo. The intermediate 4-chloro-6-(2-methoxyphenyl)pyrimidine was obtained as a pale yellow solid after chromatographic flash purification (silica gel; dichloromethane/methanol gradient 100:0 to 90:10). Yield: 1.97 g (39%).
Step 2
To a solution of the intermediate from Step 1 (100 mg; 0.453 mmol) in DMF (1 mL) was added (3-aminophenyl) methanesulfonamide (84.3 mg; 0.453 mmol). The mixture was heated for 1 h at 80°C. It was diluted with water and the solvent removed by lyophilization. The remaining yellow solid was passed through an SCX ion exchange cartridge (eluent 7N ammonia in methanol). After another lyophilization step, the pure LDC067 compound was obtained as a white powder. Yield: 119.8 mg (71%). 1H NMR (400 MHz, d6-DMSO, 300 K) δ 3.88 (s, 3H), 4.24 (s, 2H), 6.84 (bs, 2H), 7.00–7.09 (m, 2H), 7.17 (d, J = 8.4 Hz, 1H), 7.33 (t, J = 7.9 Hz, 1H), 7.42–7.47 (m, 2H), 7.62 (bs, 1H), 7.78–7.82 (m, 1H), 7.91 (dd, J = 1.7 Hz, J = 7.7 Hz, 1H), 8.68 (s, 1H), 9.76 (bs, 1H). MS (ES) C18H18N4O3S requires: 370, found: 371 (M + H)+. Purity of the compound was >95% as determined by HPLC-MS.
Cell lines and patient cells
HEK293T human embryonic kidney cells, HeLa cervix carcinoma cells, A549 human lung carcinoma cells and MCF7 human breast adenocarcinoma cells were maintained in DMEM high glucose medium (PAA Laboratories GmbH, Cölbe, Germany), HCT116 human colon carcinoma cells in modified McCoy's 5A medium (Life Technologies GmbH, Darmstadt, Germany), THP1 human monocytic leukemia cells and primary AML blast cells in RPMI1640 medium (Life Technologies). Each medium was supplemented with L-glutamine (2 mM final; Life Technologies), penicillin-streptomycin (100 U/ml-100 μg/ml final; Life Technologies) and 10% fetal bovine serum (FBS Gold; PAA). All cell lines were obtained from ATCC (via LGC Standards GmbH, Wesel, Germany) or DMSZ (Braunschweig, Germany). Cells were kept at 37°C in a humidified incubator with 5% CO2. For detachement of adherent cells 0.05% Trypsin-EDTA (Life Technologies) was used. AML leukaemic blast cells were isolated by apharesis of whole blood from a patient (AML, FAB classification M4) after obtaining written, informed consent from the patient and ethics approval (2007-390-f-S) by the Institutional Review Board of the University Hospital Muenster.
In vitro enzymic kinase assay for CDKs
The fluorescence resonance energy transfer (FRET)-based LANCE Ultra KinaSelect Ser/Thr kit (Perkin Elmer, Waltham, MA, USA) was used to determine IC50 values for various CDK inhibitors. Kinase activity and inhibition in this assay was measured as recommended by the manufacturer. Briefly, a specific ULight MBP peptide substrate (50 nM final concentration) was phosphorylated by a CDK-cyclin pair in buffer (50 mM HEPES-KOH pH 7.5, 10 mM MgCl2, 1 mM EGTA, 2 mM dithiothreitol) containing ATP at the concentration of the KM values of the individual kinases for 1 h at room temperature. Subsequently, phosphorylation was detected by addition of specific Eu-labelled anti-phospho-antibodies (2 nM), which upon binding to the phosphopeptide give rise to a FRET signal. FRET signals were recorded in a time-resolved manner in a Perkin Elmer EnVision reader. Purified cyclin-kinase pairs were obtained from the following suppliers: Carna Biosciences (Kobe, Japan; CDK1-Cyclin B1, CDK6-Cyclin D3, CDK7-Cyclin H-MAT1), ProQinase (Freiburg, Germany; CDK2-Cyclin A) and Invitrogen (Darmstadt, Germany; CDK9-Cyclin T1).
Competitive kinase binding/tracer displacement assay
The LanthaScreen Eu Kinase Binding Assay (Invitrogen) was performed for CDK-cyclin pairs (suppliers: see details mentioned earlier) to determine affinities of inhibitors binding to the ATP binding pocket (Kd determination). It is a FRET assay based on binding and displacement of an ATP-competitive tracer to the kinase of interest. Binding of an inhibitor to the kinase competes for binding with the tracer, resulting in a loss of FRET. The assays were performed under the conditions recommended by the supplier. Briefly, inhibitors were incubated for 1 h with kinase tracer 236 at the concentration corresponding to its individual Kd value for the kinase-cyclin complex, 2 nM LanthaScreen Eu-Anti-His antibody and 5 nM of the purified kinase-cyclin complex. Binding of the tracer to the kinase was measured at 340 nM excitation; 665 nm emission was used for the kinase tracer and 615 nM emission for normalization.
Kinase panel profiling
Inhibition of kinases by LDC067 was measured in a radiometric assay by Reaction Biology (Malvern, PA, USA). The assay directly measures kinase catalytic activity towards a specific substrate (Anastassiadis et al., 2011). Briefly, 10 μM LDC067, or DMSO as solvent control, were added to base reaction buffer (20 mM HEPES pH 7.5, 10 mM MgCl2, 1 mM EDTA, 0.02% Brij35, 0.02 mg·mL−1 BSA, 0.1 mM Na3VO4, 2 mM DTT, 1% DMSO) containing substrates and cofactors required by the individual kinase. 10 μCi of [γ-33P]-ATP (3000 Ci·mmol−1, 10 mCi·mL−1, Perkin Elmer) was added to the reaction mixture, and kinase reactions incubated for 120 min at room temperature. Reactions were spotted on P81 ion exchange paper (GE Healthcare Europe GmbH, Freiburg, Germany), and filters extensively washed in 0.75% phosphoric acid before radiometric quantification. Each protein kinase was measured in duplicate and its catalytic activity expressed as residual kinase activity that is the percentage of average substrate phosphorylation compared to the solvent control reaction.
In vitro transcription
Transcription reactions were conducted with the adenovirus core promoter (template Gal-ML) essentially as described previously (Boeing et al., 2010). For the kinetic elongation assay, a slightly modified immobilized Gal-ML template was used, which will be described elsewhere. For immobilization, templates were PCR amplified using a 5'-biotin-labelled primer that was purified and coupled to paramagnetic streptavidin beads (Promega GmbH, Mannheim, Germany). Reactions were conducted in 25 mM HEPES-KOH pH 8.2, 60 mM KCl, 5 mM MgCl2, 1 mM DTT, 0.01% Igepal CA-630, 5–10% glycerol, 0.5 mg·mL−1 BSA (Roche Applied Science, Mannheim Germany) and 20 units of RiboLock (Fisher Scientific Germany GmbH, Schwerte, Germany). Following transcription reactions, beads were washed, RNA eluted, precipitated and analysed on denaturing gels.
DNA microarray analysis
Gene expression profiling was conducted using high-density oligonucleotide arrays (Human Gene 1.0 ST array, Affymetrix, Santa Clara, CA, USA). Total RNA was isolated from three biological replicates of LDC067-treated (90 min) THP1 cells using the RNeasy RNA purification kit as recommended by the supplier (Qiagen GmbH, Hilden Germany). Sample labelling, hybridization, scanning, raw data extraction and robust multi-array average analysis of background-adjusted, normalized and log-transformed probe-set values were conducted by an authorized Affymetrix service provider (KFB, Regensburg, Germany). Threshold settings for regulated genes were ≥1.4-fold difference from DMSO-treated control cells, with P < 0.05. Gene ontology analysis of regulated genes was performed with DAVID (http://david.abcc.ncifcrf.gov/).
