

Abstract
The impact of pressure on the backbone 15N, 1H and 13C chemical shifts in N-terminally acetylated α-synuclein has been evaluated over a pressure range spanning from 1–2500 bar. Even while the chemical shifts fall very close to random coil values, as expected for an intrinsically disordered protein, substantial deviations in the pressure dependence of the chemical shifts are seen relative to those in short model peptides. In particular, the non-linear pressure response of the 1HN chemical shifts, which commonly is associated with the presence of low-lying "excited states", is much larger in α-synuclein than in model peptides. The linear pressure response of 1HN chemical shift, commonly linked to H-bond length change, correlates well with those in short model peptides, and is found to be anti-correlated with its temperature dependence. The pressure dependence of 13C chemical shifts shows remarkably large variations, even when accounting for residue type, and do not point to a clear shift in population between different regions of the Ramachandran map. However, a nearly universal decrease in 3JHN-Hα by 0.22 ± 0.05 Hz suggests a slight increase in population of the polyproline II region at 2500 bar. The first six residues of N-terminally acetylated synuclein show a transient ca 15% population of α-helix, which slightly diminishes at 2500 bar. The backbone dynamics of the protein is not visibly affected beyond the effect of slight increase in water viscosity at 2500 bar.
Keywords: IDP, non-uniform sampling, 15N relaxation, random coil, triple resonance
Introduction
Intrinsically disordered proteins (IDPs) are abundant in eukaryotes where they are involved in many regulatory and signaling processes. Due to their inherent structural plasticity, IDPs are able to form specific interactions with a large variety of partners, playing a pivotal role in the cellular protein-protein network.[1, 2] In depth knowledge of the structural features of IDPs is crucial to understand their functions and their connection with a range of diseases. Multi-dimensional solution NMR spectroscopy is a method of choice for analyzing the transient backbone conformations populated by IDPs and disordered regions of otherwise ordered proteins. A growing body of evidence, emerging from the study of chemical shifts, J-couplings and residual dipolar couplings, shows that IDPs populate all the favored regions of the Ramachandran plot, including the alpha, the beta and the polyproline II (PPII) regions.[3–7]
The conformational propensities of short peptides have been extensively studied over the past years as a model for disordered regions of proteins.[8, 9] Systematic measurements of 3JHN-Hα couplings revealed that the PPII conformation is dominant for most amino acids at position X in Ac-G-G-X-G-GNH2 host-guest peptides, with the highest PPII propensity measured for Ala while β-branched or aromatic residues (Val, Ile, Tyr, and Phe) have a larger β-propensity.[10] The intrinsic conformational propensities of amino acids are strongly influenced by the solvent conditions and a change in the solvent properties represents the most efficient strategy to explore the conformational free-energy landscape of peptides and disordered proteins. For example, the PPII propensity is known to decrease in low-polarity solvents,[11] while a chemical denaturant such as urea, which stabilizes extended backbone conformations, promotes the formation of PPII.[12, 13] The PPII conformation is also strongly influenced by temperature, showing an increase in PPII propensity at low temperature, while the beta conformation is favored at high temperature.[13, 14]
Despite the fact that pressure is a fundamental thermodynamic variable, as important as temperature, the effect of pressure on disordered proteins has not yet been explored. High pressure is known to unfold globular proteins due to the smaller volume occupied by the unfolded states of proteins compared to their native states.[15] High-pressure NMR has been used extensively over the last twenty years to study the response of native proteins to compression,[16–20] to characterize low-lying excited states,[21–24] and to monitor the complete unfolding of globular proteins.[19, 25–27]
In the present study we use high-pressure NMR to analyze the impact of hydrostatic pressure on human α-synuclein, a widely used model for the study of intrinsically disordered proteins. α-Synuclein is a 140-residue protein composed of three distinct regions: a positively charged N-terminal region (residues 1–60), a central hydrophobic segment (residues 61–95) often referred to as the NAC region, and a highly negatively charged and proline-rich C-terminal region (residues 96–140). The chemical shifts of α-synuclein are exceptionally close to random coil values.[28, 29] The α-helical propensity of the first six N-terminal residues is also substantially increased by N-terminal acetylation,[29] a post-translational modification in eukaryotes. Moreover, the N-terminal and hydrophobic regions of α-synuclein are known to adopt an α-helical conformation when bound to the surface of negatively charged vesicles or detergent micelles.[30–32]
To explore in detail the effect of high-pressure on the structural properties of α-synuclein, we monitored the pressure-induced changes of the 1HN, 15N, 13Cα, 13Cβ and 13C' chemical shifts and measured a nearly complete set of 3JHN-Hα couplings at 2500 bar. The change in the 15N spin relaxation was also investigated through R1ρ measurements. While the pressure-induced 1HN shifts are in close agreement with those predicted for model tetrapeptides[33] and can reasonably be explained in terms of hydrogen bond compression, additional long-range factors are likely affecting the observed 15N pressure-induced shifts. Analysis of the 13C chemical shifts and 3JHN-Hα couplings of α-synuclein suggests that pressure shifts the PPII-β equilibrium of the random coil towards increased PPII propensity.
