

ABSTRACT
Regulation of the size and abundance of membrane compartments is a fundamental cellular activity. In Saccharomyces cerevisiae, disruption of the ADP-ribosylation factor 1 (ARF1) gene yields larger and fewer Golgi cisternae by partially depleting the Arf GTPase. We observed a similar phenotype with a thermosensitive mutation in Nmt1, which myristoylates and activates Arf. Therefore, partial depletion of Arf is a convenient tool for dissecting mechanisms that regulate Golgi structure. We found that in arf1Δ cells, late Golgi structure is particularly abnormal, with the number of late Golgi cisternae being severely reduced. This effect can be explained by selective changes in cisternal maturation kinetics. The arf1Δ mutation causes early Golgi cisternae to mature more slowly and less frequently, but does not alter the maturation of late Golgi cisternae. These changes quantitatively explain why late Golgi cisternae are fewer in number and correspondingly larger. With a stacked Golgi, similar changes in maturation kinetics could be used by the cell to modulate the number of cisternae per stack. Thus, the rates of processes that transform a maturing compartment can determine compartmental size and copy number.
KEY WORDS: Organelle size, Organelle number, Golgi, Cisternal maturation, Arf
INTRODUCTION
The mechanisms that control organelle size and number are attracting increasing interest (Goehring and Hyman, 2012; Levy and Heald, 2012; Marshall, 2002; Sengupta and Linstedt, 2011). Apart from being a basic cell biological problem, this topic has clinical relevance because cancer cells display altered organelle structure (Levy and Heald, 2012; Zink et al., 2004). One way to control organelle structure is with fusion and fission events. For example, mitochondrial networks are maintained by a balance between fusion and fission (Chan, 2012; Westermann, 2010). Similarly, endosomes and yeast vacuoles undergo frequent homotypic fusion (Epp et al., 2011). Another way to control organelle structure is to alter the relative rates of membrane influx and efflux (Sengupta and Linstedt, 2011). Many of these membrane remodeling events are controlled by GTPases. Improved understanding of such processes may enable the reprogramming of organelle size in diseased cells (Marshall, 2012).
An organelle that shows great plasticity and variability is the Golgi apparatus, which consists of disk-shaped cisternae that sometimes exhibit fenestrations, tubular extensions and lateral interconnections (Warren and Rothman, 2011). The mammalian Golgi undergoes changes during differentiation or cancer progression (Kellokumpu et al., 2002; Kirk et al., 2010; Lu et al., 2001; Weller et al., 2010; Wu et al., 2000). In most eukaryotes, Golgi cisternae are arranged in polarized stacks, but in some species such as the yeast Saccharomyces cerevisiae, Golgi cisternae are dispersed throughout the cell (Mowbrey and Dacks, 2009; Preuss et al., 1992). The dispersed cisternae can be tracked individually by fluorescence microscopy (Wooding and Pelham, 1998). A typical S. cerevisiae cell contains about six to ten early Golgi cisternae plus a similar number of late or trans-Golgi network (TGN) cisternae (Papanikou and Glick, 2009). Early yeast Golgi cisternae mature to generate late Golgi cisternae, a process that appears to be evolutionarily conserved (Glick and Nakano, 2009; Losev et al., 2006; Matsuura-Tokita et al., 2006).
S. cerevisiae can be used to study the regulation of Golgi structure. In a microscopy-based screen to identify thermosensitive mutants defective in Golgi inheritance, we also identified mutants that contained fewer and larger Golgi cisternae (Rossanese et al., 2001). A similar phenotype had been described for the ADP-ribosylation factor 1 (ARF1)-deleted (arf1Δ) mutant (Gaynor et al., 1998). S. cerevisiae contains the closely related genes ARF1 and ARF2, with ARF1 accounting for ∼90% of the Golgi-associated Arf (Stearns et al., 1990a). arf1Δ mutants show abnormal Golgi structure but only mild defects in secretion (Gaynor et al., 1998; Stearns et al., 1990b). We find that a thermosensitive mutant has enlarged Golgi cisternae due to impaired function of the N-myristoyl transferase Nmt1 (Johnson et al., 1994), which activates Arf (Donaldson and Jackson, 2011; Kahn, 2009). Thus, separate lines of investigation have pointed to Arf as a key regulator of Golgi size and number.
To clarify the mechanisms that generate enlarged Golgi cisternae, we used four-dimensional (4D) fluorescence microscopy. The results indicate that during Golgi maturation, early Golgi cisternae mature more slowly and less frequently in arf1Δ cells than in wild-type cells. By contrast, after the early-to-late Golgi transition, the maturation kinetics in arf1Δ cells are essentially normal. The consequence of these selective changes is a severe reduction in the number of late Golgi cisternae. Our analysis highlights the importance of kinetic parameters for regulating the size and copy number of dynamic compartments.
RESULTS
A mutation in NMT1 results in enlarged late Golgi cisternae
We used a thermosensitive yeast mutant that has larger and fewer late Golgi cisternae, as marked by Sec7–GFP (Rossanese et al., 2001). For further analysis, we also labeled the plasma membrane with mCherry–Ras2 (Tang et al., 2009). Late Golgi cisternae were visualized with 2D projections, and with 3D rendering that represented a cisterna as a closed surface (Fig. 1A). Although Golgi cisternae are topologically complex (see below), modeling a cisterna as a closed surface enabled us to use volume as a measure of cisternal size. An alternative measure was the maximal diameter of a cisterna in the X–Y plane. Quantification revealed that on average, late Golgi cisternae in mutant cells had approximately threefold greater volume and approximately twofold greater diameter than in wild-type cells (Fig. 1B,C).
Fig. 1.
