

Abstract
Tumor-associated carbohydrate antigens (TACAs) are useful targets for the development of cancer vaccines or immunotherapies. However, a major obstacle in this application of TACAs is their poor immunogenicity. To overcome the problem, a new immunotherapeutic strategy combining synthetic vaccines made of artificial TACA derivatives and metabolic glycoengineering of cancer cells to express the artificial TACA derivatives was explored. Using a murine leukemia model FBL3 with GM3 antigen as the target, it was shown that artificial GM3 N-phenylacetyl derivative (GM3NPhAc) elicited robust antigen-specific T cell-dependent immunity and that N-phenylacetyl-d-mannosamine (ManNPhAc) as the biosynthetic precursor of GM3NPhAc selectively glycoengineered cancer cells to express GM3NPhAc both in vitro and in vivo. It was also demonstrated that GM3NPhAc-specific antisera and antibodies mediated strong cytotoxicity to ManNPhAc-treated FBL3 cell. Furthermore, vaccination with a conjugate vaccine made of GM3NPhAc followed by ManNPhAc treatment could significantly suppress tumor growth and prolong the survival of tumor-bearing mouse. These results have proved the feasibility of the new cancer immunotherapeutic strategy, as well as its efficacy to cure cancer, which is of general significance.
Keywords: Cancer immunotherapy, Cancer vaccine, Tumor-associated carbohydrate antigen, GM3 antigen, Cell metabolic glycoengineering
Introduction
Tumor-associated carbohydrate antigens (TACAs) are the unique or overexpressed glycans on the cancer cell surface, which are useful targets in the development of therapeutic cancer vaccines or cancer immunotherapies [1, 2]. However, TACAs are typically poorly immunogenic. A widely employed strategy to deal with this problem for the development of functional cancer vaccines is coupling TACAs to a protein [3, 4] or other carrier molecules [5–11] to form more immunogenic glycoconjugates that can provoke the cancer patients’ immune system for cancer therapy. Several glycoconjugate cancer vaccines have shown promising therapeutic potentials, and some, such as the keyhole limpet hemocyanin (KLH) conjugates of GM2 [12] and sTn [13] antigens, even reached the stage of Phase III clinical trials [14]. Unfortunately, so far no TACA-based cancer vaccine has met the desired treatment endpoint and overall survival rate in clinical trials. A major issue is that these vaccines usually elicit B cell-mediated immunity, instead of the desirable T cell-mediated immunity, in cancer patients [13, 15, 16], for which an important reason is that cancer patients became tolerant to the vaccines.
To overcome this problem and develop useful TACA-based cancer immunotherapies, we have recently explored a novel strategy that combines synthetic cancer vaccines made of unnatural TACA derivatives and metabolic cancer cell glycoengineering to express the designed TACA derivatives [17–21]. Its basic principle is depicted in Fig. 1 [20]. First, a glycoconjugate vaccine made of an unnatural derivative of a TACA is prepared and used to immunize animal. After a robust immune response specific for the unnatural TACA derivative is established, the animal is treated with an identically modified biosynthetic precursor of the target TACA to induce the expression of the unnatural TACA derivative in place of the natural TACA on the cancer cell surface. Subsequently, the provoked immune system will recognize and eradicate the glycoengineered tumor cells.
Fig. 1.

A new strategy for cancer immunotherapy based on cancer cell glycoengineering
Our previous studies have revealed that unnatural TACA derivatives, in particular those with unnatural N-phenylacetyl sialic acid residue, are significantly more immunogenic than the natural counterparts and can form effective vaccines that induce T cell-mediated immune response [17, 18, 22, 23]. We have also shown with several murine and human tumor cell lines that cancer cells can be metabolically glycoengineered to express unnatural TACA derivatives in place of natural TACAs by using modified monosaccharides as precursors [19, 21]. The aim of the present work is to verify in vivo the new immunotherapeutic strategy and evaluate its therapeutic efficacy to treat cancer.
For this purpose, we selected GM3, a sialylated trisaccharide TACA, as target antigen. GM3 is significantly overexpressed by a number of tumors, such as melanoma, leukemia, breast carcinoma, pulmonary cancer, and prostatic carcinoma [24–27]. In previous studies [17], we have demonstrated that unnatural GM3 derivatives, in particular N-phenylacetyl GM3 (GM3NPhAc), were much more immunogenic than the native GM3 and that conjugate vaccines made of GM3NPhAc could provoke both IgM and IgG antibody responses potentially useful for cancer immunotherapy. We have also demonstrated with a murine melanoma cell line B16F0 that cancer cells could be metabolically glycoengineered to express GM3NPhAc when it was treated with N-phenylacetyl-d-mannosamine (ManNPhAc) and that GM3NPhAc-provoked mouse antisera could selectively target metabolically glycoengineered cancer cells [19, 21]. In this research, we employed a murine leukemia cell line FBL3 to investigate the metabolically glycoengineered expression of GM3NPhAc on leukemia cell, both in vitro and in vivo, and more importantly the efficacy of the new immunotherapy, that is, immunization of mice with GM3NPhAc followed by ManNPhAc administration, to treat cancer.