Methods
N-acetylated α-synuclein was generated using a recently developed recombinant expression system that includes a plasmid for overexpression of the requisite acetylation enzyme NatB,[34] which permits bacterial expression of N-terminally acetylated and uniformly 13C/15N-enriched aS, which was subsequently purified as previously described (Maltsev et al. 2012b).
All NMR spectra were recorded at 288 K. The 15N-TROSY-HSQC spectra were recorded at a 1H frequency of 500 MHz. A total of 200* × 560* complex points were collected, for acquisition times of 164 ms and 80 ms in the 15N and 1H dimensions, respectively, using an interscan delay of 1.4 s.
3D TROSY-HNCO and TROSY-HNCACB spectra were recorded at 500 MHz with 2 scans per free induction delay. The HNCO spectra comprised 110* × 160* × 750* complex points, for acquisition times of 150.5, 144 and 107 ms in the 13C, 15N and 1H dimensions, respectively. For the HNCACB spectra, the final time domain matrices consisted of 173* × 160* × 750* complex points, corresponding to acquisition times of 144 ms (15N), 26 ms (13C) and 107 ms (1H). The 13C chemical shift was recorded using constant-time evolution. Both HNCO and HNCACB experiments were collected using a 40 % sparse sampling scheme, with the sampling schedule in the two indirect dimensions generated randomly (i.e., only 40% of the (t1,t2) pairs were actually collected). The final spectra were reconstructed as previously described[35] using the algorithm developed by Hyberts et al. [36]
The R1ρ experiments were recorded at 600 MHz on a non-acetylated α-synuclein sample. Data matrices of 320* × 1024* complex points were collected for acquisition time of 176 ms and 122 ms in the 15N and 1H dimensions, respectively. The strength of the radiofrequency spin-lock field was set to 1.3 kHz and the relaxation delays were sampled for eight different durations: 1, 20, 70, 110, 180, 230, 300 and 350 ms.
The 3JHN-Hα couplings were recorded at 800 MHz by monitoring the modulation of the cross-peak intensity from a series of constant-time 1H-15N-HMQC spectra, as previously described.[35] Eight constant time delays were used, with durations of 50, 60, 75, 95, 140, 180, 210 and 240 ms. The data matrix sizes ranged from 82* × 1024* points for the shortest constant-time duration to 402* × 1024* points for the longest duration.
For all the experiments described above, a commercial ceramic high-pressure NMR cell and an automatic pump system (Daedalus Innovations, Philadelphia, PA) were used to vary the pressure in the 1 bar to 2.5 kbar range.
Results
Effects of pressure on the 15N and HN amide chemical shifts
The pressure dependence of the 15N and HN amide chemical shifts was monitored by recording a series of 15N-TROSY-HSQC spectra of N-acetylated α-synuclein at 288 K, pH 6.0, with pressure varying from 1 bar to 2500 bar. An overlay of the 2D 15N-1H spectra recorded at four different pressures, 1 bar, 500 bar, 1250 bar and 2500 bar, shows large downfield shifts for all the amide crosspeaks with the notable exception of Gly-41, which shows an upfield HN shift with increasing pressure (Figure 1A). Figures 1B and 1C display the 15N and HN chemical shift differences between 2500 and 1 bar (Δδ2500) as a function of residue number. With a Pearson's correlation coefficient of R2 = 0.60 (rmsd=0.024 ppm), the Δδ2500(1HN) values correlate closely with the chemical shift differences predicted for the same amino acid sequence using the pressure coefficients reported by Koehler et al.[33] for model tetrapeptides (Figure 1B). By contrast, the correlation observed for Δδ2500(15N) is much weaker (R2= 0.18, rmsd=0.179 ppm; Figure 1C). The difference between the observed and predicted Δδ2500 values shows no clear correlation with residue type: for both 15N and 1HN chemical shifts, a considerable spread of Δδ2500 values is observed regardless of the nature of the amino acid (Supporting Information (SI) Figure S1A,B). Such variations narrow down slightly when taking into account the nature of the preceding residue (SI Figure S1C,D). Since the α-synuclein sequence contains seven imperfect repeats of the hexamer motif KTKEGV in the N-terminal and NAC regions, we also examined the variation of the Δδ2500 values among the six residues composing this motif (SI Figure S1E and S1F). Interestingly, a clear pattern of Δδ2500 values appears for the 1HN chemical shifts, with the largest Δδ2500(1HN) for the second residue of the motif and the smallest Δδ2500(1HN) for the fifth residue of the motif (SI Figure S1F). Residue Thr-72, the 4th residue of the sixth repeat and the sole Thr at this position (in all other repeats the 4th residue is Glu) is a distinct outlier in this pattern.
Figure 1.