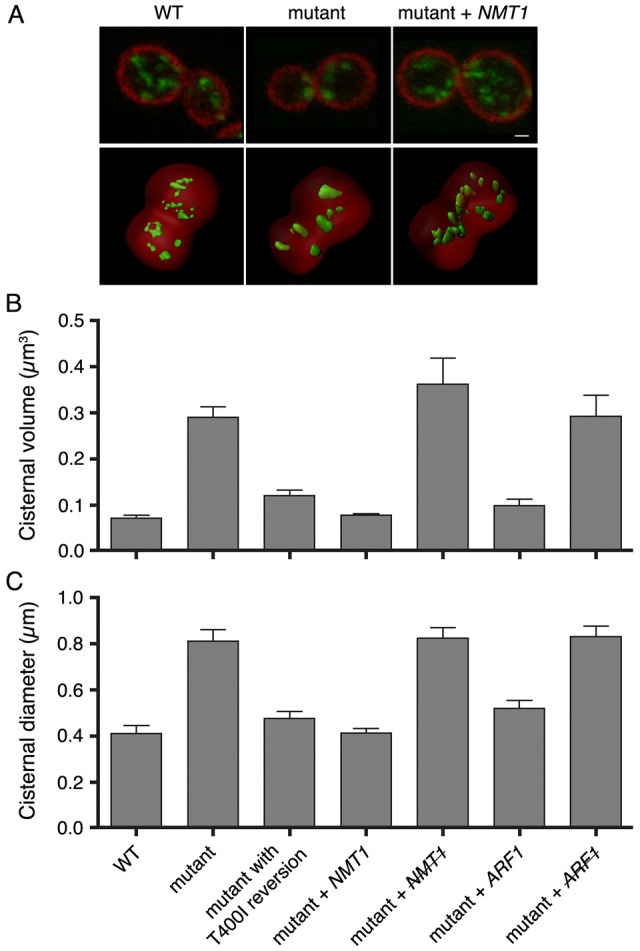
A conditional mutation in NMT1 generates enlarged late Golgi cisternae. (A) Representative images of the wild-type (WT) parental strain, the thermosensitive mutant, and the mutant transformed with a centromeric plasmid encoding NMT1. The plasma membrane was labeled with mCherry–Ras2 (red), and the late Golgi was labeled with Sec7–GFPx3 (green). The top row shows projected confocal sections of a central portion of the cell, and the bottom row shows 3D renderings. Scale bar: 1 µm. (B,C) Images of the type shown in A were quantified by rendering late Golgi cisternae as closed surfaces. The following strains were examined: the wild-type, the thermosensitive mutant, the mutant in which the nmt1 T400I mutation was reverted by gene replacement, the mutant transformed with a centromeric plasmid encoding NMT1, the mutant transformed with the plasmid encoding NMT1 and then cured of this plasmid (indicated by the strikethrough), the mutant transformed with a high copy number plasmid encoding ARF1, and the mutant transformed with the plasmid encoding ARF1 and then cured of this plasmid (indicated by the strikethrough). Sizes of the rendered late Golgi elements were quantified by measuring either (B) the average volume or (C) the average X–Y-plane diameter. Error bars indicate s.e.m.
To identify the mutated gene, a genomic library was screened for complementation of the thermosensitive growth defect (Rossanese et al., 2001). Two overlapping clones rescued the growth and Golgi size phenotypes (Fig. 1A–C). The overlapping region contained NMT1, which encodes N-myristoyl transferase (Johnson et al., 1994). Rescue of the mutant phenotypes was lost if mutant cells were cured of a plasmid carrying NMT1 (Fig. 1B,C). The nmt1 allele had a T400I mutation, and the growth and Golgi size phenotypes were rescued by reverting this mutation in the genome (Fig. 1B,C; supplementary material Fig. S1). Thus, a point mutation in NMT1 leads to enlarged late Golgi cisternae.
Golgi enlargement can be caused by reduced Arf activity
One substrate for Nmt1 is Golgi-associated Arf, a GTPase that recruits multiple effectors, including the COPI coatomer and clathrin adaptors (Donaldson and Jackson, 2011; Kahn, 2009). Because Arf activity depends on N-terminal myristoylation, we suspected that the enlarged Golgi cisternae in the nmt1 strain were due to reduced myristoylation of Arf.
This hypothesis was tested in two ways. First, we compensated for impaired Nmt1 activity in the mutant strain by overexpressing ARF1. Elevated Arf1 levels suppressed the Golgi size phenotype, and this effect was lost if the mutant cells were cured of the plasmid (Fig. 1B,C; supplementary material Fig. S1). Second, we deleted ARF1 to confirm that reduced Arf levels cause Golgi enlargement in our strain. Indeed, arf1Δ cells contained a small number of late Golgi cisternae that were often abnormally large (Fig. 2A,B). Thus, depletion or partial inactivation of Golgi-associated Arf leads to larger and fewer late Golgi cisternae.
Fig. 2.
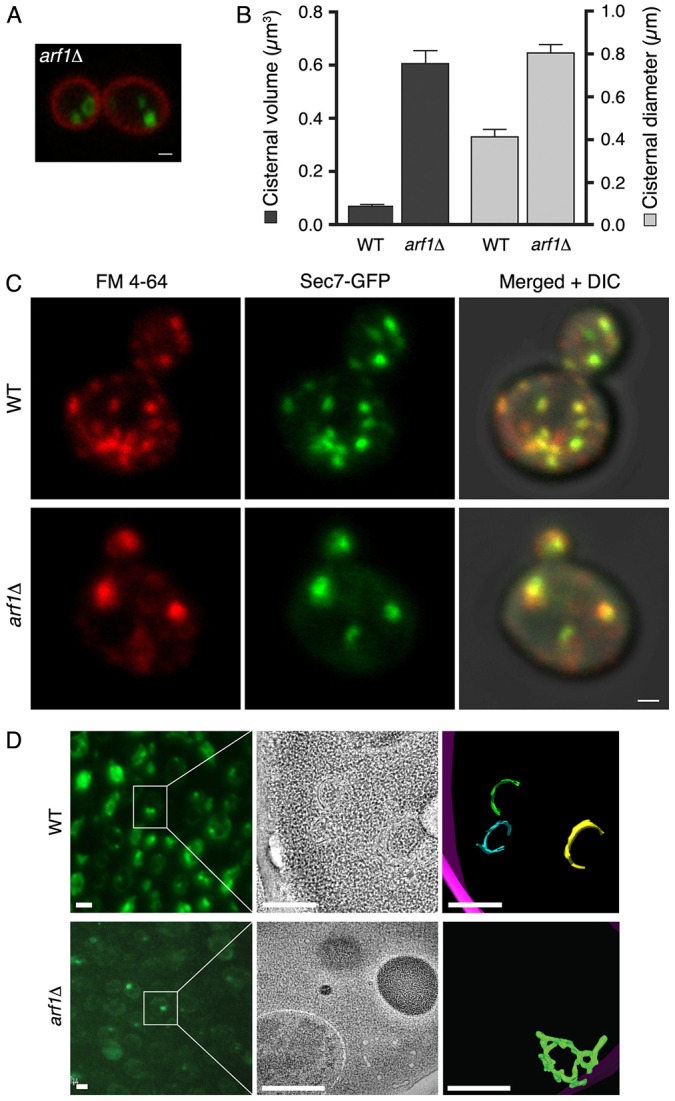
The arf1Δ mutation generates enlarged late Golgi cisternae that label with FM 4-64. (A) arf1Δ cells were imaged by fluorescence microscopy as in Fig. 1 to visualize the plasma membrane (red) and late Golgi (green). Scale bar: 1 µm. (B) The average volume and average X–Y-plane diameter of late Golgi cisternae were measured as in Fig. 1 for wild-type and arf1Δ strains. Error bars indicate s.e.m. (C) Wild-type or arf1Δ cells expressing Sec7-6xGFP were grown in SD medium at 23°C to an OD600 of 0.6, then incubated with 0.8 µM FM 4-64 for 5 minutes. SCAS was added to 4 µM, and cells were compressed beneath a coverslip. Confocal images were captured ∼2.5 minutes after SCAS addition. Scale bar: 1 µm. (D) Wild-type or arf1Δ cells expressing Sec7-GFPx3 were rapidly frozen, then freeze substituted and embedded in plastic. Left: thick sections were imaged by fluorescence microscopy to identify late Golgi cisternae. Scale bars: 2 µm. Middle: the same sections were analyzed by electron tomography to visualize membranes in tomographic slices. Scale bars: 0.5 µm. Right: membrane contours were traced to model Golgi cisternae (green, blue, yellow) and the plasma membrane (purple). Scale bars: 0.5 µm.
