

Abstract
BACKGROUND AND PURPOSE
Hypoxia causes vasodilatation of coronary arteries, but the underlying mechanisms are poorly understood. We hypothesized that hypoxia reduces intracellular Ca2+ concentration ([Ca2+]i) by opening of K channels and release of H2S.
EXPERIMENTAL APPROACH
Porcine coronary arteries without endothelium were mounted for measurement of isometric tension and [Ca2+]i, and the expression of voltage-gated K channels KV7 channels (encoded by KCNQ genes) and large-conductance calcium-activated K channels (KCa1.1) was examined. Voltage clamp assessed the role of KV7 channels in hypoxia.
KEY RESULTS
Gradual reduction of oxygen concentration from 95 to 1% dilated the precontracted coronary arteries and this was associated with reduced [Ca2+]i in PGF2α (10 μM)-contracted arteries whereas no fall in [Ca2+]i was observed in 30 mM K-contracted arteries. Blockers of ATP-sensitive voltage-gated potassium channels and KCa1.1 inhibited hypoxia-induced dilatation in PGF2α-contracted arteries; this inhibition was more marked in the presence of the Kv7 channel blockers, XE991 and linopirdine, while a KV7.1 blocker, failed to change hypoxic vasodilatation. XE991 also inhibited H2S- and adenosine-induced vasodilatation. PCR revealed the expression of KV7.1, KV7.4, KV7.5 and KCa1.1 channels, and KCa1.1, KV7.4 and KV7.5 were also identified by immunoblotting. Voltage clamp studies showed the XE991-sensitive current was more marked in hypoxic conditions.
CONCLUSION
The KV7.4 and KV7.5 channels, which we identified in the coronary arteries, appear to have a major role in hypoxia-induced vasodilatation. The voltage clamp results further support the involvement of KV7 channels in this vasodilatation. Activation of these KV7 channels may be induced by H2S and adenosine.
Keywords: potassium channels, KV7, H2S, adenosine, hypoxia, calcium, coronary, vasodilatation
Introduction
Coronary artery dilatation to hypoxia is a fast, protective response that increases flow to endangered myocardium. The hypoxic vasodilatation is of importance for vasomotor tone in the coronary microcirculation but is also present in conduit vessels (Wadsworth, 1994; Barron and Gu, 2000; Shimizu et al., 2000). While the results from clinical trials of oxygen therapy are awaited (Stub et al., 2012), the basal mechanisms linking oxygen concentration and coronary vascular tone need to be elucidated. In porcine coronary arteries, NO contributes to hypoxia-induced vasodilatation, but hypoxia still induces significant vasodilatation in arteries without endothelium (Hedegaard et al., 2011).
Hypoxia is known to have a direct effect on the vascular smooth muscle, and hypoxia-induced vasodilatation may be dependent or independent of changes in intracellular Ca2+ concentration ([Ca2+]i) (Shimizu et al., 2000; Thorne et al., 2004). Hypoxic vasodilatation independent of changes in vascular smooth muscle calcium has been ascribed to [Ca2+]i desensitization (Somlyo and Somlyo, 2003; Wardle et al., 2006), or may be mediated by force suppression due to thin filament phosphorylation of, for example heat shock protein 20 (Rembold et al., 2000; Frobert et al., 2005; Meeks et al., 2005). Hypoxia-induced vasodilatation with a reduction in smooth muscle [Ca2+]i can involve K channel opening (Lopez-Barneo et al., 2004) and/or L-type Ca2+ channel closure (Franco-Obregon and Lopez-Barneo, 1996; Smani et al., 2002). Several types of K channels including ATP-sensitive K channels (KATP) in the coronary circulation (Daut et al., 1990; Dart and Standen, 1995; Liu and Flavahan, 1997; Lee et al., 1998; Kamekura et al., 1999), large-conductance calcium-activated K channels (KCa1.1, also known as BKCa) in coronary and cerebral arteries (Gebremedhin et al., 1994; Nelson and Quayle, 1995; Lopez-Barneo et al., 2004) and voltage-gated KV channels of the KV1.5 and KV2.1 subtype in the coronary circulation (Shimizu et al., 2000; Thorne et al., 2002) have been suggested to be O2 sensors and/or involved in hypoxic vasodilatation. Thus, the type of K channel involved in hypoxia-induced coronary vasodilatation is contentious. Furthermore, KV7 channel openers were recently suggested to counteract changes induced by chronic hypoxia in the pulmonary circulation (Morecroft et al., 2009), and, therefore, these channels may also be involved in hypoxic vasodilatation. The nomenclature used to denote the K channels conforms to Br J Pharmacol's Concise Guide to PHARMACOLOGY (Alexander et al., 2013).
KV7 blockers increase vascular tone in a number of rodent blood vessels including the portal vein, aorta and mesenteric, pulmonary and cerebral arteries (Yeung and Greenwood, 2005; Joshi et al., 2006; Yeung et al., 2007; Mackie et al., 2008; Zhong et al., 2010). Experiments have also revealed that activators of KV7.2–7.5 channels, retigabine and flupirtine, cause relaxation (Yeung et al., 2007; 2008b; Mackie et al., 2008). Moreover, KV7 channels sensitive to the blocker XE991 are involved in H2S-induced relaxation in rat aorta (Schleifenbaum et al., 2010). Also H2S may act as an oxygen sensor and cause vasodilatation (Skovgaard et al., 2011; Whiteman et al., 2011). Therefore, based on these observations, we hypothesized that hypoxia reduces smooth muscle [Ca2+]i by opening K channels and the release of H2S.
To investigate this hypothesis, the following measurements were performed: (i) changes in [Ca2+]i were measured to determine whether lowering [Ca2+]i contributes to hypoxia-induced vasodilatation; (ii) the involvement of different K channels was investigated using selective K channel blockers; (iii) the presence of KV7 channels was established by PCR and immunoblotting; (iv) XE991-sensitive currents were investigated by voltage patch clamp; (v) the role of H2S was examined by inhibition of H2S synthesis in hypoxic conditions.
Methods
Hearts from Landrace-Yorkshire hogs were obtained at a local slaughterhouse. All experiments conformed to the European Convention for the Protection of Vertebrate Animals used for Experimental and Other Scientific Purposes (European Convention for the Protection of Vertebrate Animals used for Experimental and Other Scientific Purposes, 2010).
Chemicals and materials
SNP was purchased from VWR-Bie & Berntsen (Herlev, Denmark). 4-Aminopyridine (4-AP), glibenclamide, PGF2α, TEA, flupirtine, linopirdine, chromanol 293B, XE991 (10,10-bis(4-pyridinylmethyl)-9(10H)-anthracenone dihydrochloride), sodium hydrosulfide (NaHS), propargyl glycine (PPG) and amino-oxyacetate (AOA) were obtained from Sigma-Aldrich, St Louis, MO, USA. Iberiotoxin (IbTX) was purchased from Latoxan, Valence, France. The composition of PSS was (mM): NaCl 119, KCl 4.7, MgSO4 •7H2O 1.2, NaHCO3 25, KH2PO4 1.2, EDTA 0.027, glucose 10, CaCl2 1.6, adjusted to pH 7.4. In PSS containing 125 mM or 30 mM K (K125PSS and K30PSS, respectively), NaCl was replaced by KCl on an equimolar basis to achieve the desired concentration.
Isometric tension recordings
Immediately after the animals had been killed, the coronary circulation was perfused with physiological salt solution similar to PSS except that it contained glucose 5.5 mM, bubbled with 5% CO2 in O2 and was buffered with HEPES [for composition see (Pasgaard et al., 2007)]. The hearts were bathed in this solution at 5°C until the start of the experiment. The left anterior descending coronary artery was carefully dissected and cut into 2 mm segments. In all preparations, the endothelium was removed by means of either a cotton bud or rubbing with a hair. For isometric tension recordings, artery segments were mounted in a four-chamber wire myograph (Tissue Bath System 700MO, Danish Myo Technology, Aarhus, Denmark) containing PSS bubbled with 95% O2, 5% CO2 at 37°C and the artery tension was normalized according to a standard procedure (Mulvany and Warshaw, 1979). Preparation viability was examined by exposing arteries to K125PSS. Removal of the endothelial layer was evaluated by lack of response (<10% vasodilatation) to bradykinin (30 nM) in PGF2α (10 μM)-contracted arteries.
Appropriate gas composition was obtained by mixing 95% O2, 5% CO2 with 95% N2, 5% CO2 (Digamix, H. Wösthoff GmbH, Bochum, Germany or Environics 4040, Tolland, CT, USA). The organ bath was bubbled through pimpstones allowing rapid equilibration of the oxygen tension. Oxygen concentration was measured with an oxygen electrode (Unisense, Aarhus, Denmark). An oxygen concentration-response curve was made by maintaining the oxygen level until the arterial tone reached a steady state before changing to the next level.
Arteries were contracted with PGF2α (10 μM) or K30PSS. The experiments were run in parallel, blockers were administered as a pretreatment before the addition of PGF2α and PGF2α contracted artery segments served as controls. The following incubation periods and concentrations were used: 4-AP (0.5 mM), IbTX (100 nM), glibenclamide (3 μM), PPG (10 mM) and AOA (5 mM), TEA (1 and 10 mM), chromanol 293B (10 μM), XE991 (10 μM) and linopirdine (10 μM) all 30 min. Arteries were allowed to attain a stable tone before dilatation was induced by gradually reducing oxygen concentration.
Simultaneous measurements of [Ca2+]i and tension
Arterial segments were mounted in a single chamber myograph and bubbled with 20% O2, 5% CO2 in N2. A load mix was made from 1 mL DMSO, 200 μL cremophore and 4 mg pluronic acid. A total of 50 μg fura-2AM was dissolved in 20 μL of load mix and added to the chamber resulting in a final fura-2AM concentration of 6.5 μg·mL−1 as previously described (Rodriguez-Rodriguez et al., 2008). Arteries were loaded for 3 h and subsequently washed thoroughly. The myograph was placed on an inverted microscope and force and fluorescence intensities were measured simultaneously at a sampling rate of 5 Hz using a computer equipped with dedicated software (Felix, Photon Technology International, South Brunswick, NJ, USA). Excitation wavelengths of 340 and 380 nm were used and emission signals measured through a 530 nm filter. At the end of the experiment, 10 μM ionomycin was added to PSS to achieve maximal saturation of Fura-2 with calcium. Then the fluorescence was quenched using nominally calcium-free PSS and 15 mM MnCl2 to measure the background signal, which was subtracted from the recordings.
