

Abstract
Back ground
Pro-inflammatory cytokine tumor necrosis factor α (TNFα) induces β-adrenergic receptor (βAR) desensitization, but mechanisms proximal to the receptor in contributing to cardiac dysfunction are not known.
Methods and Results
Two different pro-inflammatory transgenic mouse models with cardiac overexpression of Myotrophin (a pro-hypertrophic molecule) or TNFα showed that TNFα alone is sufficient to mediate βAR desensitization as measured by cardiac adenylyl cyclase activity. M-mode echocardiography in these mouse models showed cardiac dysfunction paralleling βAR desensitization independent of sympathetic overdrive. TNFα-mediated βAR desensitization that precedes cardiac dysfunction is associated with selective upregulation of G-protein coupled receptor kinase 2 (GRK2) in both the mouse models. In vitro studies in β2 AR overexpressing HEK 293 cells showed significant βAR desensitization, GRK2 upregulation and recruitment to the βAR complex following TNFα. Interestingly, inhibition of PI3K abolished GRK2-mediated βAR phosphorylation and GRK2 recruitment upon TNFα. Furthermore, TNFα-mediated βAR phosphorylation was not blocked with βAR antagonist propranolol. Additionally, TNFα administration in transgenic mice with cardiac overexpression of Gβγ sequestering peptide βARK-ct could not prevent βAR desensitization or cardiac dysfunction showing that GRK2 recruitment to the βAR is Gβγ independent. siRNA knock down of GRK2 resulted in loss of TNFα-mediated βAR phosphorylation. Consistently, cardiomyocytes from mice with cardiac-specific GRK2 ablation normalized the TNFα-mediated loss in contractility showing that TNFα-induced βAR desensitization is GRK2 dependent.
Conclusions
TNFα-induced βAR desensitization is mediated by GRK2 and is independent of Gβγ uncovering a hitherto unknown cross-talk between TNFα and βAR function providing the underpinnings of inflammation-mediated cardiac dysfunction.
Keywords: βAR, Heart failure, TNFα, GRK2, PI3K, Inflammation, TNFR2
Introduction
Elevated levels of circulating cytokines are observed in a range of cardiac diseases including heart failure myocarditis and sepsis-induced cardiac dysfunction.1 Multiple cytokines including tumor necrosis factor α (TNFα) and interleukin-6 (IL-6) are upregulated in conditions of cardiac stress and failure.1 These cytokines contribute to left ventricular dysfunction and congestive heart failure.1 It is well known that cytokines influence cardiac mechanical function by altering intrinsic myocyte contractility and prolonged cytokine exposure leads to cardio-depressant negative inotropic effects.1 Among the cytokines, strong correlative association is found between TNFα/IL-6 and congestive heart failure.1 Studies have shown that TNFα mediates negative inotropic effects through immediate and delayed responses. Immediate effects are known to be mediated by altering intracellular Ca2+, 2 sphingolipid mediators,3 and nitric oxide (NO) derived from constitutive NO synthase (NOS).4 Delayed effects are through NO generated by inducible NOS, reactive oxygen species and/or alterations in β-adrenergic receptor (βAR) signaling.1
βARs are one of the most powerful regulators of cardiac function.5 βAR downregulation and desensitization (diminished response to catecholamines due to phosphorylation) are hallmarks of heart failure.5 Desensitization of βARs in conditions of heart failure is predominantly mediated by G-protein coupled receptor kinase 2 (GRK2, βAR kinase 1, βARK1)5 and is markedly upregulated in conditions of cardiac failure.6 Indeed, inhibition of GRK2 recruitment to the receptor complex by using GRK2 C-terminal peptide (βARK-ct) ameliorates cardiac dysfunction in mouse models of heart failure.7 Germline ablation of GRK2 results in embryonic lethality due to intrauterine heart failure.8 Generation of conditional GRK2 knockout has shown that GRK2 is a critical regulator of βAR desensitization and cardiac function.8 Cardiac-specific ablation of GRK2 resulted in enhanced basal contractility to acute βAR agonist and contrastingly has reduced cardiac responses following chronic administration of βAR agonist.8 Previous studies have shown that GRK2 associates with phosphoinositide 3-kinase gamma (PI3K γ) in the cytoplasm and recruits PI3K γ to βAR complex contributing to receptor internalization.9 Furthermore, we also have shown that GRK2 and GRK2-associated PI3K activity are significantly elevated in mouse models of heart failure10 and end-stage human heart failure9 contributing to βAR desensitization and downregulation.