Results
In vitro and in vivo studies of cancer cell metabolic glycoengineering
Metabolic glycoengineering of FBL3 cell in vitro
A murine leukemia cell line FBL3 was used to investigate in vitro the metabolically engineered expression of GM3NPhAc on cancer cell surface as a result of ManNPhAc treatment. In these studies, FBL3 cancer cells were first incubated with various concentrations of ManNPhAc for 24, 48, and 72 h, respectively, and subsequently treated with a GM3NPhAc-specific monoclonal antibody (mAb) 2H3 [19]. Finally, antibodies bound to the cancer cell surface were detected by enzyme-linked immunosorbent assay (ELISA) using alkaline phosphatase-linked goat anti-mouse IgM antibody as the secondary antibody to determine the levels of GM3NPhAc expression on the cancer cell, as reflected by OD values at 450 nm. As shown in Fig. 2, whereas incubating FBL3 cell with ManNPhAc for a short period (24 h) did not result in obvious GM3NPhAc expression, at prolonged incubation time (48 and 72 h), significant expression of GM3NPhAc (P < 0.05) on the cell surface was observed with 0.1 mM and higher concentrations of ManNPhAc. Moreover, it is evident that the GM3NPhAc expression level was dependent upon ManNPhAc concentration and incubation time, namely that higher ManNPhAc concentrations and longer incubation time constantly resulted in higher levels of GM3NPhAc expression. These results suggested that FBL3 cell did express GM3 antigen and that ManNPhAc treatment could effectively engineer FBL3 cell to express GM3NPhAc.
Fig. 2.

Expression levels of GM3NPhAc on FBL3 cells treated with ManNPhAc. After cells were incubated with 0, 0.02, 0.1, 0.5, and 2.0 mM of ManNPhAc for indicated time (24, 48, and 72 h), the cells were analyzed by ELISA using mAb 2H3 and alkaline phosphatase-linked goat anti-mouse IgM antibody as primary and secondary antibodies, respectively. GM3NPhAc levels were presented as the mean OD values at 450 nm obtained from triplicate experiments. *P < 0.05, # P < 0.001 (vs control)
Metabolic glycoengineering of FBL3 cell in vivo
Immunohistochemical (IHC) assay was used to study in vivo the glycoengineered expression of GM3NPhAc by mouse tumor and normal tissues resulting from ManNPhAc treatment. A group of five C57BL/6 mice were inoculated with FBL3 cell and treated with daily intraperitoneal (i.p.) injection of ManNPhAc. The mice were then euthanized, and their tumors, as well as the normal tissues of their lungs, livers, hearts, and kidneys, were collected and subjected to IHC assay. The GM3NPhAc-specific mAb 2H3 was utilized to stain the tissues. Figure 3 shows the representative samples of five replicated IHC experiments. Evidently, abundant GM3NPhAc antigens were present on the tumor tissues (Fig. 2, panel A), whereas GM3NPhAc was not detectable on the normal tissues of the lungs (panel B), livers (panel C), hearts (panel D), and kidneys (panel E) from the same mice. These results suggest that tumor cells were more effectively engineered to express GM3NPhAc in vivo than normal tissues, which forms the foundation for the new cancer immunotherapy to selectively target tumors.
Fig. 3.

Results of ICH assays of GM3NPhAc expression on tumor tissues, as well as on normal tissues of the lungs, liver, heart, and kidney, of mice treated with ManNPhAc. For the detection of GM3NPhAc, tissue sections were deparaffinized and stained with GM3NPhAc-specific mAb 2H3 (lyophilized powder, 200 μg/mL). Shown are representative examples of five replicated IHC staining experiments. The acquired images were captured at 200 magnifications
In vitro studies of antibody-dependent cell-mediated cytotoxicity (ADCC) and antibody-mediated complement-dependent cytotoxicity (CDC) to metabolically glycoengineered cancer cells
To study in vitro whether the GM3NPhAc-provoked immune responses or antibodies, such as mAb 2H3, are useful for cancer immunotherapy, we assessed their capacity to mediate the killing of metabolically glycoengineered cancer cells through the analysis of ADCC and antibody-mediated CDC. In these studies, cytotoxicity was expressed in cell lysis percentage determined by the lactate dehydrogenase (LDH) assay. For ADCC experiments, peritoneal macrophages isolated from healthy mouse were used as effectors, and FBL3 cells incubated with 0, 0.01, 0.02, 0.04, 0.08, and 0.16 mM of ManNPhAc were the target cells. As depicted in Fig. 4a, in the presence of mAb 2H3, mouse peritoneal macrophages started to exhibit obvious cytotoxicity to FBL3 cells treated with a low concentration of ManNPhAc (0.01 mM), and the ADCC was correlated well to the ManNPhAc concentration and reached a plateau with 0.1 mM of ManNPhAc. Moreover, we found that all of the cancer cells cultured with high concentrations (≥0.08 mM) of ManNPhAc were killed in this study, but as discussed previously [19], due to the limitation of the assessing method employed to evaluate cell lysis, only 80% cell lysis was shown at the plateau. For antibody-mediated CDC assays, rabbit complements were used as effectors. The results shown in Fig. 4b were very similar to those of the ADCC experiments, namely that obvious CDC to FBL3 cells started to occur at a low concentration of ManNPhAc (0.01 mM) and reached a plateau at ca. 0.08 mM of ManNPhAc.