PCR
Porcine arterial samples were kept in RNA later buffer RLT (Qiagen, Copenhagen, Denmark) until homogenization in 350 μL RLT with β-mercaptoethanol and 5 μL carrier RNA and homogenized for 3 min in Tissuelyser from Qiagen. The samples were centrifuged for 2 min at 13 000 x g. The isolation of total RNA from the arteries was done with the RNeasy plus mini kit by using Qiacube (Qiagen) according to the manufacturer's instructions, after which cDNA was synthesized from total RNA using SuperScriptIII reverse transcriptase, SuperAse In and random decamer primers according to the manufacturer's instructions. As KV7 channels had not previously been examined in the pig, Stratagene reference total mRNA from human ventricle (Stratagene, La Jolla, CA, USA) was included as a positive control for the expression of KV7 channels. Two microlitres of cDNA was amplified in a thermal cycler (Peqlab, Eurofins MWG Operon, Ebersberg, Germany) in a reaction (25 μL) containing 0.2 mM deoxynucleoside 5′-triphosphate mix, 0.4 μM of each primer, and 0.03 U of TaKaRa Ex Taq Hot start version DNA polymerase. A ‘hot-start’ procedure was employed (1 min at 95°C) and thermal cycling conditions were 95°C, 12 s, 56–58°C, 1 min and 70°C, 45 s for 40 cycles with a final extension of 4 min at 72°C. PCR reaction products were resolved by agarose gel electrophoresis (2.5% w v−1) and stained with ethidium bromide (0.5 μg·mL−1). Two or more sets of primers were used for each gene. The primers and expected product size are listed in Table 1. To determine the identity of the amplification products the agarose gel bands were excised and the DNA purified using a Qiaquick gel extraction kit (Quiagen). The purified PCR product was sequenced by Eurofins MWG Operon (Hedegaard and Mogensen, 2013a,b,c,d,e).
Table 1.
Primers used for PCR
| Gene | Sense | Antisense | Bp |
|---|---|---|---|
| KCNQ1 | (+) 5′-ttcctccagatcctgcggatgc-3′ | (−)5′-gttgggctcttccttacagaact-3′ | 716 |
| (+) 5′-tcatcttctcctcctacttcgtg-3′ | (−) 5′-tgttggttctcatgaaatggg-3′ | 633 | |
| KCNQ2 | (+) 5′-tctacatcctggaaatcgtgact-3′ | (−) 5′-tcagcagctccagctggttcag-3′ | 777 |
| (+) 5′-aacgtctttgccacatctgcgct-3′ | (−) 5′-cacgatctttcaaactgaccttc-3′ | 676 | |
| (+) 5′-tcggtgtctccttcttcgcgct-3′ | (−) 5′-tgtcatccacaatgtcctctcc-3′ | 579 | |
| (+) 5′-ttctacgccaccaacctctcg-3′ | (−) 5′-acagcatgtccaggtggccg-3′ | 621 | |
| KCNQ3 | (+) 5′-ccttccacttagacatgacgttc-3′ | (−) 5′-acagccataagcttcaaactcag-3′ | 865 |
| (+) 5′-ctgagtttgaagcttatggctgt-3′ | (−) 5′-acgactgattcatagaacctcca-3′ | 932 | |
| (+) 5′-gtattatgctaccaaccccaaca-3′ | (−) 5′-atatggttcattcctgggagatt-3′ | 661 | |
| (+) 5′-ggaaagctatttacccctctgaa-3′ | (−) 5′-ggaatacctgttgtcctccttct-3′ | 711 | |
| KCNQ4 | (+) 5′-ggccacctggtactactatgaca-3′ | (−) 5′-tcttcacagcaggcataacatcg-3′ | 506 |
| (+) 5′-agaagagctaccagagcgaact-3′ | (−) 5′-cgtcccatcatactgatttcatc-3′ | 314 | |
| KNCQ5 | (+) 5′-tctgcaaaaactcagggtaacat-3′ | (−) 5′-tcctccactgatacatcacactg-3′ | 944 |
| (+) 5′gccagaaacactttgagaaaaga-3′ | (−) 5′-acatcatatgggcgtaatgtttc-3′ | 619 | |
| Kcnma1 | (+) 5′-ggatgcgctcatcatcccggt-3′ | (−) 5′-ggcgcccgagtgtggtttttg-3′ | 902 |
| (+) 5′-tcctgtcagccaatcagaataat-3′ | (−) 5′-ctcttcctgcacgtacttctgtt-3′ | 909 |
The primers in bold are the ones depicted in Figure 4.
Immunoblotting
For immunoblotting of KV7.4 and KV7.5, arteries were frozen in cold acetone/dry ice (20 g/20 mL) at −78°C. For immunoblotting of KCa1.1α and KCa1.1β, the arteries were frozen at −80°C at the end of the experiment. In both cases, protein was extracted in lysis buffer using a Precellys 24 homogenizer (Bertin Technologies, Montigny-le-Bretonneux, France). The samples were exposed to three cycles at 5000 rpm for 30 s each. After homogenization, they were left on ice for 20 min before being centrifuged for 15 min at 13,000 x g at 4°C. The supernatant was transferred to a new tube and frozen at −80°C. Total protein was quantified using the Bio-Rad Protein Assay (Bio-Rad, Hercules, CA, USA). Protein lysate was mixed with sample buffer and loaded with a pre-stain marker (Bio-Rad) onto the gel. The proteins were separated by SDS-PAGE through a 4–12% Criterion XT Bis-Tris gel (Bio-Rad) at 200 V in a criterion cell (Bio-Rad). Transfer to a membrane was achieved for 1 h at 100 V in a criterion blotter (Bio-Rad). For detection of KCa1.1α and KCa1.1β, the membrane was washed for 10 min in Tris buffered saline with Tween 20 (TBS-T) and blocked in 5% skimmed milk in TBS-T for 2 h before being incubated overnight at 4°C with primary antibody against KCa1.1α (1:1000) (Alomone, Jerusalem, Israel) and KCa1.1β (1:200) (Abcam, Cambridge, UK). The membrane was washed in TBS-T before incubation with secondary antibody goat anti-rabbit IgG conjugated to HRP (Santa-Cruz Biotechnology, Santa Cruz, CA, USA) 1:4000 in TBS-T with 5% skimmed milk followed by washing. For detection of KV7.4 and KV7.5 channels, the membrane was washed for 10 min in TBS-T before it was blocked for 2 h in 0.3% I-block (Applied Biosystems, Foster City, CA, USA) and incubated overnight at 4°C with primary antibody (KV7.4 1:50 SC50417 and KV7.5 1:100 SC50416, Santa-Cruz Biotechnology), where specificity was previously tested in human embryonic kidney cells overexpressing the channels (Jepps et al., 2011; Zhang et al., 2011). The membrane was washed in PBS with Tween 20 before incubation with secondary antibody goat anti-rabbit IgG conjugated to HRP (Santa-Cruz Biotechnology) followed by washing. The membranes were developed by using the ECL-Plus kit (GE Healthcare, Copenhagen, Denmark). The blot was placed in an X-ray film cassette and developed in photographic developer.
Voltage clamp
To assess membrane conductance, smooth muscle cells from the left anterior descending coronary artery were isolated by incubation with an enzyme solution containing collagenase (1.5 mg·mL−1 type I, Worthington Biochem Corp., Lakewood, NJ, USA) in nominally Ca2+-free solution (in mM): 120 NaCl, 6 KCl, 1.2 MgCl2, 10 glucose, 10 HEPES at pH 7.3 adjusted with NaOH. A few 2–3 mm segments of coronary arteries were placed in a microtube containing an enzyme solution and incubated at 37°C for 10–15 min. Single cells were released by trituration with a polyethylene pipette into the nominally Ca2+-free solution and, then, added into Petri dish containing the bath solution. Cells attached to the bottom of Petri dish were used in the whole-cell voltage clamp experiments 20 min after isolation during next 2.5 h.
The bath solution contained (in mM): 120 NaCl, 4.2 KCl, 1.2 KH2PO4, 0.5 MgSO4, 3 NaHCO3, 1.8 CaCl2, 10 HEPES and 10 glucose at pH 7.4 adjusted with NaOH. During experiments, the oxygen tension in the bath solution was modified by gassing the solution with either 20% O2 and 5% CO2 in N2 or 5% CO2 in N2 (this gives a concentration of 1% O2) for a minimum of 20 min and continued during the whole experiment. The patch pipette solution contained (in mM) 110 potassium gluconate, 30 KCl, 5 HEPES, 0.5 MgCl2, 5 Na2ATP, 1 GTP and 0.1 EGTA at pH 7.2 adjusted with KOH (Zhong et al., 2010).
Patch pipettes were prepared from borosilicate glass (PG15OT-7.5; Harvard Apparatus, Kent, UK) and fire polished to achieve tip resistances in the range of 4–8 MΩ. Membrane current recordings were made with an Axopatch 200B amplifier (Axon Instruments, Foster City, CA, USA) in whole-cell configuration at room temperature (22–24°C). Data acquisition and analysis were done with software packages Clampex 9.0 for Windows (Axon Instruments) and Microcal Origin v.8.1 for Windows (Microcal Software, Northampton, MA, USA). Only cells with essentially no leak current (seal resistance ≥1.5 GΩ) and a low access resistance (5–10 MΩ) were used. Series resistance and capacitive current were routinely compensated for electronically. The stability of these parameters was tested regularly during the course of the experiment, that is before and after each intervention.