Elevated levels of catecholamine during cardiac stress result in abnormal βAR signaling, in part, due to GRK2-mediated receptor desensitization.11 Although cytokines cause βAR dysfunction,1 mechanisms proximal to the receptor in cytokine-induced βAR desensitization are not well understood. To determine the underlying mechanism, we have used transgenic mouse with cardiomyocyte-specific overexpression of myotrophin (Myo-Tg,12 that has elevated TNFα) or TNFα (TNFα-Tg)13 and show that these mice have significant βAR desensitization even in the absence of sympathetic overdrive. Furthermore, using HEK 293 cells stably overexpressing FLAG-β2AR (HEK-FLAG-β2AR), HL-1 cardiac myocytes and endothelial cells we show that TNFα alone is sufficient to induce βAR desensitization. Finally, using a combination of transgenic mice with cardiomyocyte-specific overexpression of βARK-ct and cardiac-specific GRK2 knockout mice (GRK2 del), we demonstrate that TNFα-induced βAR desensitization is GRK2 dependent but importantly Gβγ independent.
Methods
Experimental Animals
Transgenic mice overexpressing myotrophin (Myo-Tg,12 cardiac TNFα concentration: 4 weeks ~ 98 ± 17 pg/ml, 8 weeks ~ 159 ± 37 pg/ml and 12 weeks ~ 226 ± 53 pg/ml), TNFα (TNFα-Tg, TNFα ~ 269 ± 70 pg/ml concentration)13 or βARK-ct peptide14 (Gift from Dr. Walter J. Koch) under the control of α-MHC promoter were used for the study. See supplemental methods for GRK2 del mice and TNFR1 (TNFα receptor 1) or TNFR2 knockout mice.15 Cardiac βAR function in the βARK-ct-Tg was assessed following isoproterenol (ISO, 30 mg/kg/day for 7 days)16 or TNFα (120 μg/kg/day for 14 days)17. “n” represents number of mice used for the study and shown in figure legends. Animals were handled according to the approved protocols and animal welfare regulation of Institutional Review Board at Cleveland Clinic.
Cell culture, Treatments, Immunoprecipitation and Immunoblotting
Standard procedures for cell culture, western immunoblotting, and immunoprecipitations were followed as described previously16 (see supplemental methods). HEK 293 cells stably overexpressing FLAG-β2AR (HEK-FLAG-β2AR) in serum free media were pre-treated for one hour with cytokines TGFβ (10 ng/mL), TNFα (10 ng/mL), IL-6 (50 ng/mL) or IL-13 (50 ng/mL). ISO (10 μM) pre-treatment was for 30 minutes. Cells were re-challenged with ISO (10 μM) for 5 minutes to measure cAMP generation. Cells were pre-treated with propranolol (100 μM, for 30 minutes) prior to TNFα treatment. “n” represents the number of independent experiments and each experiment was done in triplicates. Furthermore, endothelial cells or HL-1 myocytes were treated with TNFα (10ng/ml) for receptor function analysis (See supplemental methods for culture and treatments).
Confocal Microscopy
Confocal microscopy was performed as previously described.16 HEK-FLAG-β2AR cells were plated on to cover slips and serum starved prior to ISO or TNFα treatment (see supplemental methods).
siRNA knockdown of GRK2
The 21-mer siRNA duplex 5′-GAAGTACGAGAAGCTGGAGTT-3′ was used for targeting GRK2. All-Stars negative control siRNA and siRNA against GRK2 were custom made from QIAGEN.
Total cAMP evaluation
The cAMP content from the clarified extracts was determined according to the manufacturer’s instruction using the Biotrak [3H] cAMP assay system from GE Health Care as previously described or catch point cAMP immunoassay kit (Molecular Devices; Sunnyvale, CA).16
Purification of plasma membrane, early endosomes, and late endosomes
Purification of plasma membrane, early and late endosomes were carried out as previously described16 (see supplemental methods).
βAR density and adenylyl cyclase activity
βAR density was determined by incubating 20 μg of plasma membranes with 250 pmols of [125]I-Cyanopindolol alone or along with 40 μM alprenolol for non-specific binding as previously described.16 Adenylyl cyclase assays were performed by incubating 20 μg of membranes at 37°C for 15 min with isoproterenol or NaF in 50 μl of assay mixture containing 20 mM Tris-HCl, 0.8 mM MgCl2, 2 mM EDTA, 0.12 mM ATP, 0.05 mM GTP, 0.1 mM cAMP, 2.7 mM phosphoenolpyruvate, 0.05 IU/ml myokinase, 0.01 IU/ml pyruvate kinase and [γ-32P] ATP and generated cAMP was quantified.16
Metabolic Labeling and Receptor Phosphorylation
β2AR stable cells were starved in phosphate-free media for 2 hr, treated with 100 mCi/ml of [32]Pi for 1 hr. Following stimulation, anti-Flag antibody was used for β2AR immunoprecipitation from cell lysates and immunoprecipitates were resolved by SDS-PAGE. β2AR phosphorylation was visualized by autoradiography16.