(A) Overlay of the 15N-TROSY-HSQC spectra recorded at 500 MHz, between 1 bar and 2500 bar, at 288 K, recorded on a uniformly labeled 15N/13C-enriched sample of N-acetylated α-synuclein (0.4 mM) in 20 mM sodium phosphate, pH 6.0. The chemical shifts differences between 2500 bar and 1 bar (Δδ2500) are displayed in black as a function of the protein sequence for 1HN (B) and 15N (C) and compared with the corresponding predicted chemical shift differences (red) for residues of the same type when embedded at position X in model tetrapeptide Ac-G-G-X-A-NH2.[33]
Non-linear pressure dependence of the 1HN chemical shifts
The non-linear pressure response of the 1HN chemical shifts has been extensively studied for globular proteins and has been attributed to the presence of low-lying "excited states".[21] Surprisingly, the 1HN chemical shifts reported by Koehler et al.[33] for model tetrapeptides also exhibit a significant non-linear pressure dependence. To analyze this non-linear pressure response in the case of the α-synuclein, we fitted the pressure dependence of the 1HN chemical shifts, measured at nine different pressures, ranging from 1 to 2500 bar, to a second order polynomial function (Figure 2A). Comparison of the non-linear coefficients of the 1HN pressure dependence with those reported by Koehler et al. (2012) shows that the values obtained for α-synuclein are, on average, about three times larger than the ones reported for model tetrapeptides (−(8.99 ± 3.60) × 10−9 ppm/bar2 compared to (−3.10 ± 2.19) × 10−9 ppm/bar2, respectively) (Figure 2B). The largest differences between the measured and predicted coefficients are observed in the N-terminal region (residues 6 to 36) and in the central region (residues 50 to 92). Again, no significant correlation was observed between the nature of the amino acid and the magnitude of the non-linear pressure response.
Figure 2.
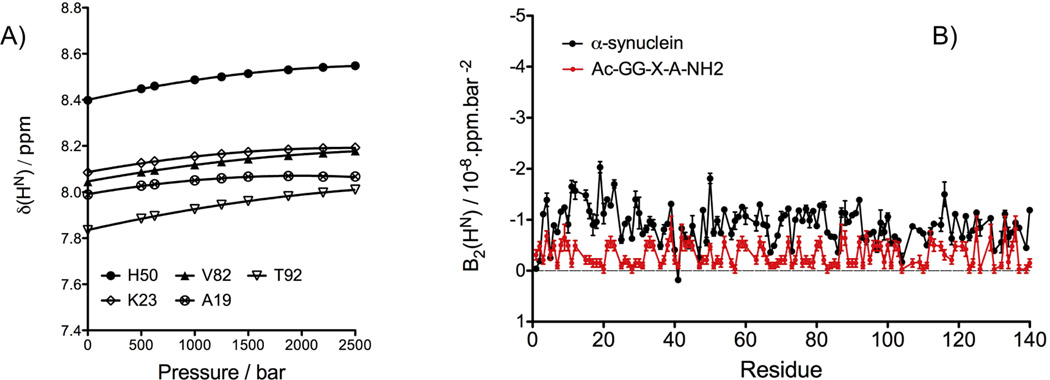
Effect of pressure on 1HN chemical shifts. (A) Pressure dependence of the 1HN chemical shifts of N-acetylated α-synuclein measured through a series of 15N-TROSY-HSQC experiments, recorded at 500 MHz, 288 K, and pressures ranging from 1 to 2500 bar. The changes of the 1HN chemical shifts with pressure (δHN(p)) are fitted to a quadratic function: δHN(p) = δ0(p0) + B1(p-p0) + B2(p-p0)2. (B) The second order coefficients (B2) are displayed as a function of the protein sequence (black) and compared with those predicted for residues of the same type when embedded in the model tetrapeptide Ac-G-G-X-A-NH2 (red).[33].
Correlation between the 1HN pressure and temperature coefficients
It has been proposed that the upfield chemical shift observed for the amide protons with increasing temperature is caused by lengthening of the hydrogen bonds, which weakens the HN electron polarization.[37, 38] Analogously, Akasaka and coworkers have interpreted the downfield shift of the amide protons with increasing pressure as resulting from a shortening of the H bond length.[39] If the residue-by-residue variation in δ(1HN) temperature and pressure coefficients indeed reflects variations in the sensitivity of the corresponding H-bonds to temperature and pressure, a correlation between these temperature and pressure coefficients is expected to be present. Although the degree of variation in δ(1HN) temperature and pressure coefficients is much smaller for an IDP than for a folded protein, we nevertheless examined the potential presence of such a correlation for acetylated α-synuclein. The pressure coefficients used here correspond to the first order coefficients extracted from the fit of the 1HN chemical shifts measured between 1 and 2500 bar, at 288K, while the temperature coefficients were calculated by a linear least square fit of the HN chemical shifts measured at 283 K, 288 K and 293 K, at atmospheric pressure. A modest correlation (R2= 0.31) with a negative slope is indeed observed between the temperature and pressure coefficients (Figure 3). Interestingly, the amide proton of Gly-41 for which the sole upfield effect of pressure was measured with a pressure coefficient of −2.8 10−5 ppm/bar, exhibits by far the smallest temperature coefficients (−1.2 ppb/K). Recently, the Gly-41 1HN also was shown to have an exceptionally large isotope shift when comparing the protein where all non-exchangeable hydrogens had been deuterated with its fully protonated form.[40] However, no structural explanation for this outlier behavior of Gly-41 1HN has emerged so far.
Figure 3.

Correlation between the 1HN pressure and temperature linear coefficients, measured for a 15N/13C enriched sample of N-acetylated α-synuclein at pH 6.0. The pressure coefficients were extracted from the fit of the 1HN chemical shifts as a function of pressure, between 1 bar and 2500 bar, at 288 K, while the temperature coefficients were calculated from a linear least squares fit of the 1HN chemical shifts at 283 K, 288 K and 293 K, under atmospheric pressure.