It was reported that arf1Δ cells also had enlarged endosomes (Gaynor et al., 1998), as judged by labeling the endocytic pathway with the dye FM 4-64 (Vida and Emr, 1995). We observed abnormally large fluorescent compartments after incubating arf1Δ cells with FM 4-64 (Fig. 2C). However, FM 4-64 is a bulk membrane marker that moves from the endocytic pathway to the late Golgi/TGN (Lewis et al., 2000), and most of the Sec7–GFP-labeled structures were also labeled with recently internalized FM 4-64 in both wild-type and arf1Δ cells (Fig. 2C). We conclude that in arf1Δ cells, the enlarged compartments containing FM 4-64 are late Golgi cisternae.
The late Golgi in arf1Δ cells consist of fenestrated cisternae
Are the late Golgi structures in arf1Δ cells large individual cisternae or clusters of smaller cisternae? To address this question, we combined fluorescence microscopy with electron tomography (Kukulski et al., 2011). While optimizing the procedure, we discovered that embedding yeast cells in Lowicryl K4M resin preserved strong GFP fluorescence while yielding sufficient contrast.
For wild-type cells, we labeled late Golgi cisternae with Sec7–GFP, and prepared ∼300-nm sections from plastic-embedded samples. Labeled cisternae were visible by fluorescence microscopy (Fig. 2D). The same structures were then analyzed by electron tomography. This method examines a single thick section, so only part of each cisterna was visible, but the results indicated that wild-type late Golgi cisternae were curved and perforated disks (Fig. 2D; supplementary material Movie 1).
For arf1Δ cells (Fig. 2D; supplementary material Movies 2, 3), partial reconstructions indicated that mutant late Golgi cisternae were large fenestrated hollow structures, presumably identical to structures previously visualized in arf1Δ cells by thin-section electron microscopy (Gaynor et al., 1998). These results confirm that partial depletion of Arf generates abnormally large late Golgi cisternae.
The changes in arf1Δ cells are more pronounced for older Golgi cisternae
Late Golgi cisternae are generated by maturation, and we sought to determine where in this pathway the arf1Δ mutation exerts its effects. First, we examined transitional endoplasmic reticulum (tER) sites, which produce COPII vesicles (Rossanese et al., 1999; Shindiapina and Barlowe, 2010). Some mutations cause coalescence of these structures (Castillon et al., 2009; Levi et al., 2010; Shindiapina and Barlowe, 2010), but we saw no consistent differences in the tER patterns of wild-type and arf1Δ cells (supplementary material Fig. S2), suggesting that the arf1Δ mutation acts at the level of the Golgi.
Next, we counted early and late Golgi cisternae by using GFP–Vrg4 as an early Golgi marker, and Sec7–DsRed as a late Golgi marker (Losev et al., 2006). Wild-type cells contained an average of nine early Golgi cisternae and seven late Golgi cisternae (Fig. 3A; Table 1). In the arf1Δ strain, early Golgi cisternae tended to be slightly enlarged (supplementary material Fig. S3) and their average number was reduced to five. For the late Golgi, the reduction in the arf1Δ strain was more pronounced, with an average of only two cisternae per cell (Fig. 3A and Table 1). Thus, as the Golgi matures, the arf1Δ mutation progressively reduces the number of cisternae.
Fig. 3.

The arf1Δ mutation differentially affects the early and late Golgi. (A) The average numbers of early Golgi cisternae, marked with GFP–Vrg4, or late Golgi cisternae, marked with Sec7–DsRed, in wild-type and arf1Δ strains. Error bars indicate s.e.m. (B) A 10-minute 4D movie was generated for an arf1Δ cell expressing Sec7-3xGFP. Fluorescence intensities were quantified for each cisterna that appeared and subsequently disappeared during the recording. The different colors represent distinct cisternae. (C) The average frequency of early-to-late Golgi transitions as indicated by loss of GFP–Vrg4 and acquisition of Sec7–DsRed in wild-type and arf1Δ strains. Error bars indicate s.e.m. (D) The average persistence time on cisternae of the early Golgi marker GFP–Vrg4 and the late Golgi marker Sec7–DsRed in wild-type and arf1Δ strains. Error bars indicate s.e.m.
Table 1. Parameters for Golgi maturation in wild-type and arf1Δ cells.

E, average number of early Golgi cisternae; L, average number of late Golgi cisternae; m, average frequency of Golgi maturation (early-to-late Golgi transitions per minute); pE, average persistence time of early Golgi cisternae (seconds); and pL, average persistence time of late Golgi cisternae (seconds).
Values are means ± s.e.m.
As determined by an two-tailed unpaired t-test, the following parameters are significantly different for wild-type versus arf1Δ cells: E (P<0.0001), L (P<0.0001), m (P<0.0001) and pE (P<0.001).
The last two columns show predicted values of E and L according to the formulas stated in the text.
Altered maturation kinetics could explain why arf1Δ cells have fewer Golgi cisternae
The effects of the arf1Δ mutation on Golgi cisternal number might be due to altered maturation kinetics. To test this idea, we devised a simple quantitative framework in which E is the average number of early Golgi cisternae per cell, L is the average number of late Golgi cisternae per cell, m is the average maturation frequency, pE is the average persistence time of early Golgi cisternae, and pL is the average persistence time of late Golgi cisternae. m is defined here as the number of early-to-late Golgi transitions per minute, i.e. the frequency at which early Golgi cisternae disappear and late Golgi cisternae appear. Because the average number of Golgi cisternae is constant at steady-state, m also refers to the frequency at which early Golgi cisternae appear. The average number of early or late Golgi cisternae should be equal to the frequency of appearance multiplied by the persistence time. Thus: E = m×pE and L = m×pL. Combining these equations yields: E/L = pE/pL.
The implications are that changes in maturation frequency or persistence time could alter the number of cisternae, and that a selective change in one of the persistence times could alter the ratio of early to late Golgi cisternae.
The arf1Δ mutation slows maturation of the early Golgi
This model was tested by measuring maturation parameters for wild-type and arf1Δ strains. An initial experiment employed arf1Δ cells expressing Sec7-GFP. In wild-type cells, Sec7–GFP is present on cisternae and then this marker is lost within ∼1.5–2 minutes (Losev et al., 2006). The same phenomenon was observed with arf1Δ cells. Because this mutant strain has so few late Golgi cisternae, we could often track every Sec7–GFP-labeled structure in a cell. Fig. 3B shows that in a representative cell over a 10-minute period, Sec7–GFP was present on each cisterna and then lost within ∼100 seconds. This result suggests that late Golgi cisternae function similarly in wild-type and arf1Δ cells (Daboussi et al., 2012).
Further analysis was performed with strains expressing GFP-Vrg4 and Sec7-DsRed (supplementary material Movie 4). Dual-color 4D movies were captured for 5 minutes, a period suitable for acquiring closely spaced Z-stacks without excessive photobleaching. To measure the maturation frequency m (Fig. 3C; Table 1), we counted the number of early-to-late Golgi transitions per minute (Losev et al., 2006). To measure the persistence times pE and pL (Fig. 3D; Table 1), we identified cisternae that acquired and then lost either GFP–Vrg4 or Sec7–DsRed.