Current-voltage (I-V) relations were determined using 500 ms step pulses to between −95 and +55 mV in increments of 10 mV from a holding potential of −75 mV. Averaged current amplitude at the last 50 ms of voltage-step pulses was used to construct I-V relations. I-V relations were determined for control conditions and after treatment with 10 μM XE991. The XE991-sensitive current was calculated by subtraction of the I-V relation after XE991 treatment from the I-V relation under control conditions.
Data analysis and statistics
Data are presented as means ± SEM. Paired t-tests were used to analyse whether incubation with different blockers altered the baseline tone, contraction and coronary artery tone remaining at 1% O2. Dilatation responses were calculated as percentage reduction of the contraction induced by either K30PSS or PGF2α (10 μM). The response curves to decreasing oxygen concentrations were compared by means of two-way ANOVA followed by a Bonferroni post hoc test or by a t-test. When the effect of a combination treatment with two different K channel blockers was compared to the effect of single channel-block treatment, the two control groups were pooled and concentration-response curves were compared by means of two-way ANOVA for concentration-dependence and effect of treatment.
In experiments where tension recordings and [Ca2+]i measurements were performed simultaneously, responses were compared by means of an unpaired t-test. The background signal at each of the excitation wavelengths was subtracted from the fluorescence intensities and the ratio F340/F380 was calculated. The change in ratio that occurred with hypoxia was expressed as a percentage of the ratio increase induced by the contracting agent (K30PSS or PGF2α). P < 0.05 was considered significant.
Results
Role of [Ca2+]i in hypoxia-induced dilatation in porcine coronary arteries
Figure 1A is a typical trace illustrating hypoxia-induced dilatation in a PGF2α-contracted coronary artery. In Figure 1B, a typical trace for hypoxia-induced dilatation in a 30 mM K-depolarized artery is depicted. In PGF2α-contracted arteries and in K30PSS-contracted arteries, a gradual reduction in oxygen concentration caused oxygen-dependent dilatation (P < 0.0001, n = 6, Figure 1C) and the coronary artery tone remaining at 1% O2 was reduced from 50 ± 8% in K30PSS to 11 ± 4% in PGF2α-contracted arteries. The contractile tension evoked by K30PSS did not differ from that induced by PGF2α (23 ± 4 Nm−1 versus 23 ± 5 Nm−1 respectively, P = 0.9, n = 6).
Figure 1.
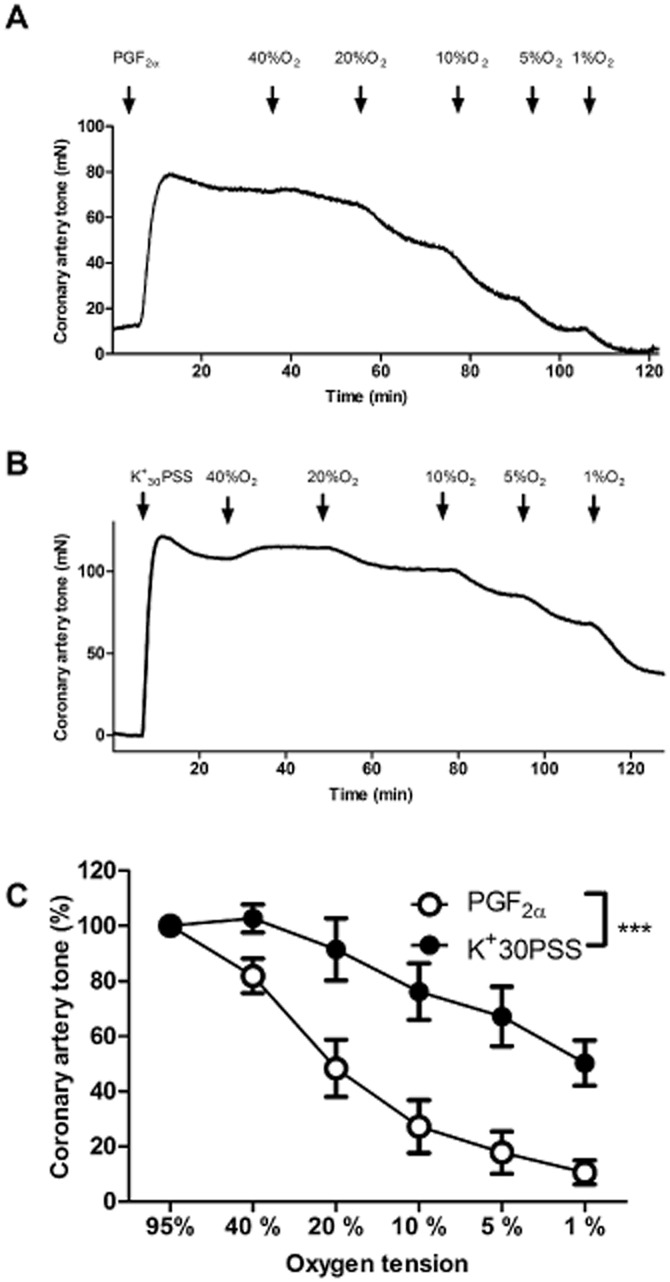
Hypoxia-evoked vasodilatation in porcine coronary artery. Original tracing illustrating the dilatation induced by decreasing O2 concentration in the organ bath (A) PGF2α (10 μM)- and (B) K30PSS-contracted porcine coronary artery without endothelium. (C) Concentration-response curves for O2 lowering in coronary arterial segments without endothelium contracted with K30PSS or PGF2α (10 μM) (n = 6, *P < 0.05 by two-way ANOVA).
Simultaneous tension recordings and [Ca2+]i measurements were performed to investigate to what extent hypoxia-induced dilatation was dependent on lowering of [Ca2+]i or rather caused by Ca2+-desensitization and/or force suppression. PGF2α (10 μM) and K30PSS caused similar increases in [Ca2+]i (Figure 2A and 2B). In PGF2α-contracted arteries, an abrupt reduction from 20 to 1% O2 lowered [Ca2+]i and induced vasodilatation (Figure 2A and 2C). However, in K30PSS-contracted arteries, hypoxia induced a smaller degree of vasodilatation than in PGF2α-contracted arteries, but without decreasing the level of [Ca2+]i (Figure 2B and 2C) suggesting that depolarization with K prevents the reduction in [Ca2+]i caused by hypoxia.
Figure 2.
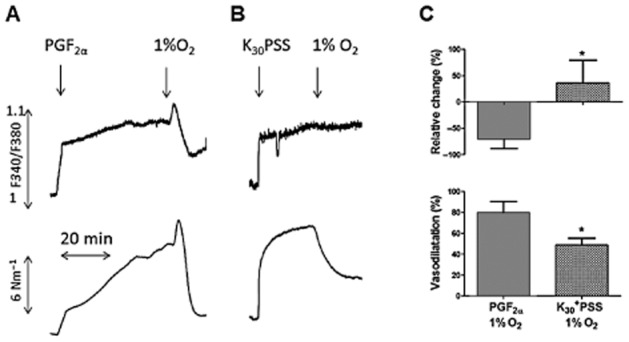
Effects of hypoxia on [Ca2+]i and tension in pig coronary artery. Effect of hypoxia on (A) PGF2α (10 μM)- and (B) 30 mM K (K30PSS)-contracted arteries without endothelium. (C): Average data showing the change in [Ca2+]i and tension in PGF2α- and K30PSS-contracted preparations in response to reducing organ bath O2 concentration from 20 to 1% (n = 6, *P < 0.05 by Student's t-test).
Effect of K channel blockers on hypoxia-induced dilatation
As the hypoxia-induced dilatation was smaller in K-contracted arteries than in those contracted with PGF2α, we investigated the effect of blocking various K channels. Incubation with the voltage-dependent K channel blocker 4-AP (0.5 mM) and the K channel blocker, TEA (10−2 M) increased baseline artery tone, but not the response to PGF2α as compared to vehicle-treated preparations. Incubation with blockers of KCa1.1 channels, TEA (10−3 M) and IbTX or a blocker of ATP-sensitive K channels, glibenclamide did not alter baseline tone or response to PGF2α as compared to parallel control preparations (Table 2).
Table 2.
Effect of K channel blockers on hypoxia-induced dilatation
| n | Baseline tone (Nm−1) | PGF2α contraction (Nm−1) | Coronary artery tone (%) remaining at 1% O2 | |
|---|---|---|---|---|
| Control | 7 | 1.2 ± 2.2 | 34.6 ± 10.5 | 22.6 ± 9.8 |
| TEA 10−3 M | 7 | 2.6 ± 2.2 | 35.1 ± 7.5 | 30.2 ± 17.4 |
| TEA 10−2 M | 7 | 18.3 ± 12.3* | 37.0 ± 10.1 | 37.0 ± 22.8 |
| Control | 12 | 0.1 ± 0.1 | 18.2 ± 1.0 | 14.1 ± 2.7 |
| 4-AP | 12 | 2.4 ± 0.8* | 19.8 ± 1.5 | 30.4 ± 2.7* |
| Control | 6 | −0.1 ± 0.1 | 24.3 ± 3.4 | 14.5 ± 1.9 |
| IbTx | 6 | 1.9 ± 1.1 | 22.0 ± 2.5 | 27.6 ± 1.9* |
| Control | 7 | 0.1 ± 0.1 | 18.6 ± 1.8 | 16.5 ± 4.4 |
| Glib | 7 | −0.1 ± 0.3 | 19.8 ± 2.8 | 20.2 ± 3.4* |
| Control | 6 | 0.9 ± 0.6 | 21.2 ± 1.8 | 18.7 ± 3.0 |
| 4-AP+IbTx | 6 | 8.6 ± 3.2* | 29.3 ± 3.5 | 34.5 ± 5.7* |
| Control | 6 | 1.0 ± 0.7 | 16.6 ± 1.1 | 14.0 ± 2.4 |
| 4-AP+Glib | 6 | 3.8 ± 1.3* | 18.3 ± 1.6 | 33.6 ± 8.7* |
| Control | 12 | −0.4 ± 0.7 | 25.5 ± 1.8 | 17.5 ± 5.2 |
| XE991 | 11 | 4.8 ± 2.7* | 18.7 ± 2.6 | 49.2 ± 2.9* |
| Linopirdine | 7 | 3.0 ± 3.1* | 19.84 ± 2.0 | 42.7 ± 2.3* |
| Control | 7 | 2.0 ± 1.4 | 26.1 ± 2.5 | 11.9 ± 4.3 |
| XE991+4-AP+IbTx | 7 | 22.3 ± 3.0* | 17.5 ± 4.0 | 62.1 ± 2.0* |
| Control | 5 | −0.5 ± 0.3 | 23.7 ± 1.7 | 26.9 ± 1.6 |
| Chromanol 293B | 5 | −0.8 ± 1.1 | 20.2 ± 4.6 | 30.0 ± 4.3 |
Effect on baseline tone, PGF2α contraction and coronary artery tone (%) remaining at 1 % O2 in the absence and presence of K channels blocker. Results are means +/− SEM. n = number of animals examined.
