

Abstract
Published biological data suggest that the methyl erythritol phosphate (MEP) pathway, a non-mevalonate isoprenoid biosynthetic pathway, is essential for certain bacteria and other infectious disease organisms. One highly conserved enzyme in the MEP pathway is 2C-methyl-D-erythritol 2,4-cyclodiphosphate synthase (IspF). Fragment-bound complexes of IspF from Burkholderia pseudomallei were used to design and synthesize a series of molecules linking the cytidine moiety to different zinc pocket fragment binders. Testing by surface plasmon resonance (SPR) found one molecule in the series to possess binding affinity equal to that of cytidine diphosphate, despite lacking any metal-coordinating phosphate groups. Close inspection of the SPR data suggest different binding stoichiometries between IspF and test compounds. Crystallographic analysis shows important variations between the binding mode of one synthesized compound and the pose of the bound fragment from which it was designed. The binding modes of these molecules add to our structural knowledge base for IspF and suggest future refinements in this compound series.
Keywords: Fragment screening, MEP pathway, IspF, Non-mevalonate, Anti-infective, SPR
Enzymes from the methyl erythritol phosphate (MEP) biosynthetic pathway are regarded to be promising targets for new anti-infective agents and herbicides.1,2 This pathway is essential in Plasmodium falciparum, Mycobacterium tuberculosis, Toxoplasma gondii, Burkholderia pseudomallei, Escherichia coli, and other infectious disease causing organisms.3–6 MEP enzymes are absent in mammalian species, which utilize the well-characterized and druggable mevalonate-dependent (MAD) pathway to synthesize isoprenoids.7 Studies on fosmidomycin have indicated clinical utility with P. falciparum infections by inhibiting the MEP enzyme IspC, thus demonstrating the druggability in this alternative route for isoprenoid production.1,8 Some cases of drug resistance have been observed for lone fosmidomycin chemotherapy, but if given in combination, fosmidomycin appears to retain its clinical activity towards P. falciparum.9 Thus novel or combination chemotherapies which target multiple MEP enzymes should minimize the possibility for resistance mutations to develop and survive in pathogenic species, with minimal off-target effects in mammalian hosts lacking the MEP pathway.
The fifth enzyme in the MEP pathway is 2C-methyl-D-erythritol-2,4-cyclo-diphosphate synthase (IspF). Sequence analysis shows over 30% identity for IspF proteins across prokaryotic and eukaryotic species.10 Structural studies show high conservation of residues essential in substrate-binding, catalysis, and other aspects of IspF enzymatic function.8 Thus compounds which inhibit the IspF enzyme from any given pathogenic organism could possess some potential for cross-species activity. Previously, we conducted fragment-based screens with IspF from B. pseudomallei (BpIspF) by nuclear magnetic resonance (NMR) spectroscopy, and by x-ray crystallography.10 Screening generated a collection of hits (Figure 1), and led to 14 fragment-bound complexes with as many as three small molecules bound in unique sites of the protein.
Figure 1.

Fragment screening hits that bind to the cytidine and zinc pockets of BpIspF.
Three distinct binding sites for fragments emerged, two of which exist in the BpIspF active site: the cytidine recognition pocket, and an adjacent pocket surrounding the catalytic zinc ion (Figure 2). Only cytosine, cytidine and cytidine phosphate derivatives were observed crystallographically in the cytidine binding pocket, despite broad chemical diversity and metabolite variants, including other close analogs of cytidine, in our screening collection.11 Conversely, a range of chemical entities with differing orientations bound in the zinc pocket. In each case, an aromatic nitrogen occupies the available metal coordination site in the fragment-bound structure.10 Structural characterization of these fragment hits provide chemical starting points as well as relative distance and orientation information, suitable to a linking strategy. We therefore set out to design and synthesize a series of molecules that connect the cytidine moiety to different zinc pocket fragment binders to form “fusion” ligands.
Figure 2.