Reverse transcription quantitative PCR (RT-qPCR)
Total RNA was prepared using Trizol reagent (Invitrogen). Reverse transcription was performed using either Superscript II reverse transcriptase (Invitrogen) or Quantitect reverse transcription kit (Qiagen) with random hexamer primers. Quantitative real-time PCR (qPCR) was carried out on the resulting cDNAs using SYBR Green PCR Master kit (Applied Biosystems, Darmstatd, Germany) according to the manufacturer's instructions, on a Step One Plus PCR system (Applied Biosystems). Relative transcript levels were determined using the comparative cycle threshold (ΔΔCT) method. Exon-intron primers were designed with the program Primer3 (http://primer3.wi.mit.edu/). Sequences are available upon request.
Chromatin immunoprecipitation (ChIP) and antibodies
ChIP was performed as previously described (Albert et al., 2010). ChIP and input DNAs were purified with the Qiaquick PCR kit (Qiagen), and qPCR was carried out as outlined above. Antibodies used included IgG control (sc-2027), anti-RNAPII, subunit RPB1 (sc-899), anti-CDK9 (sc-484; all from Santa Cruz Biotechnology, Santa Cruz Biotechnology, Dallas, TX, USA), and monoclonal rat antibodies anti-CTD Ser2-P (clone 3E10), anti-Ser5-P (3E8), and anti-Ser7-P (4E12; all kind gifts from Dirk Eick). Additional antibodies for Western blot analyses included anti-p53 phospho-serine 392 (sc-56173) and anti-Tubulin (sc-8035; Santa Cruz Biotechnology).
Apoptosis measurement
Apoptosis was measured by flow cytometry using annexin-fluorescein and propidium iodide staining according to standard protocols (BD Biosciences, Heidelberg, Germany).
Data analysis
Data are expressed as mean ± SD, and statistical significance of inhibitor effects was evaluated using Student's t-test. P-values < 0.05 were considered as statistically significant.
Materials
Flavopiridol (F3055), DRB (D1916) and staurosporine (S5921) were obtained from Sigma-Aldrich (Taufkirchen, Germany), SNS-032 from Selleck Chemicals (Houston, TX, USA). All compounds were dissolved in DMSO and stored at -20°C in the dark until use.
Results
LDC000067, a novel CDK inhibitor with high specificity for CDK9
Given the limited specificity of several widely used CDK9 inhibitors, we set out to develop highly specific ATP-competitive compounds based on a 2,4-aminopyrimidine scaffold. Specificity was increased in iterative cycles of chemical modification and validated by functional kinase assays. As an intermediate endpoint of this development, we present here the CDK9 inhibitor LDC067 (Figure 1A), which shows promising biochemical and pharmacological properties including high selectivity for CDK9, high stability as well as good tolerability and low cytotoxicity in vivo.
The increased CDK9 selectivity of LDC067 in comparison to established CDK inhibitors flavopiridol, DRB and SNS-032 (formerly BMS-387032) (Misra et al., 2004) was demonstrated via substrate phosphorylation using a FRET-based assay. Half-maximal inhibitory concentrations (IC50) for the CDK9-cyclin T1 pair are shown in Table 1. Importantly, in this assay LDC067 displayed 55/125/210/ >227/ >227-fold selectivity for CDK9 versus CDK2/1/4/6/7, while the other three compounds were much less selective (e.g. 3/ <1/7/59/20-fold for CDK9 vs. CDK2/1/4/6/7 in the case of flavopiridol). The high CDK9 selectivity of LDC067 was confirmed via another FRET-based in vitro assay where the ATP-competitive binding of LDC067, flavopiridol and DRB to CDK2-, CDK7- and CDK9-cyclin complexes was compared (Table 2). The dissociation constant of LDC067 for CDK9-cyclin T1 was 31-fold lower than for CDK2-cyclin A and nearly 500-fold lower than for the trimeric CDK7-cyclin H-MAT1 complex. Again, flavopiridol and DRB showed lower relative differences in their dissociation constants for these CDKs. As the IC50 of LDC067 for CDK2 is 55-fold and its Kd 31-fold higher than the corresponding values for CDK9, it seems unlikely that LDC067 would have significant effects on CDK2 function, if used at the concentrations required to block CDK9 function.
Table 1.
Enzymatic kinase assay
| IC50 [μM] | ||||||
|---|---|---|---|---|---|---|
| CDK1- Cyclin B1 | CDK2- Cyclin A | CDK4- Cyclin D1 | CDK6- Cyclin D3 | CDK7- Cyclin H-MAT1 | CDK9- Cyclin T1 | |
| LDC000067 | 5.513 ± 0.328 (n = 6) | 2.441 ± 0.227 (n = 6) | 9.242 ± 0.174 (n = 6) | >10 (n = 6) | >10 (n = 6) | 0.044 ± 0.010 (n = 6) |
| DRB | >10 (n = 1) | >10 (n = 1) | >10 (n = 1) | >10 (n = 1) | >10 (n = 1) | 1.942 (n = 1) |
| Flavopiridol | <0.005 (n = 3) | 0.015 ± 0.004 (n = 3) | 0.038 ± 0.008 (n = 3) | 0.305 ± 0.023 (n = 3) | 0.103 ± 0.023 (n = 3) | 0.0052 ± 0.0006 (n = 2) |
| SNS-032 | 0.052 ± 0.009 (n = 3) | 0.006 ± 0.001 (n = 3) | 0.355 ± 0.017 (n = 3) | 3.404 ± 0.139 (n = 3) | 0.068 ± 0.008 (n = 3) | 0.0014 ± 0.0005 (n = 3) |
Half-maximal inhibition (IC50) of CDK inhibitors for the indicated CDK-cyclin pairs as determined by an in vitro enzymic kinase assay using time-resolved FRET analysis. Data are expressed as mean ± SD, with number of measurements (n) indicated.
Table 2.
Kinase binding assay
| Kd [μM] | |||
|---|---|---|---|
| CDK2- Cyclin A | CDK7- Cyclin H-MAT1 | CDK9- Cyclin T1 | |
| LDC000067 | 1.014 ± 0.743 (n = 5) | 15.99 ± 8.919 (n = 5) | 0.0327 ± 0.0151 (n = 35) |
| DRB | n.d. | >10 (n = 2) | 0.614 ± 0.0269 (n = 2) |
| Flavopiridol | 0.0302 ± 0.0047 (n = 3) | 0.0963 ± 0.0114 (n = 8) | <0.005 (n = 8) |
Dissociation constants (Kd) of CDK inhibitors LDC067, DRB and flavopiridol for the indicated CDKs were determined by an in vitro kinase activity assay. All reactions contained ATP at the concentration of the KM of the individual kinases (CDK2-Cyclin A: 3 μM; CDK7-Cyclin H-MAT1: 25 μM; CDK9-Cyclin T1: 25 μM). N.d. = not determined. Data are expressed as mean ± SD, with number of measurements (n) indicated.
Further support for the selectivity of LDC067 was obtained by profiling a panel of 28 additional human recombinant non-CDK kinases for their response to LDC067. In this radiometric assay, 10 μM LDC067 or DMSO as solvent control, were added to a kinase reaction that directly measures catalytic activity of the enzyme towards a specific substrate (Anastassiadis et al., 2011). Twenty-two out of the 28 kinases showed greater than 50% residual activity in the presence of LDC067, while CDK9-cyclin T1 was almost completely blocked (only 6% residual activity; Figure 1B). In an additional experiment, comparative IC50 values for several of the less affected kinases (e.g. GSK3A, MAP4K4 or ABL2 with 9.5%, 17.5% and 27% residual activity, respectively) were determined for LDC067 versus staurosporine, a prototypical ATP-competitive kinase inhibitor that is known to bind to many kinases with high affinity but little selectivity (Karaman et al., 2008). Strikingly, IC50 values were several hundred-fold higher than the ones obtained with the non-selective inhibitor staurosporine (Table 3).
Table 3.