P<0.05 by Student's t-test.
The K channel blocker, TEA significantly inhibited dilatation induced by lowering O2 (10−3 M P < 0.05, n = 7 and 10−2 M P < 0.001, n = 7, Table 2). Furthermore, we observed an effect of TEA on adenosine-induced dilatation (10−3 M P < 0.001, n = 7 and 10−2 M P < 0.001, n = 7, data not shown). Blocking of KV channels with 4-AP, large-conductance KCa1.1 channels with IbTX or KATP channels with glibenclamide reduced hypoxia-induced dilatation (Table 2). Blocking KATP channels with glibenclamide caused a pronounced inhibition of dilatation induced by cromakalim (n = 7), an opener of KATP channels, while these dilatations were not affected by incubation with 4-AP (n = 6).
To investigate whether the effects of K channel blockers on hypoxia-induced dilatation were additive, we used a combination of different K channel blockers. The combination of 4-AP and IbTX (blockers of KV and KCa1.1 channels, respectively) significantly diminished hypoxia-induced dilatation (Table 2) as did a combination of 4-AP and glibenclamide (blockers of KV and KATP channels respectively) (Table 2). However, combinations of either 4-AP and IbTX or 4-AP and glibenclamide did not add to the blocking effect of either blocking agent alone (Table 2).
Role of KV7 channels
Blocking the KCNQ encoded KV7 channels by either XE991 (10 μM) or linopirdine (10 μM) reduced hypoxia-induced dilatation (P < 0.0001, n = 11 and P < 0.0001, n = 7 Figure 3A). Blocking KV7.1 with chromanol 293B (10 μM) failed to inhibit hypoxia-induced dilatation (Supporting Information Figure S1 and Table 2). Incubation with either XE991 (10 μM) or linopirdine (10 μM) increased baseline artery tone as compared to controls, whereas incubation with chromanol 293B did not alter baseline tone (Table 2), neither of them changed the response to PGF2α. A combination of XE991, 4-AP and IbTx showed an additional effect as compared to XE991 alone (see Table 2 and Supporting Information Figure S4).
Figure 3.
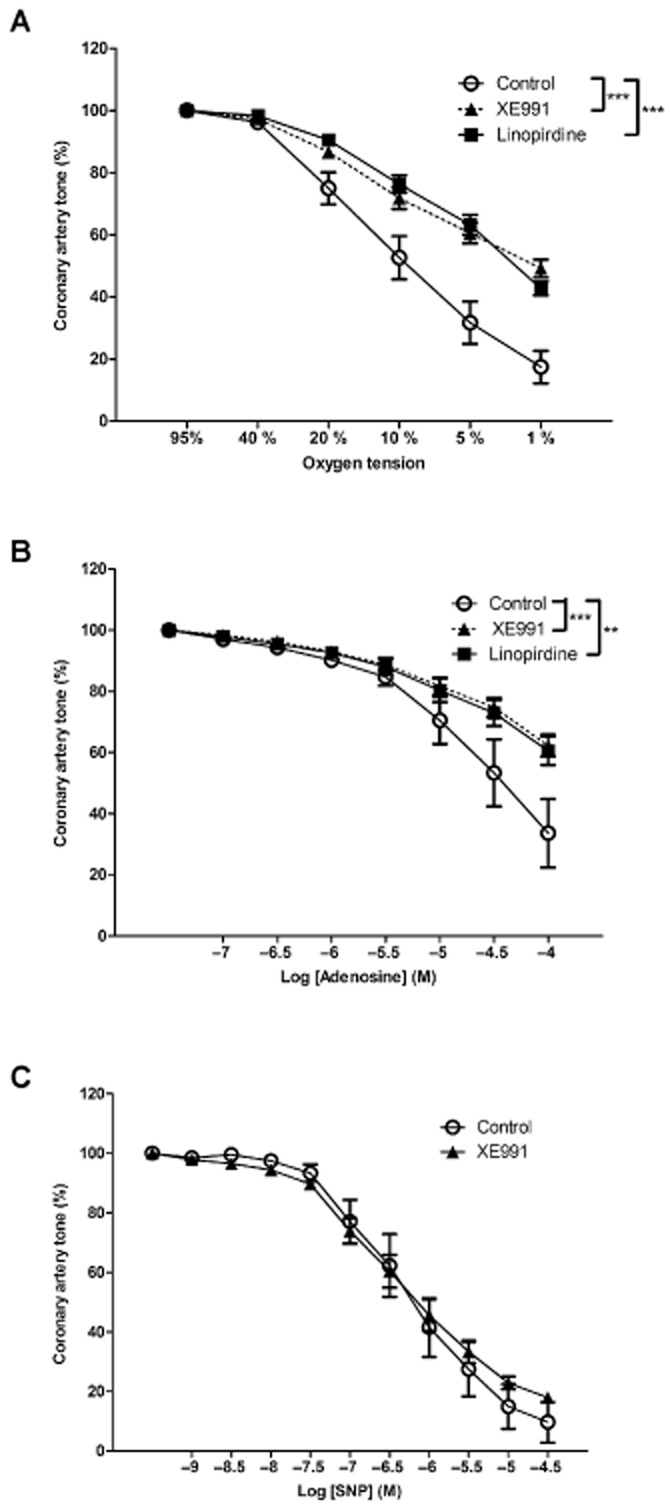
Effect of KV7 channel inhibition on hypoxia-induced dilatation in pig coronary artery. (A) Concentration-response curves for O2 lowering in coronary arterial segments without endothelium contracted with PGF2α (10 μM) in the absence and presence of the KV7.1–7.5 inhibitors XE991 (10 μM) and linopirdine (10 μM) (n = 7–12, ***P < 0.001 by two-way ANOVA). (B) Concentration-response curves for adenosine in coronary arterial segments without endothelium contracted with PGF2α (10 μM) in the absence and presence of the KV7.1–7.5 inhibitor XE991 (10 μM) and linopirdine (10 μM) (n = 5, **P < 0.01, ***P < 0.001 by two-way ANOVA). (C) Concentration-response curves for SNP in coronary arterial segments without endothelium contracted with PGF2α (10 μM) in the absence and presence of the KV7.1–7.5 inhibitors XE991 (10 μM) (n = 4, P = 0.65 by two-way ANOVA).
Adenosine-induced dilatations were also inhibited by XE991 (10 μM) and linopirdine (10 μM) (P < 0.001 n = 5 and P < 0.01 n = 5 Figure 3B) whereas chromanol 293B (10 μM) did not affect adenosine-induced dilatations (P = 0.12, n = 5 results not shown). Blockade of KV7 channels with XE991 caused a pronounced inhibition of dilatations induced by flupirtine (P < 0.001, n = 7, results not shown), an opener of KV7.2–7.5 channels, while these dilatations were not affected by incubation with chromanol 293B (P = 0.85, n = 7, results not shown). To test the selectivity of XE991, we used SNP to induce vasodilatation and found this response was not affected by the presence of XE991 (P = 0.65, n = 4, Figure 3C).
PCR and immunoblotting
We examined the presence of KV7.1–KV7.5 and KCa1.1 (BKCa) mRNA by reverse transcriptase (RT)-PCR and sequence analysis. We found expression of KV7.1, KV7.4, KV7.5 and KCa1.1 (BKCa) in porcine coronary arteries (Figure 4A, D, E and F) but we could not observe the presence of KV7.2 and KV7.3 (Figure 4B and 4C) despite using four different sets of primers directed against each of these channels. At least one of the primer sets for KV7.2 and KV7.3 resulted in a positive result in tissue from the human heart, which was used as a positive control.
Figure 4.
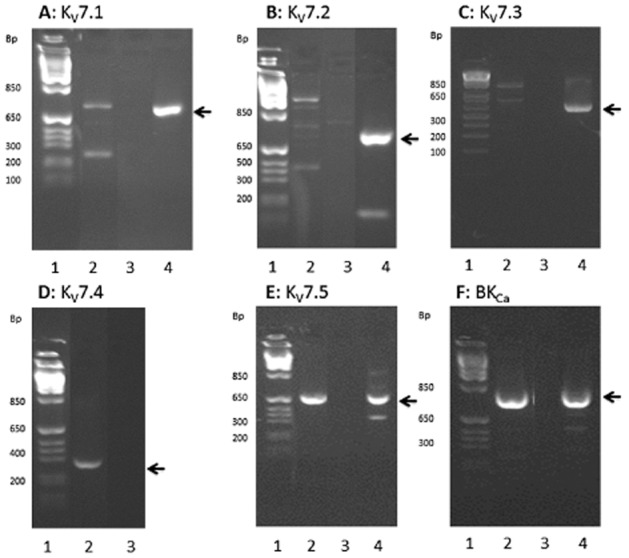
Detection of KV7 and KCa1.1 (BKCa) channels by RT-PCR. In all panels, lane 1 is a marker, lane 2 is RT-positive samples, lane 3 is RT-negative samples and lane 4 is a control with human heart. The bands analysed and confirmed as the right product are marked with an arrow. Primers can be seen in Table 1, (n = 6). (A) KV7.1 (B) KV7.2 (C) KV7.3 (D) KV7.4 (E) KV7.5 and (F) KCa1.1 (BKCa).