Echocardiography
Echocardiography was performed on lightly sedated mice at respective time points using a Vevo770 (VisualSonics) echocardiographic machine as previously described.16
Determination of plasma catecholamine levels
Plasma levels of catecholamines epinephrine and norepinephrine were measured using the catecholamine assay kit Bi-CAT EIA (17-EA613–192; Alpco Diagnostics) according to the manufacturer’s instructions as previously described.18
Myocyte isolation and contractility studies
The mice were anesthetized; the excised heart was immediately cannulated with 20 gauge needle, perfused with collagenase and contractility studies were performed as previously described.16 Contractility in the isolated myocytes were assessed using IonOptix System (Myopace, Milton, MA) (details in supplemental methods).
Statistical analysis
Results are expressed as mean +/− S.E. Data were analyzed by t test for 2-group comparisons (e.g. GRK2 densitometric analysis). For comparisons of greater than 2 groups, we used one-way ANOVA, if there was 1 independent variable (e.g. adenylyl cyclase activity vs. % FS or comparisons across samples for adenylyl cyclase assays or cAMP assays), two-way ANOVA, if there were 2 independent variables (e.g. genotype and treatments like in contractility studies) and two-way repeated measures ANOVA for matched observations over time specifically with the echocardiographic measurements. To adjust for multiple comparisons, we performed Bonferroni correction to evaluate the data. A probability value of p <0.05 was considered significant.
Results
Cardiac dysfunction and βAR desensitization in Myo-Tg mice is independent of sympathetic overdrive
Echocardiographic and morphometric analysis showed that Myo-Tg mice have progressive cardiac hypertrophy and dysfunction (4 through 36 weeks) (Supplementary Fig. 1A and Supplementary Table 1).12 To test whether progressive cardiac dysfunction in Myo-Tg mice alters cardiac βAR function, plasma membrane adenylyl cyclase activity was measured at 4, 8, 12, 16 and 36 weeks. Progressive loss in adenylyl cyclase activity was observed in Myo-Tg mice from 12 through 36 weeks compared to controls (Wt) (Fig. 1A). Importantly, a direct correlation was observed between adenylyl cyclase activity and cardiac function as measured by fractional shortening (% FS) (Fig. 1B). Despite progressive cardiac dysfunction at 4 through 16 weeks in Myo-Tg mice, no alterations of βAR density was observed in plasma membranes, early or late endosomes (Supplementary Fig. 1B & C) except at 36 weeks wherein βAR distribution was similar to a classical heart failure phenotype (Supplementary Fig. 1B & C).11
Figure 1.
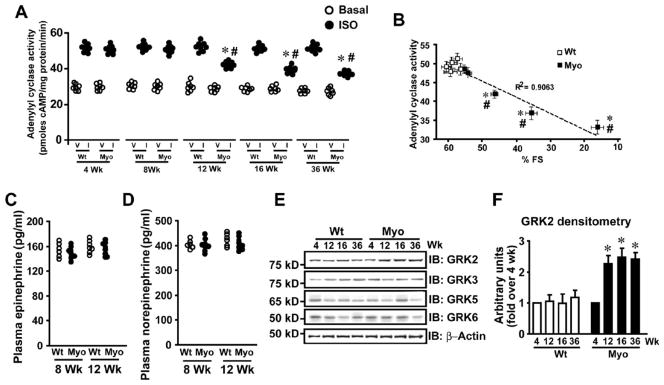
Cardiac dysfunction in Myo-Tg mice is associated with βAR desensitization and is independent of sympathetic overdrive. (A) In vitro isoproterenol (I) (closed bars) stimulated cardiac adenylyl cyclase activity compared to vehicle (V) (open bars) in the Wt and Myo-Tg mice of 4, 8, 12, 16 and 36 weeks of age (n=6–8), *p< 0.001 versus respective I Wt, #p<0.01 versus I Wt (all ages) & I Myo (4 & 8 weeks). (B) A plot of correlation between adenylyl cyclase activity and % fractional shortening (% FS) in Wt and Myo-Tg mice. *p< 0.01 versus Wt, #p<0.05 versus Myo 4 & 8 weeks. (C) Plasma epinephrine levels of Wt and Myo-Tg mice at 8 and 12 weeks (n=5–7). (D) Plasma norepinephrine levels of Wt and Myo-Tg mice at 8 and 12 weeks (n=5–7). (E) Immunoblotting for GRK 2, 3, 5, 6 and β-actin from cardiac lysates of Wt and Myo-Tg mice at 4, 12, 16, and 36 weeks of age. (F) Summary data of densitometric analysis of GRK2 (n=6–8), *p< 0.001 versus Myo-Tg at 4 weeks.