Comparison of LDC067 and staurosporine as protein kinase inhibitors
| Compound IC50 [nM] | ||
|---|---|---|
| LDC000067 | Staurosporine | |
| GSK3A | 1 460 | 4.21 |
| HGK/MAP4K4 | 820 | <1.0 |
| ABL2/ARG | 3 640 | 6.42 |
| CAMK4 | >20 000 | 28.90 |
Half-maximal inhibition (IC50) of LDC067 versus staurosporine for the indicated protein kinases as determined by an in vitro enzymic kinase FRET assay (see Table 1). Both compounds were tested in 10-dose mode, with serial dilutions starting at 1000 μM for LDC067 (measured in duplicate) and 20 μM for staurosporine (measured in triplicate). Reactions were carried out at 1 μM ATP for all kinases. Data shown are means.
LDC067 inhibits P-TEFb-dependent in vitro transcription in an ATP-competitive manner
We next asked whether LDC067 blocks transcription in a similar way to DRB and flavopiridol. In vitro transcription experiments were conducted using either soluble or immobilized promoter templates (Boeing et al., 2010) and nuclear extracts of HEK293T cells supplemented with recombinant Gal4-VP16. Transcription assays revealed a half-maximal inhibition of 4 μM for LDC067 at 100 μM ATP (Figure 1B). Using the same assay, a roughly 20-fold higher potency of flavopiridol and a 2.5-fold lower potency of DRB was revealed (Figure 1C). Nuclear extracts from four different cell lines were used to investigate the influence of ATP concentrations on LDC067-dependent inhibition of transcription activity (Figure 1D). For these and most subsequent experiments, a final concentration of 10 μM was chosen which reduces transcription levels to approximately 20–30% in vitro as well as in living cells (see below). High ATP levels (500 μM) markedly diminished the inhibitory effect of LDC067 on in vitro transcription when compared to the routinely used 60 μM ATP. ATP competition was not unexpected, because (i) LDC067 emerged from a chemical library containing scaffolds designed for binding to kinase ATP binding sites, and (ii) LDC067 competed with/displaced the tracer from the ATP binding pocket in the in vitro kinase binding assay (Table 2). We further investigated the effects of KCl or MgCl2 on the relative inhibitory potency of LDC067. While increasing the concentration of KCl from the routinely used 50 mM up to 150 mM resulted in a strong overall decrease of transcript levels, the relative inhibitory potency of LDC067 was largely unaffected (K. Baumgart, data not shown). In contrast, adding increasing amounts of MgCl2 during the transcription elongation resulted in a significant Mg2+-dependent decrease of transcriptional inhibition by LDC067 (Figure 1E).
CDK9 inhibition by LDC067 reduces Ser2-P, induces p53 activation and leads to apoptosis
Next, the effects of CDK9 inhibition by LDC067 in whole cells were investigated. One established cellular target of CDK9 is Ser2 within the RNA polymerase CTD which becomes phosphorylated (Ser2-P) during transcription elongation (Fuda et al., 2009). Previous experiments in mouse embryonic stem cells (mESCs) had shown that flavopiridol at 1 μM significantly reduced Ser2-P levels within 90 min (Rahl et al., 2010). We therefore compared the effects of LDC067 with those of flavopiridol in mESCs. Cells were treated with either 10 μM LDC067 or 1 μM flavopiridol for 90 and 180 min and subsequently analysed by Western blot. Treatment with the two compounds led to 1.6-fold and 3.4-fold reduction of Ser2-P after 90 min (Figure 2A). While Ser2-P repression remained at the same level for LDC067 after 180 min, flavopiridol treatment nearly abolished the CTD modification at the later time point. This quantitative difference might be attributed to the very high concentration of flavopiridol which could result in non-specific effects, especially at later time points (see discussion). We also compared the effects of LDC067 treatment on CTD phosphorylation to the other widely used CDK9 inhibitor DRB in HeLa cells (Figure 2B). Again, a 60 min treatment with 10 μM LDC067 resulted in 1.6-fold reduction of Ser2-P, whereas Ser5-P and Ser7-P remained largely unaffected; this reduction was similar to the one obtained with 50 μM DRB. Finally, we confirmed these results in an in vitro kinase assay using recombinant GST-CTD as substrate in HeLa nuclear extracts (Figure 2C).
Figure 2.
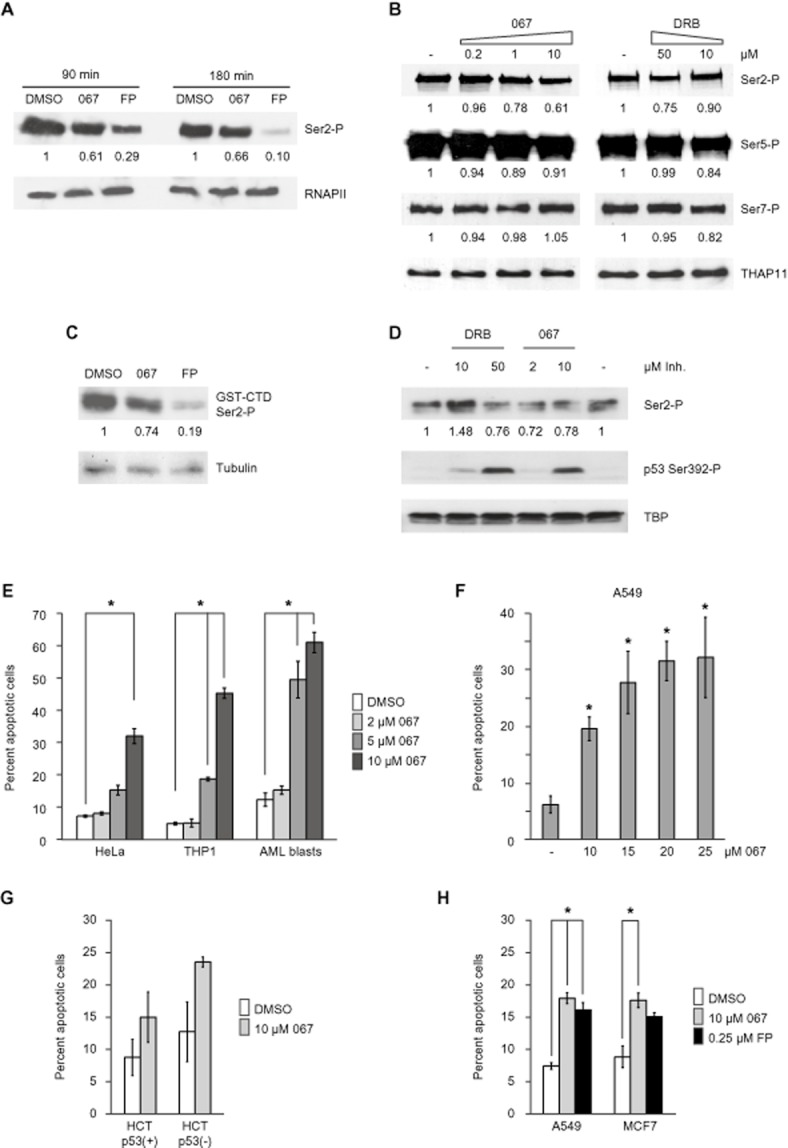
Cellular effects of CDK9 inhibition by LDC067. (A) Western blot analysis of global Ser2-P levels in mESCs treated for 90 or 180 min with DMSO, 10 μM LDC067 or 1 μM flavopiridol (FP). Ser2-P signals were quantified using ImageJ (http://rsbweb.nih.gov/ij/) and normalized to RNAPII signals. (B) Effects of CDK9 inhibitor treatment (60 min) in HeLa cells. THAP1 served as loading control. Quantification of CTD modifications was carried out as in (A). (C) Kinase assay using GST-CTD as substrate in HeLa nuclear extract treated with 10 μM LDC067 or 1 μM flavopiridol for 30 min. GST-CTD Ser2-P levels were quantified using ImageJ and Tubulin signals for normalization. (D) MCF7 cells were treated for 4 h with the indicated concentrations of CDK9 inhibitors and analysed by Western blot with the indicated antibodies. TBP served as loading control. E – H. Apoptosis induction in the indicated cell lines and patient-derived leukaemia (AML) blasts after 24 hours treatment. Apoptosis was assayed using flow cytometry of annexin V-propidium iodide double-stained cells. Signals are represented by the means ± SD (error bars) of three biological replicates, except for AML blasts from a single patient, where three independent technical replicates were analysed. *P < 0.05, significantly different from DMSO or untreated values;Student's t-tests.