The presence of KCa1.1 (BKCa) channels was demonstrated by immunoblotting, this revealed the expression of both KCa1.1 (BKCa) channel subunits in the porcine coronary arteries (Figure 5A and B). We also detected the expression of KV7.4 and KV7.5 channel proteins in tissue from porcine coronary arteries (Figure 5C and D).
Figure 5.

Detection of KCa1.1 (BKCa) and KV7 channels by immunoblotting. Immunoblot with samples from a normoxic artery (n = 3) showing the presence of (A) KCa1.1α (BKα) located at 100 kDa (B) KCa1.1β (BKβ) located at 30 kDa (C) KV7.4 located at 77 kDa and (D) KV7.5 located at 100 kDa.
Whole-cell voltage clamp studies
Further evidence for the importance of KV7 channels in hypoxia-induced vasodilatation was obtained from voltage-clamp experiments which showed that the XE991-sensitive current was more marked when bubbling with 5% CO2 in N2 (which gives approximately 1% O2 in the bath) than when bubbling with 21% O2 and 5% CO2 in N2 (Figure 6C). In Figure 6D, we show the current remaining after subtracting currents obtained after XE991-treatment from basal currents. The significantly higher XE991-sensitive current under hypoxic conditions suggests that KV7 channels are activated by hypoxia.
Figure 6.
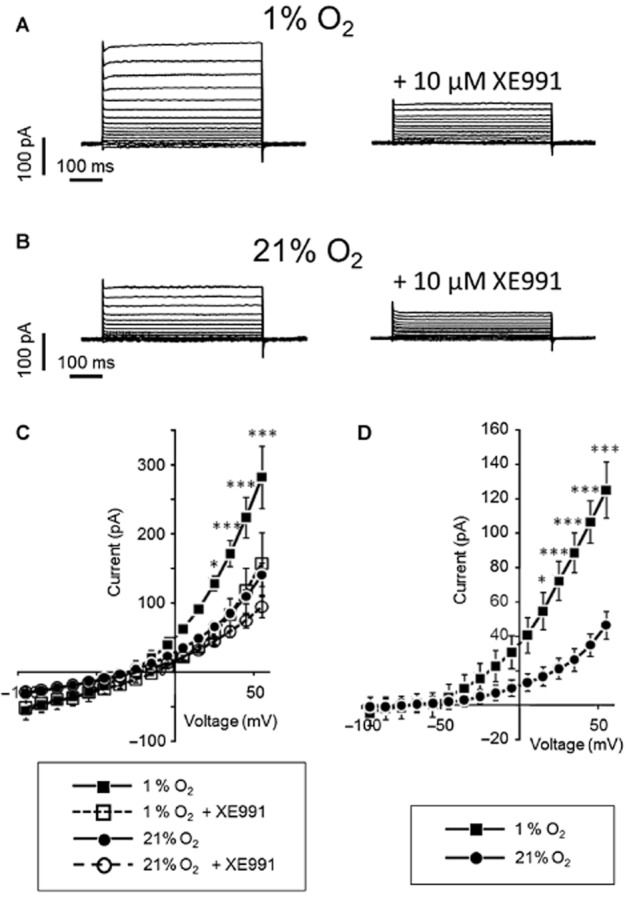
The current activated by hypoxia in the membrane of freshly isolated smooth muscle cells from pig coronary arteries is sensitive to XE991. (A): Representative traces from the whole-cell voltage-step experiments where the current was recorded by stepping membrane voltage between −95 and +55 mV. The traces were obtained in the bath solution that was virtually free from O2 (∼1% O2) by bubbling with 5% CO2 in N2. Representative traces before and after treatment with XE991 are shown. (B) Representative traces from the whole-cell voltage-step experiments similar to those in (A) but obtained in the bath solution with 21% O2. (C) Averaged current-voltage traces for whole-cell current at ∼1% O2 and at 21% O2 under control conditions and after addition of XE991. Two-way ANOVA indicates significant effect of different oxygen tensions on membrane current (F(1,12) = 8.9, P > 0.011). Bonferroni post-test indicates * and *** P < 0.05 and <0.001, n = 7. (D): Averaged current-voltage traces for XE991 currents obtained by subtraction of the current after XE991 treatment from the control current at ∼1% O2 and at 21% O2. Two-way ANOVA indicates different effect of XE991 at ∼1% O2 and at 21% O2 (F(1,12) = 8.3, P > 0.014). Bonferroni post-test indicates * and *** P < 0.05 and <0.001, n = 7.
Effect of H2S
Hydrogen sulphide (H2S) has been shown to be involved in vasodilatation responses mediated through the KV7 channels (Schleifenbaum et al., 2010). We, therefore, tested the effect of XE991 on NaHS-induced vasodilatation and found that XE991(10 μM) inhibited vasodilatation induced by NaHS (P < 0.0001, n = 6 Figure 7A). The overall relaxation curves induced by lowering oxygen were not significantly changed by incubation with a combination of PPG (10 mM) and AOA (5 mM) which respectively inhibits the H2S producing enzymes cystathionine γ-lyase and cystathionine β-synthase, when analysed with a two-way ANOVA. However, a t-test analysis of the vasodilatation induced by 1% O2 revealed that hypoxia-induced vasodilatation at 1% O2 was significantly inhibited (Figure 7B). Combining PPG, AOA and XE991 gave an additional effect compared to XE991 alone indicating that even though the effect of NaHS can be suppressed by inhibiting KV7 channels, part of the NaHS relaxation comes from pathways not affected by KV7 channel blockers.
Figure 7.
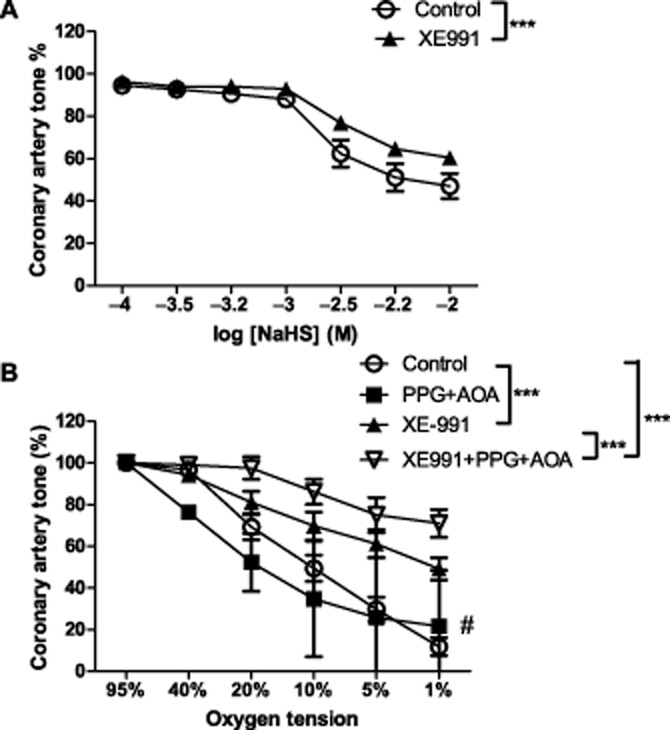
Effect of KV7 channel inhibition on NaHS-induced dilatation in pig coronary artery. (A): Concentration-response curves for NaHS in coronary arterial segments without endothelium contracted with PGF2α (10 μM) in the absence and presence of the KV7.1–7.5 inhibitor XE991 (10 μM) (n = 6, ***P < 0.0001 by two-way ANOVA) (B): Concentration-response curves for O2 lowering in coronary arterial segments without endothelium contracted with PGF2α (10 μM) in the absence and presence of the KV7.1–7.5 inhibitor XE991 (10 μM) and the inhibitors of the H2S producing enzymes PPG (10 mM) and AOA (5 mM) (n = 6–16, ***P < 0.001 by two-way ANOVA, #P < 0.05 by Student t-test).
Discussion
The main findings of the present study were that the hypoxia-induced reduction in [Ca2+]i in PGF2α-contracted arteries can be attributed to opening of one or more K channel types. The KV7.4 and KV7.5 channels, which we identified in the coronary arteries, appear to be particularly important in the hypoxia-induced vasodilatation. The hypoxic vasodilatation was inhibited by two different blockers of KV7 channels and the voltage clamp results further indicate that KV7 channels are involved in this response to hypoxia. Our findings suggest that hypoxia may act via K channel opening, which is, in part mediated by H2S.
Role of intracellular calcium and K channels in hypoxia-induced dilatation
Previous studies have shown that [Ca2+]i is reduced during hypoxia (Shimizu et al., 2000), but it has also been suggested that reduced [Ca2+]i cannot fully account for hypoxia-induced dilatation (Aalkjaer and Lombard, 1995; Shimizu et al., 2000; Thorne et al., 2001; Gu et al., 2005). We found that in PGF2α-contracted arteries, hypoxia lowered [Ca2+]i and induced dilatation. In K30PSS-contracted arteries, hypoxia did not alter [Ca2+]i but caused dilatation. In our experiments, the level of contraction induced by K30PSS or PGF2α did not differ. Since hypoxia-induced dilatation was only partially inhibited by K30PSS contraction, part of this dilatation must be caused by mechanisms independent of changes in [Ca2+]i, such as force suppression or Ca2+ desensitization. The decrease in [Ca2+]i observed in PGF2α-contracted arteries suggested the involvement of K channels, since opening of these will lead to hyperpolarization, which decreases [Ca2+]i. Blocking calcium influx may also contribute to the hypoxia-induced relaxation (Franco-Obregon and Lopez-Barneo, 1996; Smani et al., 2002), but the effect of the K channel blockers indicate that K channels are involved in the hypoxic relaxation.