Effect of pressure on 15N spin relaxation
The effect of pressure on the backbone dynamics has been described in a few cases, for globular proteins [41] and for an isolated α-helix,[42] leading to the general conclusion that rapid internal motions of the N-H vectors on the nanosecond or sub-nanosecond time scale are not affected by pressure. To extend these observations to IDPs, we measured the R1ρ relaxation rates at 600 MHz on a non-acetylated α-synuclein, at 1 bar and 2500 bar (Figure 4). A modest and rather uniform increase of ca 5% in the relaxation rates was observed at 2500 bar (<R1ρ=3.28±0.55 s−1>), compared to 1 bar (<R1ρ=3.13±0.54 s−1>), with a correlation coefficient, R2= 0.89 (rmsd=0.24 s−1) between the two data sets (Figure 4). Such a non-specific, uniform effect of pressure on the transverse relaxation rates simply reflects the small increase in the solvent viscosity[43] and suggests that the backbone dynamics of α-synuclein is not affected by pressure. The rapid internal dynamics of α-synuclein has previously been highlighted by R1 relaxation dispersion measurements.[44] Increased flexibility of the NAC region relative to the remainder of the protein, as judged by decreased 15N transverse relaxation rates, has been reported previously,[45] but is more evident in the current data due to increased sensitivity associated with the use of a cryogenic probehead used in our measurements. Our data clearly show that this increased flexibility of the NAC region is not impacted by pressure.
Figure 4.

15N R1ρ relaxation rates measured using a 1.3 kHz RF field at 60.8 MHz 15N frequency, at 1 bar (black) and 2500 bar (red), displayed as a function of the protein sequence. The sample conditions are: 0.5 mM of 15N enriched non-acetylated α-synuclein in 20 mM sodium phosphate at pH 6.0. All the experiments were performed at 288 K.
Effect of pressure on the backbone 13C chemical shifts
The pressure dependence of the 13Cα, 13Cβ and 13C' chemical shifts of N-acetylated α-synuclein was analyzed by recording HNCACB and HNCO experiments at 1 bar and 2500 bar (Figure 5A). A general upfield shift of the 13Cα chemical shifts was observed, with an average difference between 2500 and 1 bar of Δδ2500(13Cα) = −0.122 ± 0.091 ppm. Gly 13Cα resonances are an exception and show the opposite sign for the pressure dependence: Δδ2500(13Cα) = +0.124 ± 0.038 ppm (Figure 5B). A general upfield change was also observed for the 13Cβ chemical shifts (Δδ2500(13Cβ) = −0.186 ± 0.081 ppm), with the exception of Ala, which show a downfield shift: Δδ2500(13Cβ) = +0.288 ± 0.069 ppm (Figure 5C). The magnitude of the pressure induced shifts observed here for N-acetylated α-synuclein are comparable to those reported by Williamson and coworkers for the aliphatic carbons in two well-folded proteins, barnase and protein G, where an average 13C shift difference between 2000 bar and 1 bar of +0.24 ± 0.18 ppm for CH3, −0.09 ± 0.17 ppm for CH2 and −0.17 ± 0.15 ppm for the CH carbons atoms was found.[46] Surprisingly, even for α-synuclein which shows all the hallmarks of an IDP, remarkably large variations in 13Cα and 13Cβ pressure dependence remain when comparing the effects seen for residues of a given type at different locations in the protein (SI Figure S2). This observation indicates that the variation in Δδ2500(13Cα,13Cβ) values is not simply dominated by the amino acid type and instead must reflect a differential local effect in how the disordered chain reorganizes itself upon increasing pressure. Only in the case of 13Cα atoms, we noticed a slight reduction in the Δδ2500 variations when the nature of the preceding residue was considered (SI Figure S2). For the 13C' nuclei, a small but nearly universal downfield change in chemical shift was observed upon increasing the pressure to 2500 bar, with an average Δδ2500(13C') = +0.079 ± 0.076 ppm (Figure 5D). The opposite direction of the pressure-induced 13Cα and 13C' changes in chemical shift suggests that these changes do not simply reflect a change in α−helical propensity of the N-acetylated α-synuclein, as the secondary 13Cα and 13C' shifts associated with α−helix are both pronouncedly positive.
Figure 5.

Effect of pressure on chemical shifts in α-synuclein. (A) Superposition of the 2D projections of the TROSY-HNCO spectra recorded at 1 bar (black) and 2500 bar (red) on the 13C/1H plane. The chemical shifts differences between 2500 bar and 1 bar (Δδ2500) measured for the 13Cα (B), 13Cβ (C) and 13C' (D) at 288K, are displayed as a function of residue number in the α-synuclein sequence. The 13C chemical shifts were extracted from HNCACB and HNCO spectra collected at 500 MHz, using a uniformly 15N/13C-enriched sample of N-acetylated α-synuclein in 20 mM sodium phosphate, pH 6.0.