Blocking RNAPII transcription elicits a stress response that leads to activation of p53 (Ljungman et al., 1999). During this process, p53 is stabilized and becomes activated by several post-translational modifications, for example extensive phosphorylation. We therefore examined the effect of LDC067 on p53 signalling. Given that p53 is frequently mutated or inactive in tumour cells (one example is HeLa), we used the breast carcinoma cell line MCF7, in which p53 signalling is preserved. Indeed, after 4 h treatment with 10 μM LDC067 (or 50 μM DRB), the p53 protein was stabilized (data not shown) and activated, assessed by phosphorylation of Ser392 (Figure 2D), a modification that is an integral event in the induction of p53 by a diverse range of stimuli (Cox and Meek, 2010).
Various cell lines, in particular of haematopoietic origin, show pronounced sensitivity to flavopiridol- and/or DRB-induced apoptosis (Konig et al., 1997; Parker et al., 1998; Te Poele et al., 1999). Due to its pro-apoptotic effects, flavopiridol is currently under investigation in phase II clinical trials as a therapeutic agent in acute myelogenous leukaemia (AML) (Karp et al., 2012). We tested the effects of LDC067 treatment on several cancer cell lines as well as on primary patient-derived AML blasts using flow cytometry of annexin V-propidium iodide double-stained cells (Figure 2E–H). At concentrations >2 μM all cells showed a marked increase in the percentage of apoptotic cells after 24 h of exposure (Figure 2E). Apoptosis induction was most pronounced in cells of leukaemic origin such as THP1 (approximately 45% apoptotic cells at 10 μM LDC067) and patient-derived blast cells (>60% at 10 μM). Titration of LDC067 in epithelial A549 lung carcinoma cells revealed near-saturation concentrations above 15 μM, with a threefold increase in apoptosis observed at 10 μM (Figure 2F). Treatment with LDC067 of HCT116 colon carcinoma cells which either contained or lacked wild-type p53 caused a similar degree of apoptosis induction in both situations, suggesting a largely p53-independent mechanism at least in this cellular background (Figure 2G). Finally, comparison of cell death rates in LDC067-treated (10 μM) or flavopiridol-treated (0.25 μM) A549 and MCF7 cells revealed a similar degree of apoptosis induction (Figure 2H). Taken together, these data indicate that treatment of growing cells in culture with LDC067 reproduced several relevant cellular responses to CDK9 inhibition, most importantly induction of programmed cell death in leukaemia cells.
Dose-dependent inhibition of de novo RNA synthesis by LDC067
We carried out a concentration-dependent analysis of mRNA synthesis to determine quantitatively the effects of LDC067 on gene transcription in vivo. Cells were treated with the inhibitor for 90 min, RNA was extracted and mRNA analysed via RT-qPCR at several representative cellular genes, including housekeeping genes such as GAPDH and RPL3, cell cycle control genes such as p21, cyclin D1/CCND1, PLK2 or the MYC gene (Figure 3A). As newly synthesized RNA is rapidly spliced (within minutes) and mature mRNA can be stable over days, we chose to analyse de novo synthesized heterogeneous nuclear RNA using primer pairs located in neighbouring exon and intron regions. Repression of mRNA synthesis was first detectable at a concentration of 1 μM, with 50 to 80% repression obtained at a concentration of 10 μM LDC067. Half-maximal repression by LDC067 was achieved in the range of 2 to 6 μM. While relative changes were comparable, the final level of repression varied from gene to gene (Figure 3A). Notably, housekeeping genes (GAPDH, RPL3) were among the strongest responders. Similar results were obtained in several human cancer cell lines (e.g. HeLa, THP1; data not shown) as well as in mESCs (Figure 3B). Occasionally, we observed moderate stimulatory effects, for instance on Myc in mESCs, but these were restricted to low concentrations of LDC067. The underlying reasons remain unclear.
Figure 3.
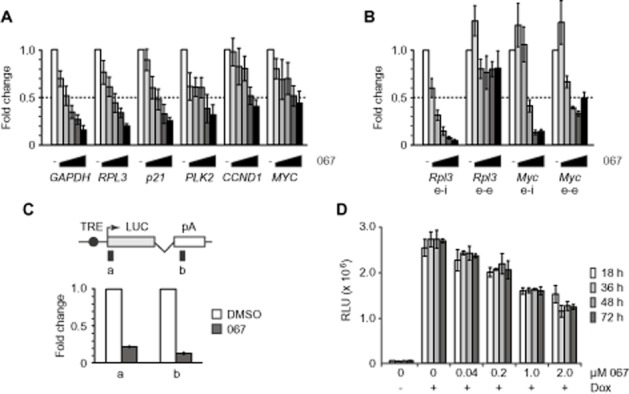
RT-qPCR analysis of de novo transcription. (A and B) Increasing concentrations of LDC067 (1/2/4/6/10 μM in A; 2.5/5/10/20/40 μM in B) were applied for 90 min and the indicated genes were analysed in A549 cells (A) or mESCs (B) using exon-intron primers (e-i) and, for the latter, also exon-exon (e-e) primers. Half-maximal inhibition is indicated by the dotted line. (C) RT-qPCR analysis of luciferase reporter gene transcripts in stably transfected HeLa cells treated with 10 μM LDC067 or DMSO for 30 min followed by 1 μg·mL−1 doxycycline (Dox) for 60 min. Scheme of the luciferase (LUC) reporter gene and primer locations are shown at the top. (D) Luciferase assay of Dox-induced HeLa reporter cells treated with increasing amounts of LDC067 for the indicated times.
De novo RNA synthesis was also measured using a tetracyclin-inducible luciferase reporter gene system in HeLa cells (Uhlmann et al., 2007; Boeing et al., 2010). Treatment with 10 μM LDC067 caused an 80 to 90% reduction of RNA synthesis (Figure 3C). Luciferase expression decreased in a concentration-dependent manner, with half-maximal repression reached at 2 μM LDC067 (Figure 3D). Luciferase repression was stable over at least 3 days, indicating that LDC067 was not metabolized to an inactive form but remained fully active inside the cells (Figure 3D). Together, these data classify LDC067 as an inhibitor of de novo RNA synthesis by RNA polymerase II that is effective at low micromolar concentrations.
P-TEFb broadly affects RNA synthesis of cellular genes
Microarray gene expression profiling was conducted on RNA isolated from cells treated for 90 min with LDC067 (Supporting Information Set S1). For this analysis, we used the leukaemia cell line THP1 transformed by MLL-AF9, as it had shown high sensitivity for CDK9 inhibition by LDC067 (cf. Figure 2E). Approximately 1300 or 4.5% of all genes were reproducibly affected by LDC067 (three biological replicates; fold ≥1.4; P ≤ 0.05), out of which 94% were down-regulated (Figure 4A). Included in the genes most affected in the latter group were the immediate early genes MYC (sevenfold down-regulated) and FOS (fivefold down). Other genes affected were M-phase regulatory kinases PLK2/3/4, chromosome segregation proteins CENPL/C1, anti-proliferative genes BTG1/2 and, consistent with the observed induction of apoptosis, MCL1, all of which were previously identified as targets of flavopiridol and/or DRB (Lam et al., 2001; Garriga et al., 2010). Interestingly, several miRNA genes, including miR-15a, miR-21, let-7f, the polycistronic MYC-controlled miR-17–92 ONCOMIR cluster (Esquela-Kerscher and Slack, 2006), and at least 106 non-coding RNA genes were shown to be down-regulated. Approximately 80% of the latter group represent small nucleolar RNAs, many of which are located in the introns of protein-coding genes that are subject to rapid splicing (Filipowicz and Pogacic, 2002). These data obtained with LDC067 are generally consistent with and extend previous data from microarray analyses using well-established CDK9 inhibitors such as flavopiridol or DRB.