Several K channels including KATP (Daut et al., 1990; Dart and Standen, 1995; Liu and Flavahan, 1997; Lee et al., 1998; Kamekura et al., 1999), KCa1.1 (BKCa;Gebremedhin et al., 1994; Nelson and Quayle, 1995; Lopez-Barneo et al., 2004) and certain KV channel subtypes KV1.5 and KV2.1 (Shimizu et al., 2000; Thorne et al., 2002) have been suggested to be involved in hypoxia-induced vasodilatation. In the present study, glibenclamide significantly inhibited the relaxations induced by the KATP opener, cromakalim, but in the same preparations only slightly inhibited the relaxations evoked by lowering oxygen. These findings agree with the observations in rat and human coronary small arteries, which also suggested that the contribution of KATP channels to hypoxic vasodilatation is negligible (Lynch et al., 2006). Although 4-AP can also inhibit KATP currents at concentrations above 0.2 mM (Beech and Bolton, 1989; Nelson and Quayle, 1995), in our experiments the concentration of 4-AP used (0.5 mM) has been shown to be relatively selective for KV channels (Nelson and Quayle, 1995). In the present study, 4-AP did not affect the cromakalim-induced relaxation, and, therefore, we believe that the reduction in hypoxic vasodilatation observed in the present study can be ascribed to inhibition of 4-AP-sensitive KV channels.
In coronary arteries with endothelium, NO release is increased and contributes to hypoxic vasodilatation (Lynch et al., 2006; Hedegaard et al., 2011) and, recently, we found that NO induces vasodilatation by activation of TEA-sensitive channels (Hedegaard et al., 2011). In the present study of porcine coronary arteries without endothelium, TEA also caused a small reduction in the hypoxia-induced vasodilatation at concentrations (10−3 M) selective for KCa1.1 (BKCa) channels (Lang and Ritchie, 1990). The observed expression of the α and β subunits of KCa1.1 (BKCa) in these arteries, and the finding that IbTX, a selective blocker of KCa1.1 channels, reduced the hypoxic vasodilatation, further indicate that smooth muscle KCa1.1 channels contribute to the hypoxic vasodilatation. The combined blockade of 4-AP-sensitive KV channels and KCa1.1 channels did not cause further inhibition of hypoxic vasodilatation. Thus, KCa1.1 channels may mediate the endothelium-dependent component, involving NO, of the hypoxic vasodilatation (Hedegaard et al., 2011). However, in porcine coronary arteries, in the absence of endothelium, our results show that KATP, 4-AP-sensitive KV and KCa1.1 channels contribute little to hypoxic vasodilatation, not enough to explain the pronounced inhibition of hypoxic vasodilatation observed in the preparations contracted by increasing the extracellular K concentration.
Role of KV7 channels in hypoxic vasodilatation
KV7 channels are involved in the regulation of vascular tone in a number of rodent blood vessels (Yeung and Greenwood, 2005; Joshi et al., 2006; Yeung et al., 2007; Mackie et al., 2008; Zhong et al., 2010) and it has also been suggested that they counteract changes induced by chronic hypoxia in the pulmonary circulation (Morecroft et al., 2009). Therefore, these channels may also be involved in hypoxic vasodilatation. Indeed, in the present study we found that two different blockers of KV7.1–7.5 channels XE991 and linopirdine inhibited the dilatation induced by hypoxia, adenosine and flupirtine, an opener of KV7.2–7.5 channels, whereas the KV7.1 inhibitor, chromanol 293B did not affect the dilatation induced by these agents. The effect of XE991 appears to be specific, since the SNP-induced relaxation was not changed in the presence of XE991.
Previous studies have shown that mRNA for KV7.1, KV7.4 and KV7.5 is readily detectable in mice portal vein, thoracic aorta, carotid and femoral artery smooth muscle (Yeung et al., 2007; 2008b). KV7.1 and KV7.5 transcripts have also been detected in adult rat aorta (Brueggemann et al., 2007) and KV7.1, KV7.4 and KV7.5 in rat mesenteric artery smooth muscle cells (Mackie et al., 2008). In human arteries, KV7.4 was shown to be expressed in all the arteries examined with variable contributions from KV7.1, KV7.3 and KV7.5 (Ng et al., 2011). We were able to detect mRNA transcripts from KV7.1, KV7.4 and KV7.5, but not KV7.2 and KV7.3, in the coronary artery, even though we tested four different sets of primers and obtained a positive result in control tissue from human heart. Our results agree with previous results showing that KV7.1, KV7.4 and KV7.5 are the dominantly expressed genes in the vasculature, and there is accumulating evidence indicating that KV7.4 and KV7.5 are responsible for the activity in the vasculature (Jepps et al., 2011). Immunoblotting showed that KV7.4 and KV7.5 channel proteins are also expressed in porcine coronary arteries. Taken together with the observations that XE991 inhibits KV7.1–7.4 with IC50 values ∼1–5 μmol·L−1 (Yeung et al., 2007; 2008a) and KV7.5 with an IC50∼60 μmol·L−1 (Jensen et al., 2005; Yeung et al., 2008a), and since we used 10 μM XE991, our results suggest that the effect we observed is likely to be mediated by KV7.4 channels.
The combination of IbTX, 4-AP and XE991 had a greater inhibitory effect on the hypoxic vasodilatation compared to XE991 alone and the effect of this combination was similar to that observed in preparations contracted with 30 mM potassium (e.g. Figure 2 versus Supporting Information Figure S4). These results suggest that XE991 has an effect on the KV7 channels, which can be separated from that induced by blocking other potassium channels.
In previous studies, resting membrane potential in pinned coronary arteries from pigs has been measured to be approximately −50 mV (Edwards et al., 2000) and PGF2α (2–6 μM) was found to depolarize the membrane to −40 to −35 mV (Thollon et al., 2002). Resting membrane potential is more depolarized in pressurized preparations, while agonist-induced depolarization is less (Schubert et al., 1996). This confirms that the XE991-sensitive currents activated above −40 mV are within the range of physiologically relevant membrane potentials.
Interestingly, in the voltage-clamp study, the membrane conductance was higher under hypoxic conditions. This hypoxia-induced current was sensitive to XE991 at a concentration that has previously been shown to inhibit KV7 currents (Yeung and Greenwood, 2005). Although recent studies have suggested that XE991 can also attenuate KV1.2/KV1.5 and KV2.1/KV9.3 channels, an overexpression study confirmed that its major effect is on KV7 channels (Zhong et al., 2010). We thus propose that members of the KV7 channel family (i.e. KV7.2-KV7.5 channels) are activated by hypoxia in coronary artery smooth muscle cells and are involved in the vasodilator response.
Role of KV7 channels in H2S and adenosine induced vasodilatation
A fall in oxygen availability during hypoxia decreases mitochondrial H2S oxidation resulting in an increase in biologically active H2S, and therefore, H2S has been suggested to mediate hypoxic vasodilatation (Skovgaard et al., 2011). Moreover, H2S has been shown to cause relaxation by activation of KATP channels and KV7 channels (Zhang et al., 2007; Schleifenbaum et al., 2010). Incubation of the porcine coronary arteries with inhibitors of enzymes synthesizing H2S attenuated the vasodilatation induced by lowering oxygen tension to 1% and the combination of these enzyme inhibitors with XE991 inhibited the hypoxic vasodilatation. In addition, the inhibitory effect of XE991 on the H2S-induced relaxation suggests that part of the KV7 activation in hypoxia may be attributed to H2S.
Another metabolite released from the myocardium in ischaemic conditions is adenosine (Decking et al., 1997). Adenosine increases cAMP and a study in renal arteries demonstrated that KV7 channels are regulated by cAMP-dependent processes (Chadha et al., 2012). This finding is in line with our observation that adenosine-induced relaxation was inhibited in the presence of XE991. Although further studies are required to reveal the source of the mediators, these results suggest that H2S and adenosine may lead to the activation of KV7 channels during hypoxia in porcine coronary arteries.
Conclusion
Our findings suggest that smooth muscle KV7.4 and 7.5 channels are the main components involved in the hypoxic vasodilatation in porcine coronary arteries with other K channels contributing to a much lesser extent. The results from the voltage clamp experiments further supported a role for KV7 channels during hypoxia. We also showed that H2S and adenosine are likely to induce the activation of KV7 channels in hypoxia. Based on these preclinical findings, it would be interesting to investigate the therapeutic potential of modulators of KV7 channels already in use. Flupirtine is approved as a treatment for pain in Europe, and retigabine has shown promising results in phase III clinical trials as an antiepileptic agent (Mackie and Byron, 2008), these openers could potentially form the basis for testing in patients with cardiac ischaemia.
Acknowledgments
The technical assistance of Margit Nielsen is highly appreciated. The study was supported by the Danish Heart Foundation and the Danish Medical Research Council.
Abbreviations
- 4-AP
4-aminopyridine
- AOA
amino-oxyacetate
- KCa1.1
large-conductance calcium-activated K channels
- [Ca2+]i
intracellular calcium
- IbTX
iberiotoxin
- K125PSS
PSS containing 125 mM K
- K30PSS
PSS containing 30 mM K
- KATP
ATP-sensitive K channels
- KV
voltage-gated K channels
- PPG
propargyl glycine
- XE991
10,10-bis(4-pyridinylmethyl)-9(10H)-anthracenone dihydrochloride
Conflict of interest
The authors state no conflict of interest.
Supporting information
Additional Supporting Information may be found in the online version of this article at the publisher's web-site:
http://dx.doi.org/10.1111/bph.12424
Figure S1 Effect of K channel inhibition with TEA on hypoxia- and adenosine-induced dilatation in pig coronary artery. (A) Concentration-response curves for O2 lowering in coronary arterial segments without endothelium contracted with PGF2α (10 μM) in the absence and presence of the K channel inhibitor TEA (1 mM and 10 mM) (n = 7, *P = 0.046, ***P < 0.001 by two-way ANOVA). (B) Concentration-response curves for adenosine in coronary arterial segments without endothelium contracted with PGF2α (10 μM) in the absence and presence of the K channel inhibitor TEA (1 mM and 10 mM) (n = 7, ***P < 0.001 by two-way ANOVA).