Since sympathetic overdrive is known to cause βAR desensitization in mouse models of heart failure,19, 20 we tested whether increased plasma epinephrine/norepinephrine levels in Myo-Tg mice could underlie the observed βAR desensitization at 12 weeks. Interestingly, no difference in plasma epinephrine/norepinephrine levels were observed at 8 or 12 weeks in Myo-Tg or Wt mice (Fig. 1C & D) showing that βAR desensitization is independent of sympathetic overdrive. Thus, βAR desensitization observed at 12 weeks in Myo-Tg mice may be due to elevated inflammatory cytokines.12
As GRKs proximally regulate βAR desensitization, we tested whether GRKs are altered in Myo-Tg mice. Significant upregulation of GRK2 was observed in Myo-Tg mice from 12 through 36 weeks compared to Wt (Fig. 1E & F) while other GRKs (GRK 3, 5 & 6) were not altered (Fig. 1E), suggesting that GRK2 may be the mediator of βAR desensitization in response to inflammatory cytokines. It is well known that inflammatory cytokines mediate βAR dysfunction causing negative inotropy1, 2 and Myo-Tg mice have elevated levels of cytokines including TNFα, TGFβ, IL-6, and IL-1312 that may underlie βAR dysfunction.
TNFα initiates βAR desensitization by upregulating GRK2
To ascertain whether cytokines desensitize βARs, HEK 293 cells stably overexpressing FLAG-β2AR (HEK-FLAG-β2AR) were pre-treated with a combination of cytokines altered in Myo-Tg mice12 (TNFα, IL-6, IL-13 and TGFβ) (Cyto) and challenged with βAR agonist isoproterenol (ISO) to measure cAMP generation. In addition to observing marked β2AR desensitization upon ISO pre-treatment (Fig. 2A), we also observed significant loss in cAMP generation following pretreatment with Cyto (Fig. 2A). To assess which of these cytokines mediate β2AR desensitization, cells were pre-treated with individual cytokines followed by ISO challenge. Significant reduction in cAMP generation was observed upon TNFα pre-treatment (Fig. 2B) suggesting that TNFα may be the specific cytokine mediating β2AR desensitization. To investigate whether TNFα mediates β2AR desensitization via GRKs, HEK-FLAG-β2AR cells were treated with individual cytokines TNFα, IL-6, IL-13 or TGFβ and assessed for expression of GRKs. Marked upregulation of GRK2 was observed in cells treated with TNFα with no appreciable changes in GRK2 expression with other cytokines (Fig. 2C & D). Furthermore, no alterations in GRK 3, 5 & 6 were observed with any of the cytokines (Fig. 2C & D) suggesting that TNFα is sufficient to upregulate GRK2 accounting for β2AR desensitization through phosphorylation.
Figure 2.

βAR desensitization is caused by TNFα mediated upregulation of GRK2. (A) cAMP generation following ISO challenge in HEK-FLAG-β2AR cells (ISO) compared to untreated control cells (C), ISO re-challenge following ISO pre-treatment (ISO+ISO) or ISO challenge following pre-treatment with combination of cytokines (TGFβ+TNFα+IL-6+IL-13=Cyto), (n=5–6), *p< 0.001 versus ISO, **p< 0.001 versus ISO. (B) cAMP generation following ISO challenge in HEK-FLAG-β2AR cells (ISO) compared to untreated control cells (C), ISO re-challenge following ISO pre-treatment (ISO/ISO), ISO challenge following TNFα pre-treatment (ISO/TNFα), ISO challenge following TGFβ pre-treatment (ISO/TGFβ), ISO challenge following IL-6 pre-treatment (ISO/IL-6) or ISO challenge following IL-13 pre-treatment (ISO/IL-13), (n=4–5), *p< 0.001 versus ISO, **p< 0.005 versus ISO, TGFβ, IL-6 & IL-13. (C) Immunoblots of GRK 2, 3, 5, and 6 following treatment of HEK-FLAG-β2AR cells with TGFβ, TNFα, IL-6 or IL-13 for 60 minutes. (D) Densitometric analysis of the same. (n=5–6), *p< 0.001 versus C. (E) FLAG-β2AR phosphorylation was visualized by confocal microscopy using anti-phospho-β2AR antibody (green) following ISO stimulation or TNFα treatment for 5 or 60 minutes. Nucleus was visualized by DAPI (blue) staining. Scale bar: 10 μm.