Fig. 4.

MAb 2H3 (a, b) and GM3NPhAc-inoculated mouse antiserum (c, d) mediated ADCC and CDC to FBL3 cells incubated with 0, 0.01, 0.02, 0.04, 0.08, and 0.16 mM of ManNPhAc, respectively. ADCC assays were performed using peritoneal macrophages of healthy mouse as effectors with an effector/target cell (FBL3) ratio of 100:1. For antibody-mediated CDC assays, rabbit complements were used as effectors. Cell lysis was evaluated by LDH assays. The results represent the mean (±SE) of triplicate experiments
We also evaluated the capacity of antisera derived from GM3NPhAc-KLH-vaccinated mice to mediate the killing of glycoengineered cancer cells. The antisera used in this research were prepared from mice according to a previously reported procedure [17]. ADCC and antibody-mediated CDC assays were carried out by the same protocols as described above except for replacing mAb 2H3 with antisera. Clearly, the anti-GM3NPhAc antisera mediated significant cytotoxicity (up to 80%) to FBL3 cells treated with various concentrations of ManNPhAc (Fig. 4c, d). The results were very similar to that obtained with mAb 2H3.
Evaluation of the mouse T cell response to GM3NPhAc-KLH vaccination
The evaluation was performed by assaying cytotoxic T lymphocyte (CTL) activity and by enzyme-linked immunosorbent spot (ELISPOT) assay of IFN-γ-releasing lymphocytes. For CTL assays, splenocytes were isolated from GM3NPhAc-KLH-immunized mice and evaluated as effector lymphocytes to elicit cancer cell lysis. As shown in Fig. 5a, the isolated splenocytes from GM3NPhAc-KLH-immunized mice exhibited significantly higher cytotoxicity to metabolically glycoengineered FBL3 cell (43 ± 2%), compared with that of the control group mice (3 ± 0.7%). IFN-γ ELISPOT assays also revealed significantly increased CTL activity in mice immunized with GM3NPhAc-KLH. As revealed in Fig. 5b, for mice immunized with GM3NPhAc-KLH, the number of IFN-γ-releasing splenocytes (93.83 ± 33.87/106) was obviously increased compared to that (39 ± 3.54/106) of mice in the control group. Immunization of mice with GM3NPhAc-KLH followed by ManNPhAc treatment, which was used to metabolically glycoengineer cancer cells to express GM3NPhAc, could further increase the number of IFN-γ-releasing splenocytes (163.83 ± 41.78/106). These results indicate that GM3NPhAc-KLH immunization combined with GM3NPhAc stimulation could establish strong T cell-mediated immunity that is important for cancer immunotherapy.
Fig. 5.

Results of the CTL and ELISPOT IFN-γ-releasing lymphocyte assays. a CTL assays: in vitro cytotoxicity of splenocytes obtained from mice treated with PBS (the control) or with GM3NPhAc-KLH toward FBL3 cells incubated with ManNPhAc. CTL responses were expressed as cell lysis rates determined by the LDH assay. *P < 0.001. b ELISPOT assays of the number of IFN-γ-releasing lymphocytes among 1 × 106 splenocytes derived from mice treated with PBS, GM3NPhAc-KLH plus PBS, and GM3NPhAc-KLH plus ManNPhAc, respectively. *P < 0.05, when compared with the control
In vivo evaluation of the new cancer immunotherapy
C57BL/6 mouse was utilized for evaluating the efficacy of the new immunotherapy to inhibit tumor growth or prolong the survival of tumor-bearing animals. For the former, the evaluation was carried out according to the experimental design outlined in Fig. 6. In the treatment group, mice were first immunized with conjugate vaccine GM3NPhAc-KLH according to the reported protocol [17]. It was shown that all of the mice invariably produced high levels of GM3NPhAc-specific total antibodies, and antibody isotype analysis revealed that the mouse antisera contained not only IgM antibodies (~45,000) but also high titers of IgG1 (~19,000), IgG2a (~32,000), and IgG3 (~25,000) antibodies, suggesting the induction of strong GM3NPhAc-specific T cell-dependent immunity. The results were consistent with that reported previously [17]. After a robust anti-GM3NPhAc immune response was affirmed, the mice were inoculated with FBL3 cell, followed by daily i.p. injection of ManNPhAc (50 mg/kg/day) for 7 days. The dosage utilized here was determined according to the cell glycoengineering results discussed above and was significantly lower than that of N-propionyl mannosamine (200 mg/kg/day) used in previous studies [28]. Meanwhile, two control groups were designed, in which the mice received GM3NPhAc-KLH plus PBS or PBS only. Tumors developed in these mice were monitored and measured regularly, and on day 56, all of the mice were euthanized, and their tumors were subjected to terminal examination.
Fig. 6.