Figure S2 Effect of K channel inhibition on hypoxia-induced dilatation in pig coronary artery. Concentration-response curves for O2 lowering in coronary arterial segments without endothelium contracted with PGF2α (10 μM) in the absence and presence of (A): 4-AP 0.5 mM (n = 12, *P < 0.001 by two-way ANOVA), (B): IbTX 100 nM (n = 6, *P < 0.001 by two-way ANOVA) or (C): glibenclamide 3 μM (n = 7, *P < 0.05 by two-way ANOVA).
Figure S3 Effect of K channel inhibition on hypoxia-induced dilatation in pig coronary artery. Concentration-response curves for O2 lowering in coronary arterial segments without endothelium contracted with PGF2α (10 μM) in the absence and presence of (A): 4-AP 0.5 mM and IbTX 100 nM (n = 6, *P < 0.01 by two-way ANOVA), (B): glibenclamide 3 μM and 4-AP 0.5 mM (n = 6, *P < 0.001 by two-way ANOVA)
Figure S4 Effect of K channel inhibition on hypoxia-induced dilatation in pig coronary artery. Concentration-response curves for O2 lowering in coronary arterial segments without endothelium contracted with PGF2α (10 μM) in the absence and presence of XE991 10 μM, 4-AP 0.5 mM and IbTX 100 nM (n = 7–8, ***P < 0.001 by two-way ANOVA).
Figure S5 Effect of KV7.1 channel inhibition on hypoxia- and adenosine-induced dilatation in pig coronary artery. (A) Concentration-response curves for O2 lowering in coronary arterial segments without endothelium contracted with PGF2α (10 μM) in the absence and presence of the KV7.1 channel inhibitor chromanol 293B (10 μM) (n = 5, P = 0.22 by two-way ANOVA). (B) Concentration-response curves for adenosine in coronary arterial segments without endothelium contracted with PGF2α (10 μM) in the absence and presence of the KV7.1 channel inhibitor chromanol 293B (10 μM) (n = 5, P = 0.45 by two-way ANOVA).
Figure S6 Detection of KCa1.1 and KV7 channels by immunoblotting. Immunoblot with samples from a normoxic artery in lane 1 and from a hypoxic artery in lane 2 and showing the presence of (A) KCa1.1α located at 100 kDa (B) KCa1.1β located at 30 kDa (C) KV7.4 located at 77 kDa and (D) KV7.5 located at 100 kDa. (E) Incubated with secondary antibody goat anti-rabbit IgG conjugated to HRP.
References
- Aalkjaer C, Lombard JH. Effect of hypoxia on force, intracellular pH and Ca2+ concentration in rat cerebral and mesenteric small arteries. J Physiol. 1995;482(Pt 2):409–419. doi: 10.1113/jphysiol.1995.sp020528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, et al. The Concise Guide to PHARMACOLOGY 2013/14: Overview. Br J Pharmacol. 2013;170:1449–1867. doi: 10.1111/bph.12444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barron JT, Gu L. Energetic effects of adenosine on vascular smooth muscle. Am J Physiol Heart Circ Physiol. 2000;278:H26–H32. doi: 10.1152/ajpheart.2000.278.1.H26. [DOI] [PubMed] [Google Scholar]
- Beech DJ, Bolton TB. Properties of the cromakalim-induced potassium conductance in smooth muscle cells isolated from the rabbit portal vein. Br J Pharmacol. 1989;98:851–864. doi: 10.1111/j.1476-5381.1989.tb14614.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brueggemann LI, Moran CJ, Barakat JA, Yeh JZ, Cribbs LL, Byron KL. Vasopressin stimulates action potential firing by protein kinase C-dependent inhibition of KCNQ5 in A7r5 rat aortic smooth muscle cells. Am J Physiol Heart Circ Physiol. 2007;292:H1352–H1363. doi: 10.1152/ajpheart.00065.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chadha PS, Zunke F, Zhu HL, Davis AJ, Jepps TA, Olesen SP, et al. Reduced KCNQ4-encoded voltage-dependent potassium channel activity underlies impaired beta-adrenoceptor-mediated relaxation of renal arteries in hypertension. Hypertension. 2012;59:877–884. doi: 10.1161/HYPERTENSIONAHA.111.187427. [DOI] [PubMed] [Google Scholar]
- Dart C, Standen NB. Activation of ATP-dependent K+ channels by hypoxia in smooth muscle cells isolated from the pig coronary artery. J Physiol Lond. 1995;483:29–39. doi: 10.1113/jphysiol.1995.sp020565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daut J, Maier-Rudolph W, von Beckerath N, Mehrke G, Günter K, Goedel-Meinen L. Hypoxic dilatation of coronary arteries is mediated by ATP-sensitive potassium channels. Science. 1990;247:1341–1344. doi: 10.1126/science.2107575. [DOI] [PubMed] [Google Scholar]
- Decking UK, Schlieper G, Kroll K, Schrader J. Hypoxia-induced inhibition of adenosine kinase potentiates cardiac adenosine release. Circ Res. 1997;81:154–164. doi: 10.1161/01.res.81.2.154. [DOI] [PubMed] [Google Scholar]
- Edwards G, Thollon C, Gardener MJ, Feletou M, Vilaine J, Vanhoutte PM, et al. Role of gap junctions and EETs in endothelium-dependent hyperpolarization of porcine coronary artery. Br J Pharmacol. 2000;129:1145–1154. doi: 10.1038/sj.bjp.0703188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- European Convention for the Protection of Vertebrate Animals used for Experimental and Other Scientific Purposes. 20 20-10-2010 Available at: http://conventions.coe.int/Treaty/en/Treaties/html/123.htm Online Source (accessed 10/20/2010)
- Franco-Obregon A, Lopez-Barneo J. Low PO2 inhibits calcium channel activity in arterial smooth muscle cells. Am J Physiol. 1996;271:H2290–H2299. doi: 10.1152/ajpheart.1996.271.6.H2290. [DOI] [PubMed] [Google Scholar]
- Frobert O, Buus CL, Rembold CM. HSP20 phosphorylation and interstitial metabolites in hypoxia-induced dilation of swine coronary arteries. Acta Physiol Scand. 2005;184:37–44. doi: 10.1111/j.1365-201X.2005.01426.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gebremedhin D, Bonnet P, Greene AS, England SK, Rusch NJ, Lombard JH, et al. Hypoxia increases the activity of Ca(2+)-sensitive K+ channels in cat cerebral arterial muscle cell membranes. Pflugers Arch. 1994;428:621–630. doi: 10.1007/BF00374586. [DOI] [PubMed] [Google Scholar]
- Gu M, Thorne GD, Wardle RL, Ishida Y, Paul RJ. Ca2+-independent hypoxic vasorelaxation in porcine coronary artery. J Physiol. 2005;562:839–846. doi: 10.1113/jphysiol.2004.073692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hedegaard ER, Mogensen S. 2013a. UNVERIFIED: Sus scrofa calcium-activated potassium channel subunit alpha-1-like mRNA, partial sequence.
- Hedegaard ER, Mogensen S. 2013b. Sus scrofa potassium voltage-gated channel member 4-like mRNA, partial sequence.
- Hedegaard ER, Mogensen S. 2013c. Sus scrofa potassium voltage-gated channel member 5 variant 1 (KCNQ5) mRNA, partial cds, alternatively spliced.
- Hedegaard ER, Mogensen S. 2013d. Sus scrofa potassium voltage-gated channel member 5 variant 2 (KCNQ5) mRNA, partial cds, alternatively spliced.
- Hedegaard ER, Mogensen S. 2013e. UNVERIFIED: Sus scrofa potassium voltage-gated channel member 1-like mRNA, partial sequence.