Effect of pressure on 3JHN-Hα
In addition to chemical shifts, 3JHN-Hα couplings are widely used as reporters for the backbone torsion angle, ϕ. We recently demonstrated that 3JHN-Hα values in α-synuclein can be measured at very high precision (<0.05 Hz).[29] Moreover, a study of the protein GB3 showed that the 3JHN-Hα values are quite insensitive to residue type, H-bonding effects, or structural variables other than ϕ, as evidenced by an RMSD of 0.4 Hz between experimental values and those predicted by a Karplus curve when using an RDC-refined high resolution X-ray structure.[47] Here, we measured 3JHN-Hα couplings at 2500 bar and compared the values with our previously published set of 3JHN-Hα couplings measured for the same protein at atmospheric pressure.[29] This is to our knowledge the first report of the effect of pressure on 3JHN-Hα couplings. Although the effects are generally small, they are almost universally negative (Δ3JHN-Hα = −0.20 ± 0.12 Hz) and in many cases exceed the uncertainty in the measurement by five standard deviations or more (Figure 6). Again, little dependence on amino acid type is observed, and the range of Δ3JHN-Hα for a given type of residue, e.g. Val or Lys, spans nearly the entire range of observed values, from 0 to −0.6 Hz. Perhaps surprisingly, even when selecting pairs of amino acids that are of the same type, considerable variation remains (SI Figure S3B), indicating that the effect of pressure extends beyond impacting the interaction between side chains on adjacent amino acids.
Figure 6.

Differences in the 3JHN-Hα couplings measured at 800 MHz between 2500 and 1 bar, at 288 K, displayed as a function of the protein sequence. The sample conditions are: 0.3 mM of 15N enriched N-acetylated α-synuclein in 20 mM sodium phosphate at pH 6.0.
Discussion
Effect of pressure on the amide chemical shifts and the hydrogen bonds length
The downfield shift observed at high pressure for the HN amide protons is commonly interpreted as resulting from the compression of the N-H⋯O=C hydrogen bond. Indeed, a close correlation between the strength of the hydrogen bond and the 1HN chemical shift has long been recognized.[39, 48] The pressure induced changes in the through-hydrogen-bond 3hJNC' scalar couplings measured in streptococcal protein G[49] and ubiquitin[50] provide another piece of direct evidence that the electronic orbital overlap associated with H bonds is indeed affected by pressure. Based on an empirical relationship between the H bond length and the 1HN chemical shift.[48] Akasaka and coworkers estimated that the N-H⋯O=C hydrogen bond distance is reduced by ca 1% at 2 kbar in globular proteins.[39] The magnitude of the pressure-induced 1HN shifts measured here for α-synuclein, on average, is very similar to that of the solvent exposed amide protons in BPTI, for which an average of −0.129 ppm/2 kbar was reported.[39] Moreover, the close agreement observed between the linear component of the pressure-induced 1HN chemical shift changes in α-synuclein and those predicted on the basis of model tetrapeptides[33] (Figure 1B), suggests that the ΔδHN shifts of α-synuclein are dominated by very local effects of pressure. On the other hand, Gly-41 is clearly an outlier to this rule, as it shows an upfield change in chemical shift with pressure (ΔδHN = −2.8×10−5 ppm/bar). It is conceivable that this residue is transiently involved in intramolecular H-bonding interactions, the population of which may be sensitive to pressure. The correlation observed between the pressure and temperature coefficients of the 1HN chemical shifts in α-synuclein (Figure 3) agrees well the idea that both temperature and pressure affect the H bond length, either by compressing (with increasing pressure) or expanding (with increasing temperature) the length of the N-H⋯O bond.
Interestingly, while the magnitude of the overall pressure-induced 1HN chemical shift changes in α-synuclein is similar to that of a small and rigid protein such as BPTI, the non-linear component of the pressure dependence of the 1HN chemical shift are considerably larger than those in the model peptides (Figure 2B), and comparable to those reported for the dihydrofolate reductase or the Ras binding domain of Ral-GDS.[21] Akasaka and Li have correlated the magnitude of the non-linear pressure response in globular proteins to the volume of internal cavities, suggesting that proteins with large internal void volumes are more likely to populate low-lying alternate conformers under the high-pressure condition.[21] Clearly, this explanation does not apply in the case of an IDP such as α-synuclein. The large non-linear HN coefficients measured here for α-synuclein most likely arise from a shift in populations sampled by the peptide chain, as reflected by the decrease of the 3JHN-Hα couplings. However, we cannot exclude a contribution arising from a perturbation of the hydration shell at high pressure.[51, 52]
The pressure-induced changes of the 15N amide chemical shifts have been described in a few cases and remain poorly understood. Akasaka and coworkers suggested that the 15N pressure coefficients are affected by site-specific variations of the backbone angles in addition to the decrease in the H-bonding distances.[16] A weak correlation was indeed found for BPTI between the magnitude of the 15N shift and the ψi-1 angle in β-sheet but not in α-helices.[16] The absence of a correlation between the pressure-induced 15N shifts measured here for α-synuclein and those predicted from model tetrapeptides, even when the nature of the preceding amino acid is considered (Figure 1C and SI Figure S1D), points to the influence of additional factors that extend beyond the amino acids sharing the peptide bond. It therefore seems clear that the Δδ2500(15N) values of α-synuclein cannot be explained solely by the compression of H-bond length to solvent, or to a strictly local interaction between the two sequential side chains, and instead must involve the differential sampling by the polypeptide backbone of the complex conformational landscape with increasing pressure.