Schematic representation of the mouse treatment schedule
The results are shown in Fig. 7a. Clearly, tumors in the treatment group (mean size on day 56: 130.32 ± 38.52 mm3) were significantly smaller (P < 0.001) than that in both control groups (418.26 ± 63.5 mm3 in PBS-only group and 345.75 ± 46.75 mm3 in GM3NPhAc-KLH/PBS group). The difference between the two control groups was not statistically significant. The results suggested that GM3NPhAc-KLH immunization combined with cell metabolic glycoengineering with ManNPhAc could effectively inhibit FBL3 tumor growth in vivo, but GM3NPhAc-KLH immunization alone did not have a significant influence on the tumor growth.
Fig. 7.

Evaluation of the new immunotherapy to treat FBL3 cancer. a Tumor sizes and growth rates in mice treated with PBS, GM3NPhAc-KLH/PBS, and GM3NPhAc-KLH/ManNPhAc, respectively. *P < 0.001, as compared to the other two treatment groups. b Survival time of tumor-bearing mice treated with PBS, GM3NPhAc-KLH/PBS, and GM3NPhAc-KLH/ManNPhAc, respectively. The average survival time of mice treated with GM3NPhAc-KLH/ManNPhAc (>40.1 days) was significantly longer than that of the other two groups (25.3 days for PBS group, 32.0 days for GM3NPhAc-KLH/PBS group)
To evaluate the efficacy of the new immunotherapy to treat cancer, we further investigated the survival of tumor-bearing animals. In this case, the mice were not subjected to terminal euthanasia after FBL3 cancer cell inoculation and ManNPhAc treatment. Again, two control groups were set, in which mice received GM3NPhAc-KLH plus PBS or PBS alone. As shown in Fig. 7b, mice in the PBS group started to die on day 19 after cancer cell challenge, and all of the mice died on day 32. However, all of the mice in the treatment group (GM3NPhAc-KLH plus ManNPhAc) were alive on day 32, and at the end of the experiment, that is on day 46, there were still 30% of mice that were surviving. The average survival time for the GM3NPhAc-KLH/ManNPhAc group (40.1 days) was significantly (P < 0.05) longer than that for both the GM3NPhAc-KLH/PBS group (32.0 days) and the PBS group (25.3 days). Furthermore, the average survival time for the GM3NPhAc-KLH/PBS group was also significantly longer than that for the PBS group (P < 0.05). These data suggested that either GM3NPhAc-KLH alone or GM3NPhAc-KLH plus ManNPhAc treatment could significantly prolong the survival of tumor-bearing mice. It was interesting to observe that the GM3NPhAc-KLH plus PBS group mice also showed prolonged survival time than the PBS-only group, which indicated that GM3NPhAc-KLH immunization alone had some inhibitory effect on tumor growth. Moreover, no remarkable treatment-related toxicities affecting the weight or the general behavior of mice were observed in these experiments.
Discussion
Immunotherapy is considered an ideal means for cancer therapy and has attracted significant attentions in recent decades. TACAs, the products of tumor cell aberrant glycosylations, are broadly expressed by various tumors and are useful targets for the development of cancer immunotherapies. However, the problem of immunotolerance to TACAs remains a main obstacle in the development of carbohydrate-based cancer vaccines. For example, GM3, a TACA overexpressed by a number of tumors, is also expressed by some normal tissues, albeit in very low concentrations, so GM3 is only weakly immunogenic. Our previous studies have shown that some unnatural GM3 derivatives, such as GM3NPhAc, could elicit robust T cell-mediated immune response and thus constitute potentially useful cancer vaccines. This discovery forms one of the important foundations for our new strategy for cancer immunotherapy based on joint application of synthetic vaccines made of unnatural TACA derivatives and cancer cell metabolic glycoengineering.
In the present work, we have demonstrated with FBL3 cell and by means of ELISA and IHC assay that ManNPhAc could be utilized to effect tumor cell metabolic glycoengineering, both in vitro and in vivo, for the expression of GM3NPhAc in place of native GM3. As a result, tumor cells may be metabolically glycoengineered for specific targeting by the immune system and for immunotherapy. It was further demonstrated that tumor cells, but not normal cells, could be effectively metabolically engineered in vivo to express GM3NPhAc (Fig. 2). These results suggest that normal tissues may not be obviously affected by ManNPhAc treatment, although some tissues may express a low level of GM3; therefore, the new immunotherapy can be rather selective. This hypothesis was supported by the fact that GM3NPhAc-KLH and ManNPhAc treatments did not cause significant side effects in mice. More importantly, our ADCC and CDC studies have shown that the GM3NPhAc-specific mAb 2H3 and the antisera derived from GM3NPhAc-KLH-inoculated mice could mediate high cytotoxicity to ManNPhAc-treated cancer cells but not to normal cells under the same conditions. These studies form the other important foundation for the new cancer immunotherapy.