To test whether TNFα treatment alters receptor phosphorylation, HEK-FLAG-β2AR cells were treated with ISO or TNFα and β2AR phosphorylation was assessed by confocal microscopy. Significant β2AR phosphorylation (green) was observed after 5 minutes of ISO treatment (positive control) (Fig. 2E, b & j). While no β2AR phosphorylation was observed by 5 minutes of TNFα (Fig. 2E, c & k), significant β2AR phosphorylation (green) was visualized by 60 minutes (Fig. 2E, d & l) suggesting that TNFα can mediate βAR phosphorylation. Consistently, significant β2AR phosphorylation with no changes in adenylate cyclase expression (Supplementary Fig. 1D) was observed in HL-1 cardiac-myoblasts following TNFα.
GRK2 mediates TNFα-induced βAR phosphorylation
To determine the mechanism underlying TNFα mediated-βAR desensitization, plasma membranes from HEK-FLAG-β2AR cells were immunoblotted for phospho-β2AR and GRK2 upon TNFα. Significant recruitment of GRK2 was observed upon TNFα (Fig. 3A, lower panel) with corresponding increase in β2AR phosphorylation as assessed by anti-phospho-β2AR antibody (Fig. 3A, upper panel). β2ARs expression was assessed by FLAG immunoblotting (Fig. 3A, middle panel). To test for recruitment of GRK2 to the βAR complex, FLAG-β2ARs were immunoprecipitated from the plasma membranes and immunoblotted for phospho-β2AR and co-immunoprecipitating GRK2. Significant GRK2 recruitment to the β2AR complex associated with a marked increase in β2AR phosphorylation (Fig. 3B, upper and lower panel) was observed with TNFα. To alternatively test whether TNFα treatment results in β2AR phosphorylation, the HEK-FLAG-β2AR cells were subjected to metabolic labeling with radio-labeled 32Pi. Following metabolic labeling, the cells were stimulated with TNFα or ISO and FLAG-β2ARs were immunoprecipitated and autoradiography was performed to assess for β2AR phosphorylation. Significant β2AR phosphorylation was observed upon TNFα when compared to untreated control cells (Fig. 3C, Supplementary Fig. 2A). In addition, mutation analysis of the GRK phosphorylation sites8, 21 showed significant decrease in TNFα mediated β2AR phosphorylation as shown by metabolic labeling studies (Figure 3D, Supplementary Fig. 2B) or by the use of anti-phospho-β2AR antibody (Supplementary Fig. 2C & D). In addition, we further tested the specificity of the anti-phospho β2AR antibody by using specific blocking peptide. Western immunoblotting showed that blocking peptide completely displaced the antibody from binding (Supplementary Fig. 3D & E) demonstrating the specificity of the anti-phospho β2AR antibody. To assess whether GRK2 mediates TNFα-induced β2AR desensitization, GRK2 was depleted by siRNA. Immunoblotting showed significant loss of TNFα-induced β2AR phosphorylation (Fig. 3E, upper panel & 3F) following GRK2 knockdown (Fig. 3E, middle panel) suggesting that β2AR desensitization upon TNFα is GRK2 dependent.
Figure 3.

βAR desensitization by TNFα is GRK2 dependent. (A) Immunoblots to assess phospho-β2AR, GRK2 and FLAG-β2AR on the plasma membranes of TNFα and ISO treated HEK-FLAG-β2AR cells, (n=4). (B) Levels of β2AR phosphorylation and GRK2 co-immunoprecipitating with FLAG-β2AR from plasma membrane fractions of HEK-FLAG-β2AR cells following TNFα or ISO, (n=4). (C) Representative autoradiograph showing β2AR phosphorylation upon TNFα or ISO treatment following metabolic [32]Pi labeling of HEK-FLAG-β2AR cells (n=4). (D) HEK 293 cells were transfected with FLAG-β2AR Wt or Serine 355/356 mutant cDNA constructs, metabolically labeled with [32]Pi, treated with TNFα. Representative autoradiograph showing β2AR phosphorylation following immunoprecipitation with anti-FLAG antibody (n=4). (E) Effect of GRK2 knock down by siRNA on phosphorylation of β2ARs following the stimulation of HEK-FLAG-β2AR cells with ISO or