Figure 4.
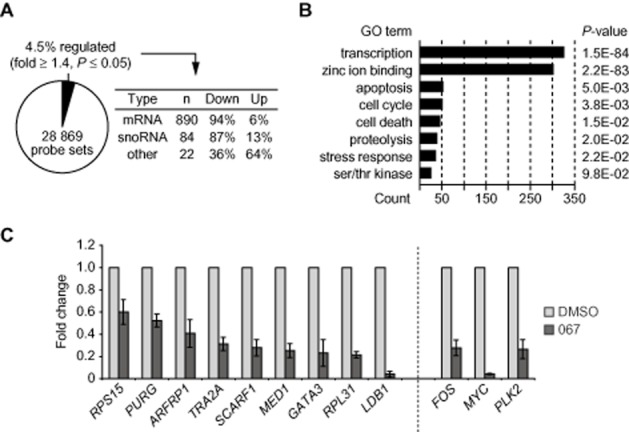
Microarray analysis of THP1 cells treated with LDC067. (A) Statistics of regulated genes. (B) Gene ontology (GO) analysis of down-regulated mRNAs. (C) RT-qPCR analysis of unspliced de novo transcripts covering exon-intron sequences of the indicated genes in control or LDC067-treated THP1 cells. Genes to the left showed unchanged steady-state mRNA levels, while genes to the right (FOS, MYC, PLK2) were significantly down-regulated in the microarray analysis.
Gene ontology analysis indicated an over-representation of transcription factors with fast-decaying mRNAs in the group of down-regulated genes (Yang et al., 2003) (Figure 4B). We hypothesized that genes with intrinsically stable mRNA might remain unaffected in the microarrays. To investigate this possibility, de novo RNA synthesis was analysed at nine genes that were shown not to be affected by LDC067 in the microarrays. This group of genes, which included the housekeeping genes RPS15, RPL31 or MED1, showed a significant decrease in their unspliced transcript levels (Figure 4C). The projection of these and the previous RT-qPCR analyses (Figure 3) suggests that CDK9 positively controls mRNA synthesis of many more genes than was initially deduced from gene expression array data, in which RNA stability appears to confer experimental bias.
CDK9 inhibition by LDC067 increases promoter-proximal RNAPII levels
CDK9 alleviates promoter-proximal pausing of RNAPII (Zhou et al., 2012). Release of the polymerase from these positions is mediated through CTD phosphorylation as well as via release of the inhibitory factors SPT5 and NELF (Yamaguchi et al., 1999). Correspondingly, CDK9 inhibition by DRB or flavopiridol results in elevated RNAPII levels at promoter-proximal pause sites (Glover-Cutter et al., 2008; Rahl et al., 2010). We next addressed whether and how transcriptional inhibition by LDC067 would correlate with the distribution of RNAPII at the prototypical paused MYC gene by conducting ChIP analysis in LDC067-treated HeLa cells (Figure 5). In line with the previous reports, LDC067 forced RNAPII to pause in a promoter-proximal position (Figure 5A). Total RNAPII, and those forms with a phosphorylated CTD at Ser5 (Figure 5D) and Ser7 (Figure 5E) moderately increased at promoter-proximal sites. Conversely, phosphorylation of the CTD at Ser2 was low at the 5'-end of the MYC gene and steadily increased towards the 3'-end (Figure 5C). The influence of LDC067 on Ser2 phosphorylation was surprisingly moderate under conditions where MYC transcription was reduced to less than 50% (compare Figure 3A). Of further note, RNAPII levels in the 3'-region of the gene remained relatively high in the presence of LDC067. Similar ChIP analyses of other genes and/or in other cells confirmed the earlier findings, that is, increased RNAPII levels predominantly at the 5'-end of genes (data not shown). Taken together, our data elucidate the cellular situation when CDK9 was inhibited by a dynamic, ATP-competitive inhibitor at intermediate non-saturating conditions. These data are likely to be relevant from a mechanistic point of view as well as for potential applications, as they illustrate the consequences of RNAPII being under limited (but not abolished) CDK9 activity.
Figure 5.
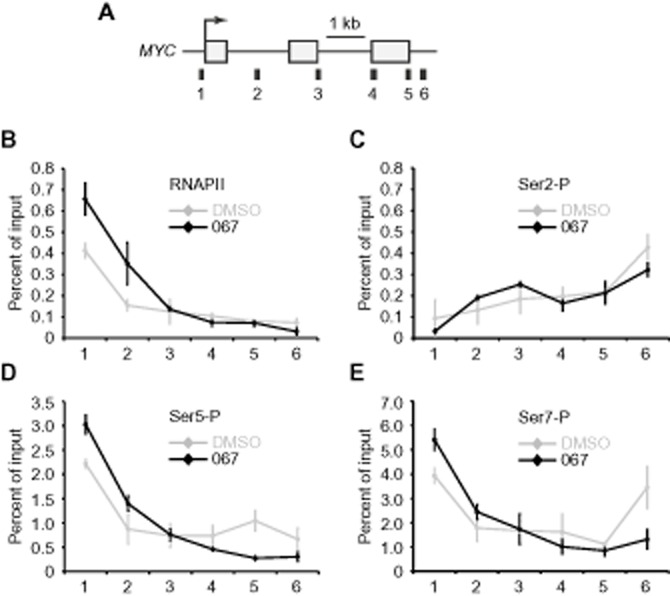
Increase of RNAPII pausing at MYC in the presence of LDC067. (A) Scheme of the human MYC gene with qPCR amplicons used for ChIP analysis indicated underneath. Distribution of RNAPII (B), Ser2-P (C), Ser5-P (D) and Ser7-P (E) was determined by ChIP of HeLa cells treated with 10 μM LDC067 or DMSO for 1 h.
Discussion and conclusions
In the current study, we introduce a novel inhibitor of CDK9 and characterize it as a highly selective binder of CDK9 that effectively interferes with the RNA synthesis of many genes. LDC067 was significantly more specific for CDK9 than both flavopiridol and DRB (Tables 1 and 2), and it had a particular advantage over the latter in that it can be used for biomedical studies in mice (B. Klebl, unpubl. obs.). High CDK9 specificity is especially important in this context because the earlier studies using DRB or flavopiridol could have been compromised by the known off-target effects on the other CTD kinases, CDK1, CDK2 and CDK7.
Using this highly specific inhibitor, we have confirmed a series of functions and mechanisms that were previously associated with CDK9, namely (i) the general requirement of CDK9 for efficient transcription in vitro (Figure 1) as well as in growing cells (Figures 3 and 4), (ii) the targeting of CTD Ser2 by CDK9 (Figure 2), (iii) the activation of the tumour suppressor p53 (Figure 2), (iv) the induction of apoptosis (Figure 2) and (v) the enhancement of RNAPII pausing (Figure 5) after selective CDK9 inhibition by LDC067. RNA analysis of whole cells carried out in the present study uncovered a critical positive role for CDK9 in the de novo RNA synthesis of all analysed genes. Relevant to therapeutic applications, LDC067 causes apoptosis in several human cancer cell lines (THP1, A549, MCF7, HeLa), as has been previously observed for other CDK9 inhibitors (Figure 2). In addition, after treatment with LDC067 apoptosis was induced in primary AML blasts isolated from leukaemia patients. Cell death following LDC067 did not depend on active p53 – a similar effect of LDC067 was observed in HCT116 cells lacking a p53 gene. Instead, we assume that cell death was related to the repression of anti-apoptotic genes such as MCL1, which was down-regulated in our microarray studies and has been reported previously in a different context (Lam et al., 2001; Ma et al., 2003). Beyond this, our data generally imply that other genes like MCL1 that encode mRNAs and proteins with short half-lives could determine the initial biological response to CDK9 inhibitors. One prominent example is MYC, whose mRNA and protein are subject to rapid turnover (see Figure 3, and data not shown). Our microarray analysis uncovered additional, novel CDK9 targets, most notably several non-coding RNAs including cancer-associated miRNA genes such as miR-15a, miR-21, let-7f (Esquela-Kerscher and Slack, 2006), and the polycystronic miR-17–92 ONCOMIR cluster (He et al., 2005). Notably, the latter is a direct target of MYC, suggesting that ONCOMIR down-regulation (2.2-fold) was mediated through transcriptional repression of MYC (O'Donnell et al., 2005).