Crystal structure of compound 3 (carbon atoms white) bound to BpIspF (PDB 3KE1) depicting the active site comprised of chains A (light green ribbon) and B (dark green ribbon). Overlaid are binding poses of fragments FOL8395 (carbon atoms cyan) and cytidine monophosphate (carbon atoms yellow) from PDB entries 3JVH and 3F0G, respectively10 Ligand oxygen atoms are colored red, nitrogen blue, and phosphorus orange. Active site residues critical for ligand binding with high conservation across pathogenic organisms are indicated. Residues 64–72 from chain A were unmodeled due to poor electron density in the diffraction data. Residues 60–63 were removed from image for clarity. Image created using PyMol 14.
Synthesis of fusion compounds began with intermediate 1, which was synthesized according to literature procedure in six steps from commercially available (+)-cytidine.12 Target fusion compounds were obtained by amide formation followed by deprotection. Two different amide coupling procedures were utilized. First, 3 was synthesized from 1 by coupling with the commercially available acid chloride 2 (Scheme 1).
Scheme 1.
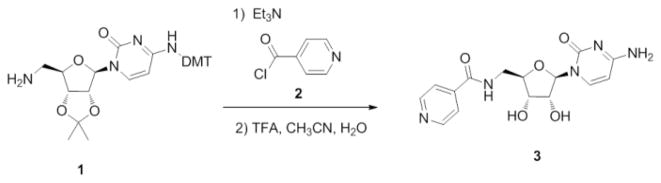
Synthesis of fusion compound 3.
Compounds 4–7 were synthesized from the amine 1 and coupled with commercially available acids using HBTU, HOBt and Et3N followed by deprotection of acetonide and DMT with trifluoroacetic acid in acetonitrile/water (Scheme 2). The final products 3–7 were purified by reverse phase HPLC to yield the products as TFA salts.
Scheme 2.
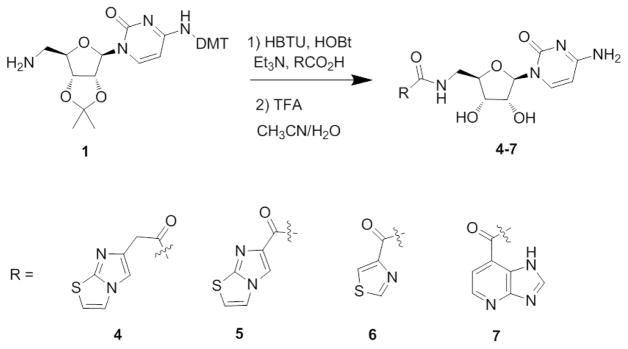
Synthesis of fusion compounds 4–7.
Dissociation constants for the fusion analogs 3–7 binding BpIspF were determined using surface plasmon resonance (SPR) similar to literature methods.12 BpIspF was immobilized using random amine, NHS/EDC coupling to a carboxylmethyl dextran SPR slide to give 3000–5000 μRIU (Refractive Index Units).
Concentration series for each compound from 2 mM to 1.7 μM (5% DMSO) were generated by serial dilution and injected over the IspF surface. Buffer-only injections were included between every compound injection at the same DMSO concentration. Data analysis was performed in scrubber (BioLogic Software), allowing double referencing (subtraction of buffer-only injection and reference flow cell). The binding responses were recorded as a function of time and at all compound concentrations, the responses reached equilibrium and dissociated rapidly, indicating that binding was reversible. Values for KD were obtained by tting the steady-state binding curves to a 1:1 Langmuir binding model.