- Hedegaard ER, Stankevicius E, Simonsen U, Frobert O. Non-endothelial endothelin counteracts hypoxic vasodilation in porcine large coronary arteries. BMC Physiol. 2011;11:8. doi: 10.1186/1472-6793-11-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jensen HS, Callo K, Jespersen T, Jensen BS, Olesen SP. The KCNQ5 potassium channel from mouse: a broadly expressed M-current like potassium channel modulated by zinc, pH, and volume changes. Brain Res Mol Brain Res. 2005;139:52–62. doi: 10.1016/j.molbrainres.2005.05.007. [DOI] [PubMed] [Google Scholar]
- Jepps TA, Chadha PS, Davis AJ, Harhun MI, Cockerill GW, Olesen SP, et al. Downregulation of kv7.4 channel activity in primary and secondary hypertension. Circulation. 2011;124:602–611. doi: 10.1161/CIRCULATIONAHA.111.032136. [DOI] [PubMed] [Google Scholar]
- Joshi S, Balan P, Gurney AM. Pulmonary vasoconstrictor action of KCNQ potassium channel blockers. Respir Res. 2006;7:1–10. doi: 10.1186/1465-9921-7-31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamekura I, Okumura K, Matsui H, Murase K, Mokuno S, Toki Y, et al. Mechanisms of hypoxic coronary vasodilatation in isolated perfused rat hearts. J Cardiovasc Pharmacol. 1999;33:836–842. doi: 10.1097/00005344-199906000-00002. [DOI] [PubMed] [Google Scholar]
- Lang DG, Ritchie AK. Tetraethylammonium blockade of apamin-sensitive and insensitive Ca2(+)-activated K+ channels in a pituitary cell line. J Physiol. 1990;425:117–132. doi: 10.1113/jphysiol.1990.sp018095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee YH, Kim JT, Kang BS. Mechanisms of relaxation of coronary artery by hypoxia. Yonsei Med J. 1998;39:252–260. doi: 10.3349/ymj.1998.39.3.252. [DOI] [PubMed] [Google Scholar]
- Liu Q, Flavahan NA. Hypoxic dilatation of porcine small coronary arteries: role of endothelium and KATP-channels. Br J Pharmacol. 1997;120:728–734. doi: 10.1038/sj.bjp.0700939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopez-Barneo J, del Toro R, Levitsky KL, Chiara MD, Ortega-Saenz P. Regulation of oxygen sensing by ion channels. J Appl Physiol. 2004;96:1187–1195. doi: 10.1152/japplphysiol.00929.2003. [DOI] [PubMed] [Google Scholar]
- Lynch FM, Austin C, Heagerty AM, Izzard AS. Adenosine and hypoxic dilation of rat coronary small arteries: roles of the ATP-sensitive potassium channel, endothelium, and nitric oxide. Am J Physiol Heart Circ Physiol. 2006;290:H1145–H1150. doi: 10.1152/ajpheart.00314.2005. [DOI] [PubMed] [Google Scholar]
- Mackie AR, Byron KL. Cardiovascular KCNQ (Kv7) potassium channels: physiological regulators and new targets for therapeutic intervention. Mol Pharmacol. 2008;74:1171–1179. doi: 10.1124/mol.108.049825. [DOI] [PubMed] [Google Scholar]
- Mackie AR, Brueggemann LI, Henderson KK, Shiels AJ, Cribbs LL, Scrogin KE, et al. Vascular KCNQ potassium channels as novel targets for the control of mesenteric artery constriction by vasopressin, based on studies in single cells, pressurized arteries, and in vivo measurements of mesenteric vascular resistance. J Pharmacol Exp Ther. 2008;325:475–483. doi: 10.1124/jpet.107.135764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meeks MK, Ripley ML, Jin Z, Rembold CM. Heat shock protein 20-mediated force suppression in forskolin-relaxed swine carotid artery. Am J Physiol Cell Physiol. 2005;288:C633–C639. doi: 10.1152/ajpcell.00269.2004. [DOI] [PubMed] [Google Scholar]
- Morecroft I, Murray A, Nilsen M, Gurney AM, MacLean MR. Treatment with the Kv7 potassium channel activator flupirtine is beneficial in two independent mouse models of pulmonary hypertension. Br J Pharmacol. 2009;157:1241–1249. doi: 10.1111/j.1476-5381.2009.00283.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mulvany MJ, Warshaw DM. The active tension-length curve of vascular smooth muscle related to its cellular components. J Gen Physiol. 1979;74:85–104. doi: 10.1085/jgp.74.1.85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nelson MT, Quayle JM. Physiological roles and properties of potassium channels in arterial smooth muscle. Am J Physiol. 1995;268:C799–C822. doi: 10.1152/ajpcell.1995.268.4.C799. [DOI] [PubMed] [Google Scholar]
- Ng FL, Davis AJ, Jepps TA, Harhun MI, Yeung SY, Wan A, et al. Expression and function of the K+ channel KCNQ genes in human arteries. Br J Pharmacol. 2011;162:42–53. doi: 10.1111/j.1476-5381.2010.01027.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pasgaard T, Stankevicius E, Jorgensen MM, Ostergaard L, Simonsen U, Frøbert O. Hyperoxia reduces basal release of nitric oxide and contracts porcine coronary arteries. Acta Physiol (Oxf) 2007;191:285–296. doi: 10.1111/j.1748-1716.2007.01745.x. [DOI] [PubMed] [Google Scholar]
- Rembold CM, Foster DB, Strauss JD, Wingard CJ, Eyk JE. cGMP-mediated phosphorylation of heat shock protein 20 may cause smooth muscle relaxation without myosin light chain dephosphorylation in swine carotid artery. J Physiol. 2000;524(Pt 3):865–878. doi: 10.1111/j.1469-7793.2000.00865.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriguez-Rodriguez R, Stankevicius E, Herrera MD, Ostergaard L, Andersen MR, Ruiz-Gutierrez V, et al. Oleanolic acid induces relaxation and calcium-independent release of endothelium-derived nitric oxide. Br J Pharmacol. 2008;155:535–546. doi: 10.1038/bjp.2008.289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schleifenbaum J, Kohn C, Voblova N, Dubrovska G, Zavarirskaya O, Gloe T, et al. Systemic peripheral artery relaxation by KCNQ channel openers and hydrogen sulfide. J Hypertens. 2010;28:1875–1882. doi: 10.1097/HJH.0b013e32833c20d5. [DOI] [PubMed] [Google Scholar]
- Schubert R, Wesselman JP, Nilsson H, Mulvany MJ. Noradrenaline-induced depolarization is smaller in isobaric compared to isometric preparations of rat mesenteric small arteries. Pflugers Arch. 1996;431:794–796. doi: 10.1007/BF02253846. [DOI] [PubMed] [Google Scholar]
- Shimizu S, Bowman PS, Thorne G, III, Paul RJ. Effects of hypoxia on isometric force, intracellular Ca(2+), pH, and energetics in porcine coronary artery. Circ Res. 2000;86:862–870. doi: 10.1161/01.res.86.8.862. [DOI] [PubMed] [Google Scholar]
- Skovgaard N, Gouliaev A, Aalling M, Simonsen U. The role of endogenous H(2)S in cardiovascular physiology. Curr Pharm Biotechnol. 2011;12:1385–1393. doi: 10.2174/138920111798280956. [DOI] [PubMed] [Google Scholar]
- Smani T, Hernandez A, Urena J, Castellano AG, Franco-Obregon A, Ordonez A, et al. Reduction of Ca(2+) channel activity by hypoxia in human and porcine coronary myocytes. Cardiovasc Res. 2002;53:97–104. doi: 10.1016/s0008-6363(01)00422-9. [DOI] [PubMed] [Google Scholar]
- Somlyo AP, Somlyo AV. Ca2+ sensitivity of smooth muscle and nonmuscle myosin II: modulated by G proteins, kinases, and myosin phosphatase. Physiol Rev. 2003;83:1325–1358. doi: 10.1152/physrev.00023.2003. [DOI] [PubMed] [Google Scholar]
- Stub D, Smith K, Bernard S, Bray JE, Stephenson M, Cameron P, et al. A randomized controlled trial of oxygen therapy in acute myocardial infarction Air Verses Oxygen In myocarDial infarction study (AVOID Study) Am Heart J. 2012;163:339–345. doi: 10.1016/j.ahj.2011.11.011. [DOI] [PubMed] [Google Scholar]
- Thollon C, Fournet-Bourguignon MP, Saboureau D, Lesage L, Reure H, Vanhoutte PM, et al. Consequences of reduced production of NO on vascular reactivity of porcine coronary arteries after angioplasty: importance of EDHF. Br J Pharmacol. 2002;136:1153–1161. doi: 10.1038/sj.bjp.0704828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thorne GD, Shimizu S, Paul RJ. Hypoxic vasodilation in porcine coronary artery is preferentially inhibited by organ culture. Am J Physiol Cell Physiol. 2001;281:C24–C32. doi: 10.1152/ajpcell.2001.281.1.C24. [DOI] [PubMed] [Google Scholar]
- Thorne GD, Conforti L, Paul RJ. Hypoxic vasorelaxation inhibition by organ culture correlates with loss of Kv channels but not Ca(2+) channels. Am J Physiol Heart Circ Physiol. 2002;283:H247–H253. doi: 10.1152/ajpheart.00569.2001. [DOI] [PubMed] [Google Scholar]
- Thorne GD, Ishida Y, Paul RJ. Hypoxic vasorelaxation: Ca2+-dependent and Ca2+-independent mechanisms. Cell Calcium. 2004;36:201–208. doi: 10.1016/j.ceca.2004.02.018. [DOI] [PubMed] [Google Scholar]
- Wadsworth RM. Vasoconstrictor and vasodilator effects of hypoxia. Trends Pharmacol Sci. 1994;15:47–53. doi: 10.1016/0165-6147(94)90109-0. [DOI] [PubMed] [Google Scholar]
- Wardle RL, Gu M, Ishida Y, Paul RJ. Ca2+-desensitizing hypoxic vasorelaxation: pivotal role for the myosin binding subunit of myosin phosphatase (MYPT1) in porcine coronary artery. J Physiol. 2006;572:259–267. doi: 10.1113/jphysiol.2005.104083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whiteman M, Le TS, Chopra M, Fox B, Whatmore J. Emerging role of hydrogen sulfide in health and disease: critical appraisal of biomarkers and pharmacological tools. Clin Sci (Lond) 2011;121:459–488. doi: 10.1042/CS20110267. [DOI] [PubMed] [Google Scholar]
- Yeung S, Greenwood IA. Electrophysiological and functional effects of the KCNQ channel blocker XE991 on murine portal vein smooth muscle cells. Br J Pharmacol. 2005;146:585–595. doi: 10.1038/sj.bjp.0706342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yeung S, Pucovsky V, Moffatt JD, Saldanha L, Schwake M, Ohya S, et al. Molecular expression and pharmacological identification of a role for K(v)7 channels in murine vascular reactivity. Br J Pharmacol. 2007;151:758–770. doi: 10.1038/sj.bjp.0707284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yeung S, Lange W, Schwake M, Greenwood IA. Expression profile and characterisation of a truncated KCNQ5 splice variant. Biochem Biophys Res Commun. 2008a;371:741–746. doi: 10.1016/j.bbrc.2008.04.129. [DOI] [PubMed] [Google Scholar]
- Yeung S, Schwake M, Pucovsky V, Greenwood IA. Bimodal effects of the Kv7 channel activator retigabine on vascular K+ currents. Br J Pharmacol. 2008b;155:62–72. doi: 10.1038/bjp.2008.231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X, Yang D, Hughes BA. KCNQ5/K(v)7.5 potassium channel expression and subcellular localization in primate retinal pigment epithelium and neural retina. Am J Physiol Cell Physiol. 2011;301:C1017–C1026. doi: 10.1152/ajpcell.00185.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Z, Huang H, Liu P, Tang C, Wang J. Hydrogen sulfide contributes to cardioprotection during ischemia reperfusion injury by opening KATP channels. Can J Physiol Pharmacol. 2007;85:1248–1253. doi: 10.1139/Y07-120. [DOI] [PubMed] [Google Scholar]
- Zhong XZ, Harhun MI, Olesen SP, Ohya S, Moffatt JD, Cole WC, et al. Participation of KCNQ (Kv7) potassium channels in myogenic control of cerebral arterial diameter. J Physiol. 2010;588:3277–3293. doi: 10.1113/jphysiol.2010.192823. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.