Our in vitro transcription analysis clearly revealed competition between LDC067 and ATP (Figure 1D). Surprisingly, CDK9 inhibition by LDC067 was also strongly dependent on magnesium (Figure 1E). Currently, we can only speculate about the underlying mechanism. Recent structure-function analyses of the closely related CDK2 demonstrated the requirement for coordination of a second Mg2+ ion in the active site to stabilize ATP-binding and to maximally enhance catalytic activity (Bao et al., 2011; Jacobsen et al., 2012). By analogy, ATP-binding in the CDK9 active site might also depend on this mechanism. As a consequence, increasing the availability of magnesium might favour ATP- over LDC067-binding to the catalytic centre of CDK9, thereby diminishing the inhibitory activity of LDC067.
Reduction of RNAPII CTD phosphorylation at Ser2 by LDC067 was moderate in comparison with the clinically evaluated flavopiridol, especially after longer drug exposure (Figure 2A). Of note, reduction in CTD phosphorylation by flavopiridol was not always directly linked with MCL1 ablation (Shapiro, 2006). On the other hand, block of MCL1 expression and concurrent induction of apoptosis is thought to be the critical mode of action for flavopiridol in chronic lymphocytic leukaemia cells (Chen et al., 2005), indicating that Ser2-P is neither the sole (nor likely the most critical) target of CDK9 inhibitors. The more severe effects of flavopiridol on Ser2-P might result from its lower specificity for CDK9. For example, the pan-specific flavopiridol could target additional CTD-Ser2 kinases such as CDK12 and/or CDK13 (Bartkowiak et al., 2010; Blazek et al., 2011). Alternatively, Ser2-P ablation by flavopiridol could simply reflect its higher affinity towards CDK9 (Table 2) (Chao et al., 2000). Improvement of the relatively low affinity of LDC067 might help to illuminate this issue in the future.
One important reason for the development of LDC067 and related compounds is that pan-CDK inhibitors such as flavopiridol exhibit a significant and dose-limiting level of cytotoxicity (Shapiro, 2006), possibly due to their equipotent inhibition of many members of the CDK family (see Table 1). This situation would be predictably different for mono- or dual-selective CDK inhibitors. Indeed, using LDC067 and analogous compounds from the series of aminopyrimidines, we could demonstrate that they exhibited a significant therapeutic window (i.e. ratio of toxic-to-efficacious dose) in cells as well as in vivo (B. Klebl, unpubl. obs.). Along the same line, a dual-selective CDK4/6 inhibitor (Fry et al., 2004) has recently yielded very encouraging results in a breast cancer trial (Guha, 2013), demonstrating that the more selective inhibition of certain members of the CDK family is a valid therapeutic principle. Taken together, our data provide a rationale for the development and optimization of clinically useful, highly selective CDK9 inhibitors based on the 2,4-aminopyrimidine scaffold, and LDC067 represents a promising lead in this context.
Acknowledgments
We thank Dirk Eick (Helmholtz Center Munich) for CTD antibodies. We are grateful to Carsten Mueller-Tidow and colleagues of the University Hospital Muenster for providing access to patient samples, and to Timothy Kelso for critical reading of the manuscript and other members of the Meisterernst laboratory for helpful discussions. This work was supported by the Ministry for Research and Technology (BMBF, grant number 0313860D to MM and 0313860B to GM) and by grants of the Westfalian Wilhelms University Muenster to MM.
Abbreviations
- LDC067
LDC000067, 3-((6-(2-methoxyphenyl)pyrimidin-4-yl)amino)phenyl) methane sulfonamide
- AML
acute myelogenous leukaemia
- CDK
cyclin-dependent kinase
- ChIP
chromatin immunoprecipitation
- CTD
carboxyterminal domain
- DRB
5,6-dichloro-1-β-D-ribofuranosyl-benzimidazole
- DSIF
DRB sensitivity-inducing factor
- FRET
fluorescence resonance energy transfer
- mESCs
mouse embryonic stem cells
- NELF
negative elongation factor
- P-TEFb
positive transcription elongation factor b
- RNAPII
RNA polymerase II
- RT-qPCR
reverse transcription-quantitative real-time PCR
- Ser2/5/7-P
CTD phospho-Ser2/5/7
Conflicts of interest
None.
Supporting information
Additional Supporting Information may be found in the online version of this article at the publisher's web-site:
http://dx.doi.org/10.1111/bph.12408
Supporting Information Set S1 Regulated genes (Fold ≥ 1.4; P ≤ 0.05) as determined by affymetrix microarray Analysis of LDC067-treated THP1 cells.
References
- Albert TK, Grote K, Boeing S, Meisterernst M. Basal core promoters control the equilibrium between negative cofactor 2 and preinitiation complexes in human cells. Genome Biol. 2010;11:R33. doi: 10.1186/gb-2010-11-3-r33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anastassiadis T, Deacon SW, Devarajan K, Ma H, Peterson JR. Comprehensive assay of kinase catalytic activity reveals features of kinase inhibitor selectivity. Nat Biotechnol. 2011;29:1039–1045. doi: 10.1038/nbt.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bao ZQ, Jacobsen DM, Young MA. Briefly bound to activate: transient binding of a second catalytic magnesium activates the structure and dynamics of CDK2 kinase for catalysis. Structure. 2011;19:675–690. doi: 10.1016/j.str.2011.02.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartkowiak B, Liu P, Phatnani HP, Fuda NJ, Cooper JJ, Price DH, et al. CDK12 is a transcription elongation-associated CTD kinase, the metazoan ortholog of yeast Ctk1. Genes Dev. 2010;24:2303–2316. doi: 10.1101/gad.1968210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bird G, Zorio DA, Bentley DL. RNA polymerase II carboxy-terminal domain phosphorylation is required for cotranscriptional pre-mRNA splicing and 3'-end formation. Mol Cell Biol. 2004;24:8963–8969. doi: 10.1128/MCB.24.20.8963-8969.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blazek D, Kohoutek J, Bartholomeeusen K, Johansen E, Hulinkova P, Luo Z, et al. The Cyclin K/Cdk12 complex maintains genomic stability via regulation of expression of DNA damage response genes. Genes Dev. 2011;25:2158–2172. doi: 10.1101/gad.16962311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boeing S, Rigault C, Heidemann M, Eick D, Meisterernst M. RNA polymerase II C-terminal heptarepeat domain Ser-7 phosphorylation is established in a mediator-dependent fashion. J Biol Chem. 2010;285:188–196. doi: 10.1074/jbc.M109.046565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bottardi S, Zmiri FA, Bourgoin V, Ross J, Mavoungou L, Milot E. Ikaros interacts with P-TEFb and cooperates with GATA-1 to enhance transcription elongation. Nucleic Acids Res. 2011;39:3505–3519. doi: 10.1093/nar/gkq1271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Byrd JC, Lin TS, Dalton JT, Wu D, Phelps MA, Fischer B, et al. Flavopiridol administered using a pharmacologically derived schedule is associated with marked clinical efficacy in refractory, genetically high-risk chronic lymphocytic leukemia. Blood. 2007;109:399–404. doi: 10.1182/blood-2006-05-020735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caracciolo V, Laurenti G, Romano G, Carnevale V, Cimini AM, Crozier-Fitzgerald C, et al. Flavopiridol induces phosphorylation of AKT in a human glioblastoma cell line, in contrast to siRNA-mediated silencing of Cdk9: implications for drug design and development. Cell Cycle. 2012;11:1202–1216. doi: 10.4161/cc.11.6.19663. [DOI] [PubMed] [Google Scholar]