Our analysis revealed a striking effect: the arf1Δ mutation selectively increased the early Golgi persistence time pE. The pE/pL ratio was ∼1 in wild-type cells but ∼2 in arf1Δ cells (Table 1). This finding could explain why the number of early Golgi cisternae, E, was similar to the number of late Golgi cisternae, L, in wild-type cells, whereas E was about twice as large as L in arf1Δ cells.
Another clear difference between the two strains is the maturation frequency, m, which was ∼2.5-fold greater in wild-type cells than in arf1Δ cells (Table 1). We infer that the entire early Golgi maturation process is slowed in arf1Δ cells, with early Golgi cisternae persisting longer and maturing less frequently than in wild-type cells.
Golgi cisternae undergo homotypic fusion
Table 1 shows a comparison of the measured values E and L, and the corresponding predicted values m×pE and m×pL. The number of late Golgi cisternae, L, agrees reasonably well with the predicted value m×pL for both wild-type and arf1Δ strains. However, the number of early Golgi cisternae, E, is higher than the predicted value m×pE for both strains.
To explore this discrepancy, we re-evaluated the rationale for using m×pE as an estimate of E. Because the spatial and temporal resolution of the 4D movies was insufficient for counting early Golgi formation events directly, we assumed that the frequency of early Golgi formation events matched the frequency of early-to-late Golgi conversion events. This assumption would be incorrect if early Golgi cisternae undergo homotypic fusion. Indeed, such homotypic fusion events take place, as illustrated in Fig. 4A and supplementary material Movie 4. A typical wild-type or arf1Δ cell exhibited about one early Golgi homotypic fusion event per minute (Fig. 4B). As a consequence of homotypic fusion, the frequency of early Golgi formation events is higher than the maturation frequency m, so m×pE is an underestimate of the number of early Golgi cisternae.
Fig. 4.

Golgi cisternae can undergo homotypic fusion. (A) Frames from a 4D movie (supplementary material Movie 4) illustrate a homotypic fusion event in an arf1Δ cell. The arrowheads mark two GFP–Vrg4-labeled early Golgi cisternae that fused before maturing into a Sec7–DsRed-labeled late Golgi cisterna. Time is shown in minutes:seconds. Scale bar: 1 µm. (B) The average number of homotypic fusion events per minute for early and late Golgi cisternae in wild-type and arf1Δ strains.
Despite the limitations caused by homotypic fusion, the quantitative framework yields two strong conclusions (supplementary material Fig. S4). First, the reason that arf1Δ cells contains more early than late Golgi cisternae is that in these cells, early Golgi cisternae persist longer than late Golgi cisternae. Second, the reason that arf1Δ cells have fewer late Golgi cisternae than wild-type cells is that arf1Δ cells have an abnormally low frequency of early-to-late Golgi transitions but a normal persistence time for the late Golgi.
DISCUSSION
Rates of membrane influx and efflux can regulate the overall size of the mammalian Golgi (Guo and Linstedt, 2006; Sengupta and Linstedt, 2011), and we find that in S. cerevisiae, kinetic parameters can also regulate Golgi copy number. Because Golgi cisternae are constantly turning over, the copy number of a Golgi compartment depends on how often the compartment forms and how long it persists. The total amount of Golgi material is presumably comparable in different strains, so when the number of Golgi cisternae in a strain is abnormally small, the average size of the cisternae is abnormally large. Thus, the rates of processes that transform a maturing compartment can dramatically influence compartmental size and copy number.
Slowed maturation of the early Golgi in arf1Δ cells helps to explain the observed delay in secretion (Gaynor et al., 1998), but the mechanism by which partial depletion of Arf alters Golgi maturation remains to be determined. A decrease in Golgi-associated Arf may compromise the recruitment or function of the COPI vesicle coat. In support of this idea, a thermosensitive COPI allele was reported to slow Golgi maturation at the restrictive temperature (Matsuura-Tokita et al., 2006). Another possibility is that Arf depletion perturbs lipid metabolism (Gaynor et al., 1998). Indeed, the arf1Δ mutation induces the unfolded protein response (Jonikas et al., 2009), which in turn alters phospholipid biosynthesis and other aspects of the secretory pathway (Travers et al., 2000). Regardless of the specific processes that are affected, it is notable that two previous studies of Golgi cisternal enlargement in S. cerevisiae both implicated Arf (Gaynor et al., 1998; Peyroche et al., 2001) and that our identification of the nmt1 mutant points to Arf as well. These findings support the idea that Arf is a regulator of Golgi maturation (Stalder and Antonny, 2013).
How might our results be relevant to organisms with a stacked Golgi? We propose that changes in the kinetics of Golgi maturation could alter the number of cisternae per stack. Golgi cisternal number is variable between different cell types – in mammals, the number of cisternae per stack ranges from about 3 to 10 depending on the cell type (Rambourg and Clermont, 1997). Large Golgi stacks typically contain multiple COPI-vesicle-producing Golgi cisternae but only one or two clathrin-coated vesicle-producing TGN cisternae (Mogelsvang et al., 2004; Staehelin and Kang, 2008). By analogy to the arf1Δ mutant, if the persistence time of Golgi cisternae increased while the persistence time of TGN cisternae remained unchanged, the result would be a selective increase in the number of Golgi cisternae (Fig. 5). Interestingly, when mammalian cells are depleted of the Rab6 GTPase, secretion slows and the number of Golgi cisternae per stack increases (Storrie et al., 2012). It is possible that Rab6 depletion increases the persistence time of Golgi cisternae. Future developments in live-cell microscopy should enable a test of our hypothesis that maturation kinetics regulate the size of Golgi stacks.
Fig. 5.
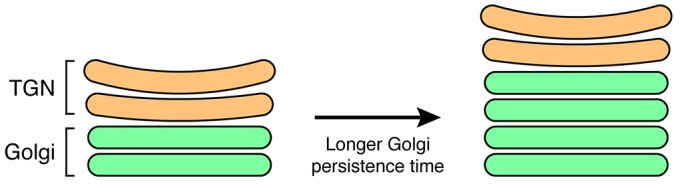
Changes in maturation kinetics could alter the number of cisternae in a stacked Golgi. A Golgi stack is represented as consisting of two classes of cisternae: the Golgi itself (green) and the trans-Golgi network (TGN; orange). These classes would correspond in S. cerevisiae to the early and late Golgi, respectively. If the persistence time of Golgi cisternae were increased without changing other parameters, the stack would contain more Golgi cisternae but the same number of TGN cisternae.
MATERIALS AND METHODS
Yeast strains and plasmids
All experiments were performed with derivatives of the haploid S. cerevisiae strain JK9-3d, which has the genotype leu2-3,112 ura3-52 rme1 trp1 his4 (Kunz et al., 1993). A mutant with enlarged late Golgi cisternae (Rossanese et al., 2001) was backcrossed four times to the parental strain, and thermosensitive growth segregated with the enlarged Golgi phenotype as a single-gene mutation through each cross. Yeast were grown in rich glucose medium (YPD) or minimal glucose medium (SD) (Sherman, 1991), with shaking at 200 r.p.m. in baffled flasks. Growth media were obtained from HiMedia Laboratories (Mumbai, India) or Difco Laboratories (Detroit, MI, USA). Wild-type strains were grown at 30°C unless otherwise indicated. Thermosensitive mutants were grown at 25°C, and then shifted to 37°C for 30 minutes before analysis. Molecular biology procedures were simulated and recorded using SnapGene software (GSL Biotech, Chicago, IL, USA; http://www.snapgene.com).