Effect of pressure on the conformational equilibrium of α-synuclein
The difference in protein volume associated with the unfolding of an α-helix is close to zero, such that α-helices in proteins are usually preserved under high-pressure conditions.[15] For example, a substantial amount of residual helical content was observed in the pressure-denatured state of SNase.[53] Imamura and Kato [54] suggested that the volume change upon unfolding (ΔVu) could even be positive (i.e. pressure promoting the formation of helical content) in the case of a small helical peptide.
The N-terminal acetylation of α-synuclein resulted in a significant, ca 15% transient population of α-helix by its six N-terminal residues, extending as far as residue 12 but at much decreased population.[29] We chose to study this N-acetylated form of the protein as it would provide direct data on the impact of pressure on the transient population of α-helix. The analysis of the 13C chemical shift changes (13Cα, 13Cβ, and 13C', Figure 5) between 1 and 2500 bar shows that the N-terminal six residues exhibit, on average, chemical shift differences of −0.211 ± 0.075 ppm (13Cα) and −0.028 ± 0.059 ppm (13C'), compared to −0.118 ± 0.034 ppm (13Cα) and + 0.083 ± 0.038 ppm (13C') for all sliding windows of six amino acids between residues 10 to 140. The 13Cβ shows a smaller average change between 1 bar and 2500 bar for the first six residues (−0.118 ± 0.074 ppm) compared to residues 10–140 (−0.189 ± 0.029 ppm). Similarly, the average decrease in 3JHN-Hα with pressure for the first six residues (<Δ3JHN-Hα> = −0.127 ± 0.076 Hz) is slightly smaller than for the remainder of the chain <Δ3JHN-Hα> = −0.221 ± 0.041 Hz (Figure 6), suggesting that relative to the fully disordered region, population of α-helix is slightly decreased at 2500 bar and therefore that the ΔVu for α-helix is negative.
Having ruled out an increased α-helical propensity at high pressure, the small decrease of the 3JHN-Hα couplings measured here for nearly all residues of α-synuclein at 2500 bar (Figure 6) can be interpreted as arising from a pressure-induced decrease of the β conformation and a corresponding increase in the conformational populations with a less extended backbone geometry, namely PPII, αL or β-turn conformations, all of which have smaller 3JHN-Hα couplings than the β conformation. Many studies have pointed out the crucial role of hydration in stabilizing PPII conformations relative to α or β conformations in peptides and proteins.[11–13, 55] Importantly, the PPII-β equilibrium in peptides is highly temperature dependent, with the amount of PPII conformations decreasing with increasing temperature.[13, 14] An increase in pressure will likely have an opposite effect to an increase in temperature, by shifting the PPII-β equilibrium toward more PPII content at high pressure. However, considering that 3JHN-Hα couplings do not uniquely report on secondary structure, further studies are needed to analyze the specific effect of pressure on PPII conformations relative to αL or β-turn conformations.
Conclusion
The NMR spectrum of α-synuclein is impacted by hydrostatic pressure to a degree that is comparable to that seen for folded proteins. While the changes in 1HN chemical shifts correlate well with values predicted from a series of short model peptides, large deviations from such model peptide behavior is seen for 15N, indicating that the pressure dependence of the α-synuclein spectrum is modulated by interactions that extend beyond the dipeptide unit for which chemical shift changes are observed. The nature of these remote interactions remains to be determined. The transient α-helical population observed for the first six N-terminal residues in the N-acetylated α-synuclein is only weakly diminished by pressure whereas the general decrease of the 3JHN-Hα couplings observed at high-pressure is suggestive of a shift in the PPII-β equilibrium towards PPII for most of the protein. Further computational studies are especially needed to explore in detail the opposite effects of heat and pressure on the PPII propensity of peptides and disordered fragments of proteins, and to explore in atomic detail which structural and hydration changes are compatible with the impact of pressure on the NMR parameters observed in our study.
Supplementary Material
Acknowledgment
This work was supported by the Intramural Research Program of the National Institute of Diabetes and Digestive and Kidney Diseases and by the Intramural Antiviral Target Program of the Office of the Director, NIH. The authors thank James Baber for technical assistance.
Footnotes
Supporting information for this article is available.