Cytidine diphosphate (CDP) was found to have a KD of 75 μM, similar to the KD determined by Ramsden for IspF from E. coli.12 Much of this binding energy appears due to polar interactions between the phosphates of CDP and the catalytic zinc of IspF, as demonstrated in E. coli crystal structures of IspF bound to the enzymatic product.13 Our fusion series molecules generated a range of KD from 70 to 200 μM (Table 1) without the diphosphate group of CDP present in the final molecule. Interestingly, each molecule in the fusion series gave exactly 3-fold higher molecular weight-adjusted RUmax, compared to CDP and several fragment hits tested by SPR. This is strongly indicative of 3:1 stoichiometric binding for the fusion series (Table 1), a result consistent with binding to each of the three active sites in the trimeric structure of BpIspF. Binding to only one active site per trimer is also observed crystallographically for certain fragments.10 However, this result does not correlate with affinity, nor explain the 1:1 binding observed for CDP. We speculate that the apparent 1:1 binding observed for CDP and certain fragments may be due to differential affinity across the three active sites in the BpIspF trimer. Three-fold symmetry is not preserved in any crystal structures solved for BpIspF, due to slight structural differences in each protomer of the trimer in the crystal lattice.10 Thus the 3:1 versus 1:1 binding observed by SPR may represent binding affinity differences across the three active sites of the IspF trimer in solution for various small molecules.
Table 1.
Dissociation constant (KD) values of five fusion compounds (3–7), two fragments, and CDP for BpIspF as determined by SPR. Binding stoichiometry estimates were determined by molecular weight-normalized RUmax values for dose-response binding curves. Docking scores for the fusion series are also reported using BpIspF (PDB ID:3Q8H) as the starting model.
| Compound | KD (μM) | SPR-based stoichiometry [ligand: holoenzyme] | Docking Score |
|---|---|---|---|
| CDP | 75 | 1:1 | N.A. |
| 3 | 180 | 3:1 | 8.11 |
| 4 | 148 | 3:1 | 8.34 |
| 5 | 70 | 3:1 | 8.91 |
| 6 | 200 | 3:1 | 7.13 |
| 7 | 90 | 3:1 | 8.45 |
| FOL717 | 135 | 1:1 | N.A. |
| FOL955 | > 500 | 1:1 | N.A. |
To further characterize the binding interactions of our lead series, we determined crystal structures for two of the five fusion compounds bound to BpIspF: compound 3 (Figure 2; PDB: 3KE1) and compound 5 (Figure 3; PDB: 3Q8H). Similar to IspF from other species, BpIspF is trimeric, with three active sites each straddling a protomer-protomer interface in the holoenzyme. A single catalytic zinc is observed in each active site for these structures, consistent with previous data.10 A second metal is implicated in the catalytic mechanism of IspF, but was not observed in the electron density of either structure.13 Electron density for compound 3 is visible in all three active sites of the BpIspF trimer, showing full occupancy in each site. Similarly, electron density for compound 5 appears in each of the three active sites. Although the cytidine moiety is fully occupied in all three sites, the biaryl moiety of compound 5 has partial occupancy in one site. This observed differential occupancy, and the lack of three-fold crystallographic symmetry for trimeric BpIspF, further suggest differential binding affinity across the three active sites. Such differences are most likely manifested in dynamic differences, atomic motions difficult to capture crystallographically, but consistent with binding stoichiometry differences observed by SPR.
Figure 3.

Crystal structure of compound 5 (carbon atoms white) bound to BpIspF (PDB 3Q8H) depicting the active site comprised of chains A (light green) and B (dark green). Overlaid are binding poses of fragments FOL717 (carbon atoms cyan) and cytidine monophosphate (carbon atoms yellow) from PDB entries 3IKF and 3F0G, respectively10 Ligand oxygen atoms are colored red, nitrogen blue, phosphorus orange, and sulfur gold. When compound 5 is bound, a water molecule (red asterisk) occupies the available zinc coordination site. Active site residues critical for ligand binding with high conservation across pathogenic organisms are highlighted. Residues 60–67 removed from image for clarity. Image created using PyMol 14.
For compound 3, the orientation of the cytidine group is preserved, nearly identical to that of the free cytidine when bound (Figure 2). The cytidine moiety of fusion compound 5 contacts the backbone of one monomer at Ala102, Pro105 and Ala108, with the 2′- and 3′-ribosyl hydroxyls making hydrogen bonds with the Asp58 side chain of the other monomer. The same interactions are preserved in fusion compound 5, but with a puckered ribosyl ring (Figure 3). Both fusion compounds bind across a protomer-protomer interface within the active site, as is the case with CDP and the native substrate binding. This cross-protomer binding provides a structural basis for an increase in the structural integrity of the active site after each fusion compound is bound.