Integrating or centromeric plasmids were transformed into yeast using the lithium acetate method (Gietz and Woods, 2002), and chromosomal gene replacements were performed using the pop-in/pop-out method (Rothstein, 1991). SEC7 was tagged by gene replacement with three or six tandem copies of monomeric EGFP (mEGFP) (Zacharias et al., 2002) as previously described (Rossanese et al., 2001). Additional integrating constructs were used to overexpress either Sec7-3xmEGFP or Sec7-6xDsRed.M1 (Losev et al., 2006). Vrg4 was tagged at its N-terminus with sGFP by gene replacement (Losev et al., 2006). Sec13 was tagged at its C-terminus with EGFP by gene replacement (Rossanese et al., 1999). Ras2 was tagged at its N-terminus with mCherry (Shaner et al., 2004) as follows. The RAS2 gene plus flanking sequences were amplified using forward primer 5′-TCGATGAATTCTAGCTCTCGGGCGAATATC-3′ and reverse primer 5′-TCTAAGGGGAAAGAGAAGCTTG-3′. The PCR product was digested with EcoRI and HindIII and subcloned at the same sites in the centromeric URA3 plasmid YCplac33 (Gietz and Sugino, 1988). A BamHI-NotI cassette was then introduced by primer-directed mutagenesis at the RAS2 start codon. mCherry was amplified with primers 5′-tggtaGGATCCATGGTGAGCAAGGGCGAGGA-3′ and 5′-tgttcGCGGCCGCaCTTGTACAGCTCGTCCATGC-3′, and the PCR product was subcloned as a BamHI–NotI fragment. A BamHI–HindIII fragment encoding mCherry–Ras2 was then subcloned into the TRP1 integrating plasmid YIplac204-T/C to drive strong constitutive expression from the TPI1 promoter (Losev et al., 2006). This construct was linearized with EcoRV for integration at the TRP1 locus.
To complement the nmt1 mutation, the wild-type NMT1 gene with its 5′ and 3′ control regions was amplified with primers 5′-AAGTATAAGCTTGCTATCGTTTACAAATTAAA-3′ and 5′-CGCTGGGTACCATTTTTTCACTTCGTCAATTGAC-3′, and subcloned as HindIII–KpnI fragment into YCplac33. To overexpress ARF1, this gene with its 5′ and 3′ control regions was amplified with primers 5′-TGCACAGTTGAATTCTCG-3′ and 5′-GATGAAAGCTTCTGCAGTTGTCAGTC-3′, and subcloned as an EcoRI–HindIII fragment into the high-copy episomal URA3 plasmid YEplac195 (Gietz and Sugino, 1988). Where indicated, strains were cured of the URA3 plasmids by growth on plates containing 1 mg/ml 5-fluoroorotic acid (United States Biological, Salem, MA, USA).
To revert the T400I mutation in the nmt1 mutant, a fragment of the wild-type NMT1 gene was amplified with primers 5′-GCCACCAAGCTTACACCGGAAGATATATCTGA-3′ and 5′-CGCTGGGTACCATTTTTTCACTTCGTCAATTGAC-3′, and subcloned as a HindIII–KpnI fragment into a modified URA3 integrating plasmid YIplac211 (Gietz and Sugino, 1988) that had been mutagenized to remove the EcoRI site. One of the two EcoRI sites in the NMT1 fragment was then removed by changing codon 293 from GAA to GAG. The final construct was linearized at the unique EcoRI site for integrative gene replacement, and the reverted NMT1 allele was confirmed by PCR amplification and sequencing of genomic DNA. To delete the ARF1 gene, the kanMX cassette was amplified with the following primers to append sequences that matched the regions flanking the ARF1 open reading frame: 5′-ATTGAAGGTATAAGAAAGAACTCAAACAGGTTTAATAGAATTAAAACGTACGCTGCAGGTCGAC-3′ and 5′-TTCATTTAGTTTATACAAGCGTATTTGATCCATATTCTAGAATTTATCGATGAATTCGAGCTCG-3′. The resulting fragment was transformed into cells, which were plated on YPD medium containing 250 µg/ml G418 (Sigma-Aldrich, St. Louis, MO, USA) to select for double-crossover replacement of the ARF1 open reading frame (Wach et al., 1994).
Fluorescence image capture and processing
To capture static images of fluorescently tagged tER sites, live yeast cells were compressed beneath a coverslip and viewed using a Zeiss Axioplan 2 microscope with a 100× 1.4 NA objective. A single image plane was captured with a Hamamatsu digital camera. To quantify the number of tER sites, Z-stacks were captured with a Zeiss LSM 780 confocal microscope. Each confocal 3D data set was opened with Imaris software (Bitplane, Zurich, Switzerland). The spot detection option was used, with the threshold set to detect even the smallest fluorescent tER sites. The number of spots was then counted in randomly chosen full-size ‘mother’ cells.
To capture static images of cells labeled with FM 4-64, dye in a 1 mM DMSO stock solution was added to a log-phase culture at 23°C. After a brief incubation, non-internalized dye was quenched by adding excess SCAS (4-sulfonato calix[8]arene, sodium salt; Biotium, Hayward, CA, USA) from a 1 mM aqueous stock solution. Cells were then mixed with 3.0 µm polystyrene beads (Polysciences, Warrington, PA, USA), compressed beneath a coverslip that had been siliconized with Sigmacote (Sigma-Aldrich), and viewed with a Zeiss LSM 710. GFP fluorescence was excited with a 488-nm laser and collected between 495 and 550 nm, and simultaneously, FM 4-64 fluorescence was excited with a 561-nm laser and collected between 650 and 760 nm. A 3D image volume spanning an entire cell was captured using a pixel size of 40 nm and Z-stack spacing of 0.47 µm. Image stacks were converted to 16-bit and average projected using ImageJ (http://rsbweb.nih.gov/ij/).
Confocal imaging was performed with either a Leica SP5 or a Zeiss LSM 780 for 4D imaging, or a Zeiss LSM 510 META or LSM 710 for single time-point measurements, equipped with 100× or 63× 1.4 NA objectives. Cells grown to log phase at 25°C or 30°C in nonfluorescent or minimally fluorescent SD medium were immobilized on glass-bottomed dishes (Cell E&G, Houston, TX, USA) using concanavalin A (Sigma-Aldrich) as previously described (Bevis et al., 2002), and were imaged at room temperature. Single- or dual-color data sets were obtained using separate excitation and capture of red and green signals, with a pinhole of 1.0–1.2 AU and with line averaging of 4 to improve the signal-to-noise ratio. Transmitted light images were captured in the blue channel. The pixel size was 70–90 nm. Optical sections were 0.25–0.40 µm apart, and ∼15–20 optical sections were collected to span an entire cell. Z-stacks were collected at intervals of 2–4 seconds. To limit photodamage, laser illumination was minimized and confocal scans were carried out as quickly as possible.