References
- 1.Haynes C, Oldfield CJ, Ji F, Klitgord N, Cusick ME, Radivojac P, Uversky VN, Vidal M, Iakoucheva LM. PLoS Comput. Biol. 2006;2:890–901. doi: 10.1371/journal.pcbi.0020100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Uversky VN, Oldfield CJ, Dunker AK. Annual Review of Biophysics. 2008;37:215–246. doi: 10.1146/annurev.biophys.37.032807.125924. [DOI] [PubMed] [Google Scholar]
- 3.Jensen MR, Salmon L, Nodet G, Blackledge M. J. Am. Chem. Soc. 2010;132:1270-+. doi: 10.1021/ja909973n. [DOI] [PubMed] [Google Scholar]
- 4.Camilloni C, De Simone A, Vranken WF, Vendruscolo M. Biochemistry. 2012;51:2224–2231. doi: 10.1021/bi3001825. [DOI] [PubMed] [Google Scholar]
- 5.Bernado P, Blanchard L, Timmins P, Marion D, Ruigrok RWH, Blackledge M. Proc. Natl. Acad. Sci. U. S. A. 2005;102:17002–17007. doi: 10.1073/pnas.0506202102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Mukrasch MD, Markwick P, Biernat J, von Bergen M, Bernado P, Griesinger C, Mandelkow E, Zweckstetter M, Blackledge M. J. Am. Chem. Soc. 2007;129:5235–5243. doi: 10.1021/ja0690159. [DOI] [PubMed] [Google Scholar]
- 7.Ozenne V, Schneider R, Yao M, Huang J-r, Salmon L, Zweckstetter M, Jensen MR, Blackledge M. J. Am. Chem. Soc. 2012;134:15138–15148. doi: 10.1021/ja306905s. [DOI] [PubMed] [Google Scholar]
- 8.Shi ZS, Chen K, Liu ZG, Kallenbach NR. Chem. Rev. (Washington, DC, U. S.) 2006;106:1877–1897. doi: 10.1021/cr040433a. [DOI] [PubMed] [Google Scholar]
- 9.Hagarman A, Measey TJ, Mathieu D, Schwalbe H, Schweitzer-Stenner R. J. Am. Chem. Soc. 2010;132:540–551. doi: 10.1021/ja9058052. [DOI] [PubMed] [Google Scholar]
- 10.Shi ZS, Chen K, Liu ZG, Ng A, Bracken WC, Kallenbach NR. Proc. Natl. Acad. Sci. U. S. A. 2005;102:17964–17968. doi: 10.1073/pnas.0507124102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Liu ZG, Chen K, Ng A, Shi ZS, Woody RW, Kallenbach NR. J. Am. Chem. Soc. 2004;126:15141–15150. doi: 10.1021/ja047594g. [DOI] [PubMed] [Google Scholar]
- 12.Li W, Qin M, Tie Z, Wang W. Physical Review E. 2011;84 doi: 10.1103/PhysRevE.84.041933. Article Number 041933. [DOI] [PubMed] [Google Scholar]
- 13.Elam WA, Schrank TP, Campagnolo AJ, Hilser VJ. Biochemistry. 2013;52:949–958. doi: 10.1021/bi301435p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Shi ZS, Olson CA, Rose GD, Baldwin RL, Kallenbach NR. Proc. Natl. Acad. Sci. U. S. A. 2002;99:9190–9195. doi: 10.1073/pnas.112193999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Royer CA. Biochimica Et Biophysica Acta-Protein Structure and Molecular Enzymology. 2002;1595:201–209. doi: 10.1016/s0167-4838(01)00344-2. [DOI] [PubMed] [Google Scholar]
- 16.Akasaka K, Li H, Yamada H, Li RH, Thoresen T, Woodward CK. Protein Sci. 1999;8:1946–1953. doi: 10.1110/ps.8.10.1946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Refaee M, Tezuka T, Akasaka K, Williamson MP. J. Mol. Biol. 2003;327:857–865. doi: 10.1016/s0022-2836(03)00209-2. [DOI] [PubMed] [Google Scholar]
- 18.Fu Y, Kasinath V, Moorman VR, Nucci NV, Hilser VJ, Wand AJ. J. Am. Chem. Soc. 2012;134:8543–8550. doi: 10.1021/ja3004655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Prehoda KE, Mooberry ES, Markley JL. Biochemistry. 1998;37:5785–5790. doi: 10.1021/bi980384u. [DOI] [PubMed] [Google Scholar]
- 20.Vajpai N, Nisius L, Wiktor M, Grzesiek S. Proc. Natl. Acad. Sci. U. S. A. 2013;110:1578–1578. doi: 10.1073/pnas.1212222110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Akasaka K, Li H. Biochemistry. 2001;40:8665–8671. doi: 10.1021/bi010312u. [DOI] [PubMed] [Google Scholar]
- 22.Akasaka K. Chem. Rev. (Washington, DC, U. S.) 2006;106:1814–1835. doi: 10.1021/cr040440z. [DOI] [PubMed] [Google Scholar]
- 23.Kuwata K, Li H, Yamada H, Legname G, Prusiner SB, Akasaka K, James TL. Biochemistry. 2002;41:12277–12283. doi: 10.1021/bi026129y. [DOI] [PubMed] [Google Scholar]
- 24.Kitahara R, Yamada H, Akasaka K. Biochemistry. 2001;40:13556–13563. doi: 10.1021/bi010922u. [DOI] [PubMed] [Google Scholar]
- 25.Roche J, Caro JA, Norberto DR, Barthe P, Roumestand C, Schlessman JL, Garcia AE, Garcia-Moreno E B, Royer CA. Proc. Natl. Acad. Sci. U. S. A. 2012;109:6945–6950. doi: 10.1073/pnas.1200915109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kamatari YO, Smith LJ, Dobson CM, Akasaka K. Biophys. Chem. 2011;156:24–30. doi: 10.1016/j.bpc.2011.01.009. [DOI] [PubMed] [Google Scholar]