- Chao SH, Price DH. Flavopiridol inactivates P-TEFb and blocks most RNA polymerase II transcription in vivo. J Biol Chem. 2001;276:31793–31799. doi: 10.1074/jbc.M102306200. [DOI] [PubMed] [Google Scholar]
- Chao SH, Fujinaga K, Marion JE, Taube R, Sausville EA, Senderowicz AM, et al. Flavopiridol inhibits P-TEFb and blocks HIV-1 replication. J Biol Chem. 2000;275:28345–28348. doi: 10.1074/jbc.C000446200. [DOI] [PubMed] [Google Scholar]
- Chen R, Keating MJ, Gandhi V, Plunkett W. Transcription inhibition by flavopiridol: mechanism of chronic lymphocytic leukemia cell death. Blood. 2005;106:2513–2519. doi: 10.1182/blood-2005-04-1678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cox ML, Meek DW. Phosphorylation of serine 392 in p53 is a common and integral event during p53 induction by diverse stimuli. Cell Signal. 2010;22:564–571. doi: 10.1016/j.cellsig.2009.11.014. [DOI] [PubMed] [Google Scholar]
- Esquela-Kerscher A, Slack FJ. Oncomirs – microRNAs with a role in cancer. Nat Rev Cancer. 2006;6:259–269. doi: 10.1038/nrc1840. [DOI] [PubMed] [Google Scholar]
- Filipowicz W, Pogacic V. Biogenesis of small nucleolar ribonucleoproteins. Curr Opin Cell Biol. 2002;14:319–327. doi: 10.1016/s0955-0674(02)00334-4. [DOI] [PubMed] [Google Scholar]
- Fisher RP. Secrets of a double agent: CDK7 in cell-cycle control and transcription. J Cell Sci. 2005;118:5171–5180. doi: 10.1242/jcs.02718. [DOI] [PubMed] [Google Scholar]
- Fry DW, Harvey PJ, Keller PR, Elliott WL, Meade M, Trachet E, et al. Specific inhibition of cyclin-dependent kinase 4/6 by PD 0332991 and associated antitumor activity in human tumor xenografts. Mol Cancer Ther. 2004;3:1427–1438. [PubMed] [Google Scholar]
- Fuda NJ, Ardehali MB, Lis JT. Defining mechanisms that regulate RNA polymerase II transcription in vivo. Nature. 2009;461:186–192. doi: 10.1038/nature08449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujinaga K, Irwin D, Huang Y, Taube R, Kurosu T, Peterlin BM. Dynamics of human immunodeficiency virus transcription: P-TEFb phosphorylates RD and dissociates negative effectors from the transactivation response element. Mol Cell Biol. 2004;24:787–795. doi: 10.1128/MCB.24.2.787-795.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garriga J, Grana X. Cellular control of gene expression by T-type cyclin/CDK9 complexes. Gene. 2004;337:15–23. doi: 10.1016/j.gene.2004.05.007. [DOI] [PubMed] [Google Scholar]
- Garriga J, Xie H, Obradovic Z, Grana X. Selective control of gene expression by CDK9 in human cells. J Cell Physiol. 2010;222:200–208. doi: 10.1002/jcp.21938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glover-Cutter K, Kim S, Espinosa J, Bentley DL. RNA polymerase II pauses and associates with pre-mRNA processing factors at both ends of genes. Nat Struct Mol Biol. 2008;15:71–78. doi: 10.1038/nsmb1352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guha M. Blockbuster dreams for Pfizer's CDK inhibitor. Nat Biotechnol. 2013;31:187. doi: 10.1038/nbt0313-187a. [DOI] [PubMed] [Google Scholar]
- He L, Thomson JM, Hemann MT, Hernando-Monge E, Mu D, Goodson S, et al. A microRNA polycistron as a potential human oncogene. Nature. 2005;435:828–833. doi: 10.1038/nature03552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hou T, Ray S, Brasier AR. The functional role of an interleukin 6-inducible CDK9.STAT3 complex in human gamma-fibrinogen gene expression. J Biol Chem. 2007;282:37091–37102. doi: 10.1074/jbc.M706458200. [DOI] [PubMed] [Google Scholar]
- Isel C, Karn J. Direct evidence that HIV-1 Tat stimulates RNA polymerase II carboxyl-terminal domain hyperphosphorylation during transcriptional elongation. J Mol Biol. 1999;290:929–941. doi: 10.1006/jmbi.1999.2933. [DOI] [PubMed] [Google Scholar]
- Ivanov D, Kwak YT, Guo J, Gaynor RB. Domains in the SPT5 protein that modulate its transcriptional regulatory properties. Mol Cell Biol. 2000;20:2970–2983. doi: 10.1128/mcb.20.9.2970-2983.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacobsen DM, Bao ZQ, O'brien P, Brooks CL, 3rd, Young MA. Price to be paid for two-metal catalysis: magnesium ions that accelerate chemistry unavoidably limit product release from a protein kinase. J Am Chem Soc. 2012;134:15357–15370. doi: 10.1021/ja304419t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanazawa S, Okamoto T, Peterlin BM. Tat competes with CIITA for the binding to P-TEFb and blocks the expression of MHC class II genes in HIV infection. Immunity. 2000;12:61–70. doi: 10.1016/s1074-7613(00)80159-4. [DOI] [PubMed] [Google Scholar]
- Karaman MW, Herrgard S, Treiber DK, Gallant P, Atteridge CE, Campbell BT, et al. A quantitative analysis of kinase inhibitor selectivity. Nat Biotechnol. 2008;26:127–132. doi: 10.1038/nbt1358. [DOI] [PubMed] [Google Scholar]
- Karp JE, Garrett-Mayer EL, Estey EH, Rudek MA, Smith BD, Greer JM, et al. Randomized phase II study of two schedules of flavopiridol given as timed sequential therapy with cytosine arabinoside and mitoxantrone for adults with newly diagnosed, poor-risk acute myelogenous leukemia. Haematologica. 2012;97:1736–1742. doi: 10.3324/haematol.2012.062539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Konig A, Schwartz GK, Mohammad RM, Al-Katib A, Gabrilove JL. The novel cyclin-dependent kinase inhibitor flavopiridol downregulates Bcl-2 and induces growth arrest and apoptosis in chronic B-cell leukemia lines. Blood. 1997;90:4307–4312. [PubMed] [Google Scholar]
- Lam LT, Pickeral OK, Peng AC, Rosenwald A, Hurt EM, Giltnane JM, et al. Genomic-scale measurement of mRNA turnover and the mechanisms of action of the anti-cancer drug flavopiridol. Genome Biol. 2001;2:RESEARCH0041. doi: 10.1186/gb-2001-2-10-research0041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J, Gilmour DS. Promoter proximal pausing and the control of gene expression. Curr Opin Genet Dev. 2011;21:231–235. doi: 10.1016/j.gde.2011.01.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lis JT, Mason P, Peng J, Price DH, Werner J. P-TEFb kinase recruitment and function at heat shock loci. Genes Dev. 2000;14:792–803. [PMC free article] [PubMed] [Google Scholar]