Furthermore, we have demonstrated that vaccinating mice with GM3NPhAc-KLH could elicit functional CTLs that killed tumor cells efficiently and could also elicit IFN-γ-releasing splenocytes as determined by ELISPOT assay. The induction of functional CTLs and IFN-γ-releasing lymphocytes is a solid proof for antigen-specific T cell-mediated immunity, which is critical for effective cancer immunotherapy. Ultimately, we have verified that the new cancer immunotherapy, which combines GM3NPhAc-KLH vaccination with ManNPhAc treatment, could significantly inhibit tumor growth and prolong the survival time of cancer-bearing mice. Thus, the results have proved the feasibility of the novel cancer immunotherapy, as well as its efficacy to cure cancer. It was further observed that GM3NPhAc-KLH vaccination alone could also inhibit tumor growth and prolong the survival time of tumor-bearing mice, but to a lesser extent than that with GM3NPhAc/ManNPhAc treatment. This may result from nonspecific reactions of the immune system that was stimulated by vaccination.
In conclusion, we have proved that GM3NPhAc-KLH could stimulate robust T cell-mediated immunity in mice and that cancer cell FBL3 could be effectively and selectively glycoengineered in vivo through i.p. injection of ManNPhAc to the mouse. We have also demonstrated in vitro that the stimulated immune system could selectively target and kill metabolically glycoengineered cancer cells and in vivo that treatment of the cancer-bearing mice with GM3NPhAc-KLH and ManNPhAc could significantly suppress tumor growth and prolong animal survival. These results have verified the promise of the new immunotherapeutic strategy to circumvent the immunotolerance problem as well as its potential for effective cancer therapy. Currently, additional animal studies of the new immunotherapy and detailed investigations to optimize the treatment schemes are underway. It is worth pointing out that this new immunotherapeutic strategy is not limited to GM3 antigen and the related tumors; it is theoretically applicable to other tumors expressing sialo-TACAs, which are rather common.
Materials and methods
Reagents
GM3NPhAc-KLH and GM3NPhAc-HSA conjugates, GM3NPhAc-specific mAb 2H3, and ManNPhAc were prepared according to previously reported methods [17, 19]. The LDH assay kit was purchased from Takara Bio Inc. Freund’s complete adjuvant and rabbit complement sera were purchased from Sigma–Aldrich. Cell culture media RPMI-1640 and the Dulbecco’s modified Eagle’s medium (DMEM), and bovine fetal serum (FBS) were purchased from Gibco. Alkaline phosphatase-linked goat anti-mouse Kappa, IgM, IgG1, IgG2a, and IgG3 antibodies were purchased from Immunology Consultants Laboratory, Inc., Newberg, USA. p-Nitrophenyl phosphate (PNPP) substrate was purchased from Sigma.
Cell lines and animals
FBL3 is an F-mulv-induced leukemia cell line of C57BL mouse and was purchased from ATCC. If not otherwise specified, in vitro culturing of cells was performed in RPMI-1640 supplemented with 10% heat-inactivated FBS, 100 U/mL of penicillin, and 100 μg/mL of streptomycin at 37°C. Female C57BL/6 mice of 6–8 weeks of age were used in this study and were allowed for free access to food and water. The animal experiments and protocols were approved by the animal use and care committee.
Analysis of cell metabolic glycoengineering by cellular ELISA
After cells were cultured in RPMI-1640 containing various concentrations (0, 0.02, 0.1, 0.5, and 2.0 mM) of ManNPhAc for 24, 48, and 72 h, respectively, the cells were trypsinized, counted, and loaded onto 96-well microplates (1.5 × 104 cells/well in 100 μL medium), and the plates were incubated at 37°C overnight. The cells were then washed with DMEM and treated with 100 μL of anti-GM3NPhAc mAb 2H3 (1:10 diluted cell culture supernatant) in DMEM at 37°C for 2 h. The cells were washed again and incubated with 100 μL of goat anti-mouse IgM antibody (1:1000 in RPMI-1640) at 37°C for 1 h. Thereafter, the cells were washed, and to the washed cells was added PNPP, followed by incubation at room temperature for 30 min and colorimetric readout using a microplate reader at 450 nm wavelength. These experiments were repeated three times, and the mean values of the triplicate experiments were presented.
Immunization and immunotherapy of mice
Several groups of female C57BL/6 mice (five per group) were vaccinated on day 1 by subcutaneous (s.c.) injection of a mixture of the Freund’s complete adjuvant (0.1 mL) and the conjugate vaccine GM3NPhAc-KLH (containing 2 μg of GM3NPhAc, in 0.1 mL of PBS). In the control groups, mice received a mixture of the Freund’s complete adjuvant and GM3-KLH conjugate (containing 2 μg of GM3, in 0.1 mL of PBS) or PBS only, respectively. These mice were then immunologically boosted twice with the same preparations 2 and 3 weeks after the initial immunization, that is, on day 14 and day 21, respectively. The mice were bled by tail vein on day 0 prior to the initial immunization and on day 28 after the second booster immunization, and the blood samples were clotted to prepare mouse sera. The day 0 mouse sera and sera from mice treated with PBS only were used as controls, while the day 28 sera were used to evaluate the immunological responses of mice to GM3NPhAc-KLH and GM3-KLH by ELISA of antigen-specific antibody titers. In the meantime, on day 28, approximately 5 × 105 FBL3 cells in 0.1 mL of PBS were injected into the right flank of each mouse, which was followed by daily i.p. injection of ManNPhAc (50 mg/kg/day in 0.1 mL of PBS) or PBS (0.1 mL) for 7 days. On day 56, the mice were euthanized for a series of analysis described below.