Fluorescence micrographs were assembled using Adobe Photoshop, with uniform adjustments of brightness where appropriate. Projected movies of 4D confocal data sets were generated and quantified using ImageJ as follows. To remove shot noise, individual optical sections were processed with a custom plugin that implemented a 3D version of a 3×3 hybrid median filter (Hammond and Glick, 2000). The processed optical sections were then average projected and corrected for exponential photobleaching (Bevis et al., 2002), and the fluorescence and transmitted light channels were merged. Quantification of late Golgi fluorescence intensities was performed as previously described (Losev et al., 2006).
For measurements of cisternal size, a confocal 3D data set was opened with Imaris. After the image was cropped, the surface fill option was used to define a closed surface around each fluorescent structure of average diameter at least 0.5 µm, with objects being smoothed using a filter radius of 0.05 µm. The rendered 3D objects were then analyzed to obtain the desired parameters. To calculate the average volume of late Golgi cisternae, 60 cells were imaged for each strain, and all of the rendered cisternae were measured. The corresponding volume measurements of early Golgi cisternae were performed on 20 cells for each strain. To calculate the average X–Y-plane diameter, 10 cells were imaged for each strain, and the maximal X–Y-plane diameter was measured for three or four representative cisternae from each cell.
For measurements of maturation and homotypic fusion parameters, a confocal 4D data set was opened with Imaris. The image was exported to Fiji (http://fiji.sc/Fiji), and shot noise was removed by processing individual optical sections with a custom plug-in that implemented a 3D version of a 3×3 hybrid median filter with a tight mean. The processed optical sections were then corrected for exponential photobleaching, and the fluorescence and transmitted light channels were merged. This processed image was imported back into Imaris for visual inspection and manual quantification.
Quantitative analysis of maturation and homotypic fusion
All measurements were performed using the same set of 20 dual-color 5-minute 4D movies, with 10 movies each for wild-type and arf1Δ strains expressing GFP-Vrg4 and Sec7-DsRed. A single full-size ‘mother’ cell from each movie was analyzed using Imaris. To count early and late Golgi cisternae in a cell, the numbers of green and red spots in each movie were recorded at 10 successive time points and averaged, with a spot being recorded only if it persisted for at least two consecutive time points. To measure the maturation frequency, the number of maturation events in each movie was counted during each of minutes 2, 3 and 4, with a cisterna being tracked for additional time if needed to verify the maturation event. To measure the persistence times of early and late Golgi cisternae, 20 cisternae that could be seen to acquire and then lose either the early or the late Golgi marker were chosen at random from the 10 movies, and the fluorescence duration for each cisterna was measured. To measure the frequency of homotypic fusion, the number of early or late Golgi fusion events was counted during a 2-minute interval for each movie. Cisternae that underwent homotypic fusion were excluded from the measurements of persistence times.
Correlative fluorescence microscopy and electron tomography
Wild-type or arf1Δ strains carried a Sec7-3xmEGFP gene replacement plus an integrating vector that drove mild (∼3×) overexpression of Sec7-3xmEGFP (Losev et al., 2006). A 100 ml culture was grown in SD medium at 30°C to mid-log phase with shaking. Cells were then subjected to high-pressure freezing, freeze substitution, and embedding in plastic as described previously (Levi et al., 2010), except that glutaraldehyde was omitted during freeze substitution and the resin was Lowicryl K4M.
Embedded samples were trimmed and cut with a Leica EM UC6 ultramicrotome to produce ∼300 nm sections, which were placed on 200 mesh London Finder Formvar/carbon-coated copper grids (Electron Microscopy Sciences, Hatfield, PA, USA). To provide fiducial markers for both light and electron microscopy, 15 nm colloidal gold was applied to both sides of the grid as described previously (Levi et al., 2010). For light microscopy, a grid was placed on a glass slide with the resin side up, and a 22×22 mm number 1.5 glass coverslip with a 10-µl drop of 500 mM Na+HEPES, pH 7.5 was inverted onto the grid. After capture of fluorescence and differential interference contrast micrographs on a Zeiss Axioplan 2 with a 1.4 NA objective, the grid was retrieved and blotted dry. For embedded wild-type cells, the samples were post-stained for 8 minutes with 2% uranyl acetate in 70% methanol, followed by a water rinse, followed by 5 minutes with Reynold's lead citrate. Cells that showed promising morphologies and fluorescence patterns were analyzed by electron tomography as described previously (Levi et al., 2010). Golgi membrane contours were traced in the tomograms, and the structures were modeled.
Supplementary Material
Footnotes
Competing interests
The authors declare no competing interests.
Author contributions
O.W.R. provisionally identified the nmt1 mutant. M.B. and P.I. created yeast strains and acquired images for the nmt1 analysis, created the ARF1 overexpression strain, and re-created and modified arf1Δ strains for Golgi maturation studies. M.B., P.I., K. Pandya, B.K.J. and C.S. carried out quantitative analyses of the nmt1 strains. M.B. performed kinetic studies of Golgi maturation in wild-type and arf1Δ cells by collecting 4D movies and analyzing the data. M.B., P.I., K. Pandya, B.K.J., A.G. and K. Pawar measured persistence times, cisternal numbers, and other Golgi parameters. E.P. analyzed tER structure, and worked with K.J.D. on initial kinetic studies of arf1Δ cells. E.P. and J.A. performed correlative fluorescence and electron tomography. K.J.D. analyzed FM 4-64 localization. B.S.G. and D.B. supervised the project and assembled the manuscript and figures.
Funding
This work was supported by the Department of Biotechnology [grant number 102/IFD/SAN/2282/2012-2013 to D.B and C.S.]; the Council of Scientific and Industrial Research [doctoral fellowship Ref. no. 20-6/2009 (i) EU-IV to M.B.]; the Council of Scientific and Industrial Research grant [grant number 37(1553)112/EMR-II to K. Pandya]; an Advanced Centre for Treatment, Research and Education in Cancer doctoral fellowship to A.G., B.K.J. and P.I.; the National Institutes of Health [grant numbers R01 GM061156 and R01 GM104010 to B.S.G.]; and a National Institutes of Health training grant [grant number T32 GM007183 to K.J.D. and O.W.R.]. Deposited in PMC for release after 12 months.