- Liu X, Shi S, Lam F, Pepper C, Fischer PM, Wang S. CDKI-71, a novel CDK9 inhibitor, is preferentially cytotoxic to cancer cells compared to flavopiridol. Int J Cancer. 2012;130:1216–1226. doi: 10.1002/ijc.26127. [DOI] [PubMed] [Google Scholar]
- Ljungman M, Zhang F, Chen F, Rainbow AJ, McKay BC. Inhibition of RNA polymerase II as a trigger for the p53 response. Oncogene. 1999;18:583–592. doi: 10.1038/sj.onc.1202356. [DOI] [PubMed] [Google Scholar]
- Loyer P, Trembley JH, Katona R, Kidd VJ, Lahti JM. Role of CDK/cyclin complexes in transcription and RNA splicing. Cell Signal. 2005;17:1033–1051. doi: 10.1016/j.cellsig.2005.02.005. [DOI] [PubMed] [Google Scholar]
- Ma Y, Cress WD, Haura EB. Flavopiridol-induced apoptosis is mediated through up-regulation of E2F1 and repression of Mcl-1. Mol Cancer Ther. 2003;2:73–81. [PubMed] [Google Scholar]
- Malumbres M, Barbacid M. Mammalian cyclin-dependent kinases. Trends Biochem Sci. 2005;30:630–641. doi: 10.1016/j.tibs.2005.09.005. [DOI] [PubMed] [Google Scholar]
- Malumbres M, Barbacid M. Cell cycle, CDKs and cancer: a changing paradigm. Nat Rev Cancer. 2009;9:153–166. doi: 10.1038/nrc2602. [DOI] [PubMed] [Google Scholar]
- Marshall NF, Price DH. Purification of P-TEFb, a transcription factor required for the transition into productive elongation. J Biol Chem. 1995;270:12335–12338. doi: 10.1074/jbc.270.21.12335. [DOI] [PubMed] [Google Scholar]
- Martinez-Rucobo FW, Cramer P. Structural basis of transcription elongation. Biochim Biophys Acta. 2013;1829:9–19. doi: 10.1016/j.bbagrm.2012.09.002. [DOI] [PubMed] [Google Scholar]
- Martinez-Rucobo FW, Sainsbury S, Cheung AC, Cramer P. Architecture of the RNA polymerase-Spt4/5 complex and basis of universal transcription processivity. EMBO J. 2011;30:1302–1310. doi: 10.1038/emboj.2011.64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Misra RN, Xiao HY, Kim KS, Lu S, Han WC, Barbosa SA, et al. N-(cycloalkylamino)acyl-2-aminothiazole inhibitors of cyclin-dependent kinase 2. N-[5-[[[5-(1,1-dimethylethyl)-2-oxazolyl]methyl]thio]-2-thiazolyl]-4- piperidinecarboxamide (BMS-387032), a highly efficacious and selective antitumor agent. J Med Chem. 2004;47:1719–1728. doi: 10.1021/jm0305568. [DOI] [PubMed] [Google Scholar]
- Nguyen VT, Kiss T, Michels AA, Bensaude O. 7SK small nuclear RNA binds to and inhibits the activity of CDK9/cyclin T complexes. Nature. 2001;414:322–325. doi: 10.1038/35104581. [DOI] [PubMed] [Google Scholar]
- O'donnell KA, Wentzel EA, Zeller KI, Dang CV, Mendell JT. c-Myc-regulated microRNAs modulate E2F1 expression. Nature. 2005;435:839–843. doi: 10.1038/nature03677. [DOI] [PubMed] [Google Scholar]
- Parker BW, Kaur G, Nieves-Neira W, Taimi M, Kohlhagen G, Shimizu T, et al. Early induction of apoptosis in hematopoietic cell lines after exposure to flavopiridol. Blood. 1998;91:458–465. [PubMed] [Google Scholar]
- Peterlin BM, Price DH. Controlling the elongation phase of transcription with P-TEFb. Mol Cell. 2006;23:297–305. doi: 10.1016/j.molcel.2006.06.014. [DOI] [PubMed] [Google Scholar]
- Rahl PB, Lin CY, Seila AC, Flynn RA, McCuine S, Burge CB, et al. c-Myc regulates transcriptional pause release. Cell. 2010;141:432–445. doi: 10.1016/j.cell.2010.03.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shapiro GI. Cyclin-dependent kinase pathways as targets for cancer treatment. J Clin Oncol. 2006;24:1770–1783. doi: 10.1200/JCO.2005.03.7689. [DOI] [PubMed] [Google Scholar]
- Shim EY, Walker AK, Shi Y, Blackwell TK. CDK-9/cyclin T (P-TEFb) is required in two postinitiation pathways for transcription in the C. elegans embryo. Genes Dev. 2002;16:2135–2146. doi: 10.1101/gad.999002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Te Poele RH, Okorokov AL, Joel SP. RNA synthesis block by 5, 6-dichloro-1-beta-D-ribofuranosylbenzimidazole (DRB) triggers p53-dependent apoptosis in human colon carcinoma cells. Oncogene. 1999;18:5765–5772. doi: 10.1038/sj.onc.1202961. [DOI] [PubMed] [Google Scholar]
- Uhlmann T, Boeing S, Lehmbacher M, Meisterernst M. The VP16 activation domain establishes an active mediator lacking CDK8 in vivo. J Biol Chem. 2007;282:2163–2173. doi: 10.1074/jbc.M608451200. [DOI] [PubMed] [Google Scholar]
- Wada T, Takagi T, Yamaguchi Y, Ferdous A, Imai T, Hirose S, et al. DSIF, a novel transcription elongation factor that regulates RNA polymerase II processivity, is composed of human Spt4 and Spt5 homologs. Genes Dev. 1998;12:343–356. doi: 10.1101/gad.12.3.343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang S, Fischer PM. Cyclin-dependent kinase 9: a key transcriptional regulator and potential drug target in oncology, virology and cardiology. Trends Pharmacol Sci. 2008;29:302–313. doi: 10.1016/j.tips.2008.03.003. [DOI] [PubMed] [Google Scholar]
- Wang W, Yao X, Huang Y, Hu X, Liu R, Hou D, et al. Mediator MED23 regulates basal transcription in vivo via an interaction with P-TEFb. Transcription. 2013;4:39–51. doi: 10.4161/trns.22874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wohlbold L, Larochelle S, Liao JC, Livshits G, Singer J, Shokat KM, et al. The cyclin-dependent kinase (CDK) family member PNQALRE/CCRK supports cell proliferation but has no intrinsic CDK-activating kinase (CAK) activity. Cell Cycle. 2006;5:546–554. doi: 10.4161/cc.5.5.2541. [DOI] [PubMed] [Google Scholar]
- Yamaguchi Y, Takagi T, Wada T, Yano K, Furuya A, Sugimoto S, et al. NELF, a multisubunit complex containing RD, cooperates with DSIF to repress RNA polymerase II elongation. Cell. 1999;97:41–51. doi: 10.1016/s0092-8674(00)80713-8. [DOI] [PubMed] [Google Scholar]
- Yang E, Van Nimwegen E, Zavolan M, Rajewsky N, Schroeder M, Magnasco M, et al. Decay rates of human mRNAs: correlation with functional characteristics and sequence attributes. Genome Res. 2003;13:1863–1872. doi: 10.1101/gr.1272403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Z, Zhu Q, Luo K, Zhou Q. The 7SK small nuclear RNA inhibits the CDK9/cyclin T1 kinase to control transcription. Nature. 2001;414:317–322. doi: 10.1038/35104575. [DOI] [PubMed] [Google Scholar]
- Zhou Q, Li T, Price DH. RNA polymerase II elongation control. Annu Rev Biochem. 2012;81:119–143. doi: 10.1146/annurev-biochem-052610-095910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu Y, Pe'ery T, Peng J, Ramanathan Y, Marshall N, Marshall T, et al. Transcription elongation factor P-TEFb is required for HIV-1 tat transactivation in vitro. Genes Dev. 1997;11:2622–2632. doi: 10.1101/gad.11.20.2622. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.