Immunohistochemical assay of GM3NPhAc expression on tumor and normal tissues
After the groups of mice treated with ManNPhAc were killed, their tumors, lungs, spleens, hearts, and kidneys were harvested and subjected to IHC assay. For IHC assays, the harvest tissues and organs were fixed in formalin, embedded in paraffin, and then sectioned. The sections were deparaffinized and treated with anti-GM3NPhAc mAb 2H3 at 4°C overnight [19]. After 3 times of washing with PBS containing 0.02% Tween 20, the slides were incubated with the horseradish peroxidase-labeled polymer-conjugated secondary antibody and then stained with diaminobenzidene (DAB). Finally, the slides were rinsed in distilled water and counterstained with hematoxylin. Thereafter, the sections were dehydrated in ethanol, cleared with xylene, and observed under a microscope.
ADCC assay
FBL3 cells were cultured with 0, 0.01, 0.02, 0.04, 0.08, and 0.16 mM of ManNPhAc for 72 h before they were loaded onto 96-well microplates (1.5 × 104 cells/well). The plates were incubated at 37°C overnight, washed by DMEM, and then treated with mAb 2H3 (1:10 dilution in DMEM, 100 μL) or with antiserum derived from GM3NPhAc-KLH-immunized mice (1:20 dilution in DMEM) at 37°C for 2 h. Thereafter, peritoneal macrophages isolated from healthy mice were introduced to each well as effectors with an effector/target (FBL3) cell ratio of 100:1. Then, the plates were incubated at 37°C for another 18 h before the cell supernatants were isolated and used to detect cell lysis by means of LDH assay according to the manufacture’s protocol. Briefly, after each plate was centrifugated at 300×g for 5 min, 20 μL of the cell-free supernatants was carefully transferred to the corresponding wells of another 96-well plate containing 80 μL of PBS and 100 μL of LDH assay reagents in each well. The plates were incubated at room temperature for 30 min. The absorptions (A) of these plates were read at 490 nm wavelength using a microplate reader. In the meantime, spontaneous LDH release and maximum LDH release of the cells were obtained by replacing the mAb or antisera with 100 μL of DMEM or 100 μL of 1% tritone X-100 in PBS, respectively, in the above-described protocol. The percentage of cell lysis was calculated by the following equation:
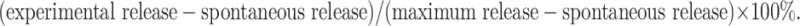 |
Antibody-mediated CDC assay
The experimental procedure for CDC assay was exactly the same as that of ADCC assay, except that after mAb 2H3 or anti-GM3NPhAc serum was introduced, the rabbit complement serum (1:10 in DMEM), rather than peritoneal macrophages, was employed to target cancer cells and mediate cell lysis that was also analyzed by LDH assay.
CTL assay
After the two groups of mice that were treated with GM3NPhAc-KLH/FBL3 cells and PBS/FBL3 cells were killed, their spleens were harvested for the preparation of splenocytes after lysis of the red blood cells with 0.84% ammonium chloride. These splenocytes were cultured in complete medium containing GM3NPhAc-KLH (2 μg/mL) for 24 h and then used as effector cells for CTL assays. The splenocytes were co-cultured with FBL3 cells (target cells) incubated with ManNPhAc (0.1 mM) at 100:1 in RPMI 1640 plus 10% fetal bovine serum for 24 h. Finally, the effector cell-mediated cytotoxicities to target cells were determined by LDH assay as described above. The spontaneous and maximum LDH release values were determined by incubating cancer cells in DMEM alone or in DMEM containing 1% Triton X-100, respectively, in the absence of splenocytes.
ELISPOT assay
The IFN-γ-producing splenocytes in mice inoculated with vaccine and FBL3 cell were analyzed by ELISPOT assay according to the manufacturer’s protocol (Dakewe). In brief, after the mice treated with GM3NPhAc-KLH and FBL3 cell were killed on day 56, the suspensions of their splenocytes were prepared as described above. Then, 1 × 106 splenocytes in 100 μL of DMEM were added to ImmunoSpot plates that were precoated with anti-IFN-γ antibody specific for IFN-γ ELISPOT. After incubation at 37°C for 18 h, the plates were washed with deionized water and then with PBS containing 0.05% Tween 20 three times. The plates were incubated with biotin-conjugated anti-IFN-γ mAb at 37°C for 1 h, followed by washing with buffer (PBS + 0.05% Tween 20) three times. Streptavidin–alkaline phosphatase was added to each well of the plates, which were incubated for 1 h at room temperature. Thirty microliters of the activator solution (supplied in ImmunoSpot kit, Dakewe) was added to each well of the plates for spot development at room temperature for 30 min. The reaction was quenched by washing with water. After being air dried, the number of spots in each well of the plates was counted using a microplate reader. These experiments were repeated in triplicate.