- 27.Kitahara R, Akasaka K. Proc. Natl. Acad. Sci. U. S. A. 2003;100:3167–3172. doi: 10.1073/pnas.0630309100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bermel W, Bertini I, Felli IC, Lee YM, Luchinat C, Pierattelli R. J. Am. Chem. Soc. 2006;128:3918–3919. doi: 10.1021/ja0582206. [DOI] [PubMed] [Google Scholar]
- 29.Maltsev AS, Ying JF, Bax A. Biochemistry. 2012;51:5004–5013. doi: 10.1021/bi300642h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Davidson WS, Jonas A, Clayton DF, George JM. J. Biol. Chem. 1998;273:9443–9449. doi: 10.1074/jbc.273.16.9443. [DOI] [PubMed] [Google Scholar]
- 31.Ulmer TS, Bax A, Cole NB, Nussbaum RL. J. Biol. Chem. 2005;280:9595–9603. doi: 10.1074/jbc.M411805200. [DOI] [PubMed] [Google Scholar]
- 32.Bodner CR, Dobson CM, Bax A. J. Mol. Biol. 2009;390:775–790. doi: 10.1016/j.jmb.2009.05.066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Koehler J, Erlach MB, Crusca E, Jr, Kremer W, Munte CE, Kalbitzer HR. Materials. 2012;5:1774–1786. [Google Scholar]
- 34.Johnson M, Coulton AT, Geeves MA, Mulvihill DP. PLoS One. 2010;5 doi: 10.1371/journal.pone.0015801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kuboniwa H, Grzesiek S, Delaglio F, Bax A. J. Biomol. NMR. 1994;4:871–878. doi: 10.1007/BF00398416. [DOI] [PubMed] [Google Scholar]
- 36.Hyberts SG, Milbradt AG, Wagner AB, Arthanari H, Wagner G. J. Biomol. NMR. 2012;52:315–327. doi: 10.1007/s10858-012-9611-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Baxter NJ, Williamson MP. J. Biomol. NMR. 1997;9:359–369. doi: 10.1023/a:1018334207887. [DOI] [PubMed] [Google Scholar]
- 38.Hong J, Jing Q, Yao L. J. Biomol. NMR. 2013;55:71–78. doi: 10.1007/s10858-012-9689-3. [DOI] [PubMed] [Google Scholar]
- 39.Li H, Yamada H, Akasaka K. Biochemistry. 1998;37:1167–1173. doi: 10.1021/bi972288j. [DOI] [PubMed] [Google Scholar]
- 40.Maltsev AS, Ying JF, Bax A. J. Biomol. NMR. 2012;54:181–191. doi: 10.1007/s10858-012-9666-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Sareth S, Li H, Yamada H, Woodward GK, Akasaka K. FEBS Lett. 2000;470:11–14. doi: 10.1016/s0014-5793(00)01283-7. [DOI] [PubMed] [Google Scholar]
- 42.Orekhov VY, Dubovskii PV, Yamada H, Akasaka K, Arseniev AS. J. Biomol. NMR. 2000;17:257–263. doi: 10.1023/a:1008346414720. [DOI] [PubMed] [Google Scholar]
- 43.Bett KE, Cappi JB. Nature. 1965;207:620-&. [Google Scholar]
- 44.Bertini I, Gupta YK, Luchinat C, Parigi G, Schlorb C, Schwalbe H. Angewandte Chemie-International Edition. 2005;44:2223–2225. doi: 10.1002/anie.200462344. [DOI] [PubMed] [Google Scholar]
- 45.Bussell R, Eliezer D. J. Biol. Chem. 2001;276:45996–46003. doi: 10.1074/jbc.M106777200. [DOI] [PubMed] [Google Scholar]
- 46.Wilton DJ, Kitahara R, Akasaka K, Williamson MP. J. Biomol. NMR. 2009;44:25–33. doi: 10.1007/s10858-009-9312-4. [DOI] [PubMed] [Google Scholar]
- 47.Vogeli B, Ying JF, Grishaev A, Bax A. J. Am. Chem. Soc. 2007;129:9377–9385. doi: 10.1021/ja070324o. [DOI] [PubMed] [Google Scholar]
- 48.Wagner G, Pardi A, Wuthrich K. J. Am. Chem. Soc. 1983;105:5948–5949. [Google Scholar]
- 49.Li H, Yamada H, Akasaka K, Gronenborn AM. J. Biomol. NMR. 2000;18:207–216. doi: 10.1023/a:1026537609584. [DOI] [PubMed] [Google Scholar]
- 50.Nisius L, Grzesiek S. Nature Chem. 2012;4:711–717. doi: 10.1038/nchem.1396. [DOI] [PubMed] [Google Scholar]
- 51.Smolin N, Winter R. BBA-Proteins Proteomics. 2006;1764:522–534. doi: 10.1016/j.bbapap.2006.01.003. [DOI] [PubMed] [Google Scholar]
- 52.Sumi T, Sekino H. Phys. Chem. Chem. Phys. 2011;13:15829–15832. doi: 10.1039/c1cp21347d. [DOI] [PubMed] [Google Scholar]
- 53.Panick G, Malessa R, Winter R, Rapp G, Frye KJ, Royer CA. J. Mol. Biol. 1998;275:389–402. doi: 10.1006/jmbi.1997.1454. [DOI] [PubMed] [Google Scholar]
- 54.Imamura H, Kato M. Proteins-Structure Function and Bioinformatics. 2009;75:911–918. doi: 10.1002/prot.22302. [DOI] [PubMed] [Google Scholar]
- 55.Garcia AE. Polymer. 2004;45:669–676. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.