Supplementary material available online at http://jcs.biologists.org/lookup/suppl/doi:10.1242/jcs.140996/-/DC1
References
- Bevis B. J., Hammond A. T., Reinke C. A., Glick B. S. (2002). De novo formation of transitional ER sites and Golgi structures in Pichia pastoris. Nat. Cell Biol. 4, 750–756 10.1038/ncb852 [DOI] [PubMed] [Google Scholar]
- Castillon G. A., Watanabe R., Taylor M., Schwabe T. M., Riezman H. (2009). Concentration of GPI-anchored proteins upon ER exit in yeast. Traffic 10, 186–200 10.1111/j.1600-0854.2008.00857.x [DOI] [PubMed] [Google Scholar]
- Chan D. C. (2012). Fusion and fission: interlinked processes critical for mitochondrial health. Annu. Rev. Genet. 46, 265–287 10.1146/annurev-genet-110410-132529 [DOI] [PubMed] [Google Scholar]
- Daboussi L., Costaguta G., Payne G. S. (2012). Phosphoinositide-mediated clathrin adaptor progression at the trans-Golgi network. Nat. Cell Biol. 14, 239–248 10.1038/ncb2427 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donaldson J. G., Jackson C. L. (2011). ARF family G proteins and their regulators: roles in membrane transport, development and disease. Nat. Rev. Mol. Cell Biol. 12, 362–375 10.1038/nrm3117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Epp N., Rethmeier R., Krämer L., Ungermann C. (2011). Membrane dynamics and fusion at late endosomes and vacuoles – Rab regulation, multisubunit tethering complexes and SNAREs. Eur. J. Cell Biol. 90, 779–785 10.1016/j.ejcb.2011.04.007 [DOI] [PubMed] [Google Scholar]
- Gaynor E. C., Chen C. Y., Emr S. D., Graham T. R. (1998). ARF is required for maintenance of yeast Golgi and endosome structure and function. Mol. Biol. Cell 9, 653–670 10.1091/mbc.9.3.653 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gietz R. D., Sugino A. (1988). New yeast-Escherichia coli shuttle vectors constructed with in vitro mutagenized yeast genes lacking six-base pair restriction sites. Gene 74, 527–534 10.1016/0378-1119(88)90185-0 [DOI] [PubMed] [Google Scholar]
- Gietz R. D., Woods R. A. (2002). Transformation of yeast by lithium acetate/single-stranded carrier DNA/polyethylene glycol method. Methods Enzymol. 350, 87–96 10.1016/S0076-6879(02)50957-5 [DOI] [PubMed] [Google Scholar]
- Glick B. S., Nakano A. (2009). Membrane traffic within the Golgi apparatus. Annu. Rev. Cell Dev. Biol. 25, 113–132 10.1146/annurev.cellbio.24.110707.175421 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goehring N. W., Hyman A. A. (2012). Organelle growth control through limiting pools of cytoplasmic components. Curr. Biol. 22, R330–R339 10.1016/j.cub.2012.03.046 [DOI] [PubMed] [Google Scholar]
- Guo Y., Linstedt A. D. (2006). COPII-Golgi protein interactions regulate COPII coat assembly and Golgi size. J. Cell Biol. 174, 53–63 10.1083/jcb.200604058 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hammond A. T., Glick B. S. (2000). Raising the speed limits for 4D fluorescence microscopy. Traffic 1, 935–940 [PubMed] [Google Scholar]
- Johnson D. R., Bhatnagar R. S., Knoll L. J., Gordon J. I. (1994). Genetic and biochemical studies of protein N-myristoylation. Annu. Rev. Biochem. 63, 869–914 10.1146/annurev.bi.63.070194.004253 [DOI] [PubMed] [Google Scholar]
- Jonikas M. C., Collins S. R., Denic V., Oh E., Quan E. M., Schmid V., Weibezahn J., Schwappach B., Walter P., Weissman J. S. et al. (2009). Comprehensive characterization of genes required for protein folding in the endoplasmic reticulum. Science 323, 1693–1697 10.1126/science.1167983 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kahn R. A. (2009). Toward a model for Arf GTPases as regulators of traffic at the Golgi. FEBS Lett. 583, 3872–3879 10.1016/j.febslet.2009.10.066 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kellokumpu S., Sormunen R., Kellokumpu I. (2002). Abnormal glycosylation and altered Golgi structure in colorectal cancer: dependence on intra-Golgi pH. FEBS Lett. 516, 217–224 10.1016/S0014-5793(02)02535-8 [DOI] [PubMed] [Google Scholar]
- Kirk S. J., Cliff J. M., Thomas J. A., Ward T. H. (2010). Biogenesis of secretory organelles during B cell differentiation. J. Leukoc. Biol. 87, 245–255 10.1189/jlb.1208774 [DOI] [PubMed] [Google Scholar]
- Kukulski W., Schorb M., Welsch S., Picco A., Kaksonen M., Briggs J. A. (2011). Correlated fluorescence and 3D electron microscopy with high sensitivity and spatial precision. J. Cell Biol. 192, 111–119 10.1083/jcb.201009037 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kunz J., Henriquez R., Schneider U., Deuter-Reinhard M., Movva N. R., Hall M. N. (1993). Target of rapamycin in yeast, TOR2, is an essential phosphatidylinositol kinase homolog required for G1 progression. Cell 73, 585–596 10.1016/0092-8674(93)90144-F [DOI] [PubMed] [Google Scholar]
- Levi S. K., Bhattacharyya D., Strack R. L., Austin J. R. I., 2nd, Glick B. S. (2010). The yeast GRASP Grh1 colocalizes with COPII and is dispensable for organizing the secretory pathway. Traffic 11, 1168–1179 10.1111/j.1600-0854.2010.01089.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levy D. L., Heald R. (2012). Mechanisms of intracellular scaling. Annu. Rev. Cell Dev. Biol. 28, 113–135 10.1146/annurev-cellbio-092910-154158 [DOI] [PubMed] [Google Scholar]
- Lewis M. J., Nichols B. J., Prescianotto-Baschong C., Riezman H., Pelham H. R. B. (2000). Specific retrieval of the exocytic SNARE Snc1p from early yeast endosomes. Mol. Biol. Cell 11, 23–38 10.1091/mbc.11.1.23 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Losev E., Reinke C. A., Jellen J., Strongin D. E., Bevis B. J., Glick B. S. (2006). Golgi maturation visualized in living yeast. Nature 441, 1002–1006 10.1038/nature04717 [DOI] [PubMed] [Google Scholar]
- Lu Z., Joseph D., Bugnard E., Zaal K. J. M., Ralston E. (2001). Golgi complex reorganization during muscle differentiation: visualization in living cells and mechanism. Mol. Biol. Cell 12, 795–808 10.1091/mbc.12.4.795 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marshall W. (2002). Size control in dynamic organelles. Trends Cell Biol. 12, 414–419 10.1016/S0962-8924(02)02341-3 [DOI] [PubMed] [Google Scholar]
- Marshall W. F. (2012). Organelle size control systems: from cell geometry to organelle-directed medicine. Bioessays 34, 721–724 10.1002/bies.201200043 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuura-Tokita K., Takeuchi M., Ichihara A., Mikuriya K., Nakano A. (2006). Live imaging of yeast Golgi cisternal maturation. Nature 441, 1007–1010 10.1038/nature04737 [DOI] [PubMed] [Google Scholar]
- Mogelsvang S., Marsh B. J., Ladinsky M. S., Howell K. E. (2004). Predicting function from structure: 3D structure studies of the mammalian Golgi complex. Traffic 5, 338–345 10.1111/j.1398-9219.2004.00186.x [DOI] [PubMed] [Google Scholar]