Evaluation of the in vivo efficacy of the new cancer immunotherapy to treat cancer
After the inoculation of mice with FBL3 cells, the tumor size in each mouse of all the therapeutic and control groups was monitored and measured regularly with a caliper before their euthanasia on day 56. The size of each tumor was terminally determined. Tumor size was calculated using the formula 0.4 × (A2 × B) (where B represents the largest diameter and A the diameter perpendicular to B). On the other hand, some other groups of the mice with the same treatment schemes (both the therapeutic and the control groups) were not euthanized and used to evaluate the impact of the immunotherapy on the survival of animals suffering from cancer. These animals were kept under close observation until they died of cancer or their tumors reached the size of 700 mm3. The animals of the latter were then euthanized but also considered as died of cancer.
Statistical methods
Unpaired Student’s t tests were performed to analyze the tissue GM3NPhAc expression, tumor size, and ELISPOT data. Kaplan–Meier analyses were preformed to evaluate the data of animal survival experiments. P < 0.05 is considered as statistically significant.
Acknowledgments
This work was supported in part by the National Institutes of Health of USA (R01CA095142), National Natural Science Foundation of China (30728032), and Non-governmental International Science and Technology Cooperation Project of Shanghai, China (10410703100).
Contributor Information
Junping Zhang, Phone: +86-21-81871328, FAX: +86-21-81871328, Email: jpzhang08@hotmail.com.
Zhongwu Guo, Phone: +1-313-577-2557, FAX: +1-313-577-8822, Email: zwguo@chem.wayne.edu.
References
- 1.Dube DH, Bertozzi CR. Glycans in cancer and inflammation-potential for therapeutics and diagnostics. Nat Rev Drug Discov. 2005;4:477–488. doi: 10.1038/nrd1751. [DOI] [PubMed] [Google Scholar]
- 2.Wang Q, Guo Z. Recent development in carbohydrate-based cancer vaccines. Curr Opin Chem Biol. 2009;13:608–617. doi: 10.1016/j.cbpa.2009.08.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Livingston PO. Approaches to augmenting the immunogenicity of melanoma gangliosides: from the whole melanoma cells to ganglioside-KLH conjugate vaccines. Immunol Rev. 1995;145:147–156. doi: 10.1111/j.1600-065X.1995.tb00080.x. [DOI] [PubMed] [Google Scholar]
- 4.Danishefsky SJ, Allen JR. From the laboratory to the clinic: a retrospective on fully synthetic carbohydrate-based anticancer vaccines. Angew Chem Int Ed. 2000;39:837–863. doi: 10.1002/(SICI)1521-3773(20000303)39:5<836::AID-ANIE836>3.0.CO;2-I. [DOI] [PubMed] [Google Scholar]
- 5.Buskas T, Ingale S, Boons GJ. Towards a fully synthetic carbohydrate-based anticancer vaccine: synthesis and immunological evaluation of a lipidated glycopeptide containing the tumor-associated Tn antigen. Angew Chem Int Ed. 2005;44:5985–5988. doi: 10.1002/anie.200501818. [DOI] [PubMed] [Google Scholar]
- 6.Renaudet O, BenMohamed L, Dasgupta G, Bettahi I, Dumy P. Towards a self-adjuvanting multivalent B and T cell epitope containing synthetic glycolipopeptide cancer vaccine. ChemMedChem. 2008;3:737–741. doi: 10.1002/cmdc.200700315. [DOI] [PubMed] [Google Scholar]
- 7.Ingale S, Wolfert MA, Buskas T, Boons GJ. Increasing the antigenicity of synthetic tumor-associated carbohydrate antigens by targeting Toll-like receptors. ChemBioChem. 2009;10:455–463. doi: 10.1002/cbic.200800596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wilkinson BL, Day S, Malins LR, Apostolopoulos V, Payne RJ. Self-adjuvanting multicomponent cancer vaccine candidates combining per-glycosylated MUC1 Glycopeptides and the toll-like receptor 2 agonist Pam3CysSer. Angew Chem Int Ed. 2011;50:1635–1639. doi: 10.1002/anie.201006115. [DOI] [PubMed] [Google Scholar]
- 9.Tang S, Wang Q, Guo Z. Synthesis of a monophosphoryl derivative of Escherichia coli lipid A and its efficient coupling to a tumor-associated carbohydrate antigen. Chem Eur J. 2010;16:1319–1325. doi: 10.1002/chem.200902153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wang Q, Zhou Z, Tang S, Guo Z (2012) Carbohydrate-monophosphoryl lipid A conjugates are fully synthetic self-adjuvanting cancer vaccines eliciting robust immune responses in the mouse. ACS Chem Biol 7. doi:10.1021/cb200358r [DOI] [PMC free article] [PubMed]
- 11.Wang Q, Guo Z (2009) Synthesis of a monophosphoryl lipid A derivative and its coupling to a tumor-associated carbohydrate antigen for cancer vaccine development. Chem Commun 5536–5537 [DOI] [PMC free article] [PubMed]