The pyridyl nitrogen in compound 3 is bonded directly to the zinc ion in IspF, recapitulating that of the original fragment FOL8395 (Figure 2). This data supports the importance of zinc binding groups for inhibitor design, with interactions that can substitute for the highly polar metal-phosphate contact made with the native substrate. However, the crystal structure for compound 5 reveals bonding to the catalytic zinc ion through a bridging water molecule, a different pose from the original fragment FOL717 crystallized with BpIspF (Figure 3). It is notable that compound 5 loses direct contact with catalytic zinc, yet generates the strongest KD in the series, similar to that of CDP (Table 1). Hence the increase in binding affinity most likely arises from hydrophobic interactions between the bicyclic heteroaromatic group of compound 5 and the erythritol pocket above zinc in the BpIspF active site. This is reflected in the relatively high affinity generated for FOL717, the fragment from which it was designed (Table 1).
To further probe the hydrophobic interactions in our fusion series, molecular docking studies were conducted using SYBYL X-2.0. The structure of PDB entry 3Q8H, was prepared for docking studies using Surflex-Dock Geom by removal of ligand and waters. Fusion compounds were then docked using standard parameters to acquire docking scores and correlate these to the binding affinity as determined by SPR (Table 1). Comparison of predicted and experimental binding modes of compounds provide information on the reliability of docking models, and highlight areas to target for compound design. As shown in Figure 4, the cytosine portion of the docked fusion molecules overlap well with the crystallographic binding conformation. There is difference between the ribose portions in the docked and experimental binding modes, and further differences observed in the orientation of the amide bond. The heterocyclic group. tends to occupy a lipophilic pocket created by Ile 59, Phe63 and Leu78 (Figure 4). Overall, docking scores indicate compound 5 to have the highest binding affinity and compound 6 as the weakest binder, which compares well with the KD values determined by SPR (Table 1).
Figure 4.

Docked poses of compound 5 (magenta) and compound 7 (orange) overlaid on the crystal structure for compound 5 (white) bound to BpIspF (green) (non-carbon ligand atoms are colored as in Figure 3). Docking poses show favorable binding near a lipophilic pocket created by Ile59, Phe63 and Leu78, supported by crystallographic data. Residues 65 – 67 removed for clarity. Image created using PyMol 14.
All five fusion compounds were tested for anti-bacterial activity against Burkholderia thailandensis and anti-malarial activity against Plasmodium falciparum D6, C235, W2 strains according to published procedures.15,16 The fusion compounds were determined to be inactive at the highest concentrations tested, 1 mM for anti-bacterial assay and 40 μM for anti-malarial assay (data not shown). This inactivity may be due to the negative LogP of these compounds. Further optimization of other series based upon the fragment hits is in progress.
In this paper we present our initial effort in structure-based lead design based upon fragment hits. The fusion molecules connect the cytidine and zinc binding groups. The binding modes of the fusion molecules add to our structural knowledge base for BpIspF and will help in the design of new IspF inhibitors based upon fragment screening hits.
Supplementary Material
Acknowledgments
We acknowledge Northern Illinois University for supporting this work. This project has been funded in part with Federal funds from the National Institute of Allergy and Infectious Diseases, National Institutes of Health, Department of Health and Human Services, under Contract Nos. HHSN272200700057C and HHSN272201200025C. The authors would like to acknowledge Dr. Peggy Cotter and Mathew Byrd at the University of North Carolina for antibacterial assay. Dr. Richard Sciotti and Patricia Lee and the Walter Reed Army Institute for Research for anti-malarial assays. X-ray coordinates for crystal structures of IspF bound to compound 3 (PDB ID 3KE1) and compound 5 (PDB ID 3Q8H) have been deposited with the RCSB and are available in the Protein Data Bank (www.pdb.org).