- Mowbrey K., Dacks J. B. (2009). Evolution and diversity of the Golgi body. FEBS Lett. 583, 3738–3745 10.1016/j.febslet.2009.10.025 [DOI] [PubMed] [Google Scholar]
- Papanikou E., Glick B. S. (2009). The yeast Golgi apparatus: insights and mysteries. FEBS Lett. 583, 3746–3751 10.1016/j.febslet.2009.10.072 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peyroche A., Courbeyrette R., Rambourg A., Jackson C. L. (2001). The ARF exchange factors Gea1p and Gea2p regulate Golgi structure and function in yeast. J. Cell Sci. 114, 2241–2253 [DOI] [PubMed] [Google Scholar]
- Preuss D., Mulholland J., Franzusoff A., Segev N., Botstein D. (1992). Characterization of the Saccharomyces Golgi complex through the cell cycle by immunoelectron microscopy. Mol. Biol. Cell 3, 789–803 10.1091/mbc.3.7.789 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rambourg A., Clermont Y. (1997). Three-dimensional structure of the Golgi apparatus in mammalian cells. The Golgi Apparatus Berger E G, Roth J, ed37–61Basel: Birkhäuser Verlag [Google Scholar]
- Rossanese O. W., Soderholm J., Bevis B. J., Sears I. B., O'Connor J., Williamson E. K., Glick B. S. (1999). Golgi structure correlates with transitional endoplasmic reticulum organization in Pichia pastoris and Saccharomyces cerevisiae. J. Cell Biol. 145, 69–81 10.1083/jcb.145.1.69 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rossanese O. W., Reinke C. A., Bevis B. J., Hammond A. T., Sears I. B., O'Connor J., Glick B. S. (2001). A role for actin, Cdc1p, and Myo2p in the inheritance of late Golgi elements in Saccharomyces cerevisiae. J. Cell Biol. 153, 47–62 10.1083/jcb.153.1.47 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rothstein R. (1991). Targeting, disruption, replacement, and allele rescue: integrative DNA transformation in yeast. Methods Enzymol. 194, 281–301 10.1016/0076-6879(91)94022-5 [DOI] [PubMed] [Google Scholar]
- Sengupta D., Linstedt A. D. (2011). Control of organelle size: the Golgi complex. Annu. Rev. Cell Dev. Biol. 27, 57–77 10.1146/annurev-cellbio-100109-104003 [DOI] [PubMed] [Google Scholar]
- Shaner N. C., Campbell R. E., Steinbach P. A., Giepmans B. N. G., Palmer A. E., Tsien R. Y. (2004). Improved monomeric red, orange and yellow fluorescent proteins derived from Discosoma sp. red fluorescent protein. Nat. Biotechnol. 22, 1567–1572 10.1038/nbt1037 [DOI] [PubMed] [Google Scholar]
- Sherman F. (1991). Getting started with yeast. Methods Enzymol. 194, 3–21 10.1016/0076-6879(91)94004-V [DOI] [PubMed] [Google Scholar]
- Shindiapina P., Barlowe C. (2010). Requirements for transitional endoplasmic reticulum site structure and function in Saccharomyces cerevisiae. Mol. Biol. Cell 21, 1530–1545 10.1091/mbc.E09-07-0605 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Staehelin L. A., Kang B. H. (2008). Nanoscale architecture of endoplasmic reticulum export sites and of Golgi membranes as determined by electron tomography. Plant Physiol. 147, 1454–1468 10.1104/pp.108.120618 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stalder D., Antonny B. (2013). Arf GTPase regulation through cascade mechanisms and positive feedback loops. FEBS Lett. 587, 2028–2035 10.1016/j.febslet.2013.05.015 [DOI] [PubMed] [Google Scholar]
- Stearns T., Kahn R. A., Botstein D., Hoyt M. A. (1990a). ADP ribosylation factor is an essential protein in Saccharomyces cerevisiae and is encoded by two genes. Mol. Cell. Biol. 10, 6690–6699 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stearns T., Willingham M. C., Botstein D., Kahn R. A. (1990b). ADP-ribosylation factor is functionally and physically associated with the Golgi complex. Proc. Natl. Acad. Sci. USA 87, 1238–1242 10.1073/pnas.87.3.1238 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Storrie B., Micaroni M., Morgan G. P., Jones N., Kamykowski J. A., Wilkins N., Pan T. H., Marsh B. J. (2012). Electron tomography reveals Rab6 is essential to the trafficking of trans-Golgi clathrin and COPI-coated vesicles and the maintenance of Golgi cisternal number. Traffic 13, 727–744 10.1111/j.1600-0854.2012.01343.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang X., Punch J. J., Lee W. L. (2009). A CAAX motif can compensate for the PH domain of Num1 for cortical dynein attachment. Cell Cycle 8, 3182–3190 10.4161/cc.8.19.9731 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Travers K. J., Patil C. K., Wodicka L., Lockhart D. J., Weissman J. S., Walter P. (2000). Functional and genomic analyses reveal an essential coordination between the unfolded protein response and ER-associated degradation. Cell 101, 249–258 10.1016/S0092-8674(00)80835-1 [DOI] [PubMed] [Google Scholar]
- Vida T. A., Emr S. D. (1995). A new vital stain for visualizing vacuolar membrane dynamics and endocytosis in yeast. J. Cell Biol. 128, 779–792 10.1083/jcb.128.5.779 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wach A., Brachat A., Pöhlmann R., Philippsen P. (1994). New heterologous modules for classical or PCR-based gene disruptions in Saccharomyces cerevisiae. Yeast 10, 1793–1808 10.1002/yea.320101310 [DOI] [PubMed] [Google Scholar]
- Warren G., Rothman J. (2011). The Golgi 322.Cold Spring Harbor, NY: Cold Spring Harbor Laboratory Press [Google Scholar]
- Weller S. G., Capitani M., Cao H., Micaroni M., Luini A., Sallese M., McNiven M. A. (2010). Src kinase regulates the integrity and function of the Golgi apparatus via activation of dynamin 2. Proc. Natl. Acad. Sci. USA 107, 5863–5868 10.1073/pnas.0915123107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Westermann B. (2010). Mitochondrial dynamics in model organisms: what yeasts, worms and flies have taught us about fusion and fission of mitochondria. Semin. Cell Dev. Biol. 21, 542–549 10.1016/j.semcdb.2009.12.003 [DOI] [PubMed] [Google Scholar]
- Wooding S., Pelham H. R. B. (1998). The dynamics of golgi protein traffic visualized in living yeast cells. Mol. Biol. Cell 9, 2667–2680 10.1091/mbc.9.9.2667 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu C. C., Yates J. R., 3rd, Neville M. C., Howell K. E. (2000). Proteomic analysis of two functional states of the Golgi complex in mammary epithelial cells. Traffic 1, 769–782 10.1034/j.1600-0854.2000.011004.x [DOI] [PubMed] [Google Scholar]
- Zacharias D. A., Violin J. D., Newton A. C., Tsien R. Y. (2002). Partitioning of lipid-modified monomeric GFPs into membrane microdomains of live cells. Science 296, 913–916 10.1126/science.1068539 [DOI] [PubMed] [Google Scholar]
- Zink D., Fischer A. H., Nickerson J. A. (2004). Nuclear structure in cancer cells. Nat. Rev. Cancer 4, 677–687 10.1038/nrc1430 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.