- 12.Chapman PB, Morrissey DM, Panageas KS, Hamilton WB, Zhan C, Destro AN, Williams L, Israel RJ, Livingston PO. Induction of antibodies against GM2 ganglioside by immunizing melanoma patients using GM2-keyhole limpet hemocyanin + QS21 vaccine: a dose-response study. Clin Cancer Res. 2000;6:874–879. [PubMed] [Google Scholar]
- 13.Holmberg LA, Sandmaier BM. Vaccination with Theratope (STn-KLH) as treatment for breast cancer. Expert Rev Vaccines. 2004;3:655–663. doi: 10.1586/14760584.3.6.655. [DOI] [PubMed] [Google Scholar]
- 14.Franco A. Glycoconjugates as vaccines for cancer immunotherapy: clinical trials and future directions. Anticancer Agents Med Chem. 2008;8:86–91. doi: 10.2174/187152008783330888. [DOI] [PubMed] [Google Scholar]
- 15.Krug LM, Ragupathi G, Hood C, Kris MG, Miller VA, Allen JR, Keding SJ, Danishefsky SJ, Gomez J, Tyson L, Pizzo B, Baez V, Livingston PO. Vaccination of patients with small-cell lung cancer with synthetic fucosyl GM-1 conjugated to keyhole limpet hemocyanin. Clin Cancer Res. 2004;10:6094–6100. doi: 10.1158/1078-0432.CCR-04-0482. [DOI] [PubMed] [Google Scholar]
- 16.Slovin SF, Ragupathi G, Musselli C, Olkiewicz K, Verbel D, Kuduk SD, Schwarz JB, Sames D, Danishefsky S, Livingston PO, Scher HI. Fully synthetic carbohydrate-based vaccines in biochemically relapsed prostate cancer: clinical trial results with alpha-N-acetylgalactosamine-O-serine/threonine conjugate vaccine. J Clin Oncol. 2003;21:4292–4298. doi: 10.1200/JCO.2003.04.112. [DOI] [PubMed] [Google Scholar]
- 17.Pan YB, Chefalo P, Nagy N, Harding CV, Guo Z. Synthesis and immunological properties of N-modified GM3 antigens as therapeutic cancer vaccines. J Med Chem. 2005;48:875–883. doi: 10.1021/jm0494422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wu J, Guo Z. Improving the antigenicity of sTn antigen by modification of its sialic acid residue for development of glycoconjugate cancer vaccines. Bioconjugate Chem. 2006;17:1537–1544. doi: 10.1021/bc060103s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wang Q, Zhang J, Guo Z. Efficient glycoengineering of GM3 on melanoma cell and monoclonal antibody-mediated selective killing of the glycoengineered cancer cell. Bioorg Med Chem. 2007;15:7561–7567. doi: 10.1016/j.bmc.2007.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Guo Z. Glycoengineering of cell surface sialic acid and its application to cancer immunotherapy. In: Guo Z, Boons G-J, editors. Carbohydrate-based vaccines and immunotherapies. New Jersey: Hoboken; 2009. pp. 313–331. [Google Scholar]
- 21.Chefalo P, Pan Y, Nagy N, Harding C, Guo Z. Effective metabolic engineering of GM3 on tumor cells by N-phenylacetyl-d-mannosamine. Biochemistry. 2006;45:3733–3739. doi: 10.1021/bi052161r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Yu S, Wang Q, Zhang J, Wu Q, Guo Z (2011) Synthesis and evaluation of protein conjugates of GM3 derivatives carrying modified sialic acids as highly immunogenic cancer vaccine candidates, MedChemComm 2. doi:10.1039/c1031md00033k [DOI] [PMC free article] [PubMed]
- 23.Wang Q, Guo Z (2011) Synthetic and immunological studies of sTn derivatives carrying substituted phenylacetylsialic acids as cancer vaccine candidates. ACS Med Chem Lett 2. doi:10.1021/ml100313d [DOI] [PMC free article] [PubMed]
- 24.Tsuchida T, Saxton RE, Morton DL, Irie RF. Gangliosides of human melanoma. J Natl Cancer Inst. 1987;78:45–54. doi: 10.1093/jnci/78.1.45. [DOI] [PubMed] [Google Scholar]
- 25.Hirabayashi Y, Hamaoka A, Matsumoto M, Matsubara T, Tagawa M, Wakabayashi S, Taniguchi M. Syngeneic monoclonal antibody against melanoma antigen with interspecies cross-reactivity recognizes GM3, a prominent ganglioside of B16 melanoma. J Biol Chem. 1985;260:13328–13333. [PubMed] [Google Scholar]
- 26.Ollila DW, Kelley MC, Gammon G, Morton DL. Overview of melanoma vaccines: active specific immunotherapy for melanoma patients. Semin Surg Oncol. 1998;14:328–336. doi: 10.1002/(SICI)1098-2388(199806)14:4<328::AID-SSU9>3.0.CO;2-4. [DOI] [PubMed] [Google Scholar]
- 27.Morton DL, Barth A. Vaccine therapy for malignant melanoma. CA Cancer J Clin. 1996;46:225–244. doi: 10.3322/canjclin.46.4.225. [DOI] [PubMed] [Google Scholar]
- 28.Liu T, Guo Z, Yang Q, Sad S, Jennings HJ. Biochemical engineering of surface α(2–8)polysialic acid for immunotargeting cancer cells. J Biol Chem. 2000;275:32832–32836. doi: 10.1074/jbc.C000573200. [DOI] [PubMed] [Google Scholar]