Footnotes
Supplementary data associated with this article can be found, in the online version, at
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References and notes
- 1.Jomaa H, Wiesner J, Sanderbrand S, Altincicek B, Weidemeyer C, Hintz M, Turbachova I, Eberl M, Zeidler J, Lichtenthaler HK, Soldati D, Beck E. Science. 1999;285:1573. doi: 10.1126/science.285.5433.1573. [DOI] [PubMed] [Google Scholar]
- 2.Odom AR. PLoS Pathog. 2011;7:e1002323. doi: 10.1371/journal.ppat.1002323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Rohmer M, Knani M, Simonin P, Sutter B, Sahm H. Biochem J. 1993;295(Pt 2):517. doi: 10.1042/bj2950517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Eisenreich W, Bacher A, Arigoni D, Rohdich F. Cell Mol Life Sci. 2004;61:1401. doi: 10.1007/s00018-004-3381-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Missinou MA, Borrmann S, Schindler A, Issifou S, Adegnika AA, Matsiegui PB, Binder R, Lell B, Wiesner J, Baranek T, Jomaa H, Kremsner PG. Lancet. 2002;360:1941. doi: 10.1016/S0140-6736(02)11860-5. [DOI] [PubMed] [Google Scholar]
- 6.Wiesner J, Borrmann S, Jomaa H. Parasitol Res. 2003;90(Suppl 2):S71. doi: 10.1007/s00436-002-0770-9. [DOI] [PubMed] [Google Scholar]
- 7.Slater EE, MacDonald JS. Drugs. 1988;36(Suppl 3):72. doi: 10.2165/00003495-198800363-00016. [DOI] [PubMed] [Google Scholar]
- 8.Wiesner J, Henschker D, Hutchinson DB, Beck E, Jomaa H. Antimicrob Agents Chemother. 2002;46:2889. doi: 10.1128/AAC.46.9.2889-2894.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ruangweerayut R, Looareesuwan S, Hutchinson D, Chauemung A, Banmairuroi V, Na-Bangchang K. Malar J. 2008;7:225. doi: 10.1186/1475-2875-7-225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Begley DW, Hartley RC, Davies DR, Edwards TE, Leonard JT, Abendroth J, Burris CA, Bhandari J, Myler PJ, Staker BL, Stewart LJ. J Struct Funct Genomics. 2011;12:63. doi: 10.1007/s10969-011-9102-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Davies DR, Mamat B, Magnusson OT, Christensen J, Haraldsson MH, Mishra R, Pease B, Hansen E, Singh J, Zembower D, Kim H, Kiselyov AS, Burgin AB, Gurney ME, Stewart LJ. J Med Chem. 2009;52:4694. doi: 10.1021/jm900259h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ramsden NL, Buetow L, Dawson A, Kemp LA, Ulaganathan V, Brenk R, Klebe G, Hunter WN. J Med Chem. 2009;52:2531. doi: 10.1021/jm801475n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Steinbacher S, Kaiser J, Wungsintaweekul J, Hecht S, Eisenreich W, Gerhardt S, Bacher A, Rohdich F. J Mol Biol. 2002;316:79. doi: 10.1006/jmbi.2001.5341. [DOI] [PubMed] [Google Scholar]
- 14.The PyMOL Molecular Graphics System. DeLano Scientific LLC; Palo Alto, CA, USA: 2008. [Google Scholar]
- 15.Wangtrakuldee P, Byrd MS, Campos CG, Henderson MW, Zhang Z, Clare M, Masoudi A, Myler PJ, Horn JR, Cotter PA, Hagen TJ. ACS Med Chem Lett 2013. 2013;4:699. doi: 10.1021/ml400034m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Milner E, Gardner S, Moon J, Grauer K, Auschwitz J, Bathurst I, Caridha D, Gerena L, Gettayacamin M, Johnson J, Kozar M, Lee P, Leed S, Li Q, McCalmont W, Melendez V, Roncal N, Sciotti R, Smith B, Sousa J, Tungtaeng A, Wipf P, Dow G. J Med Chem. 2011;54:6277. doi: 10.1021/jm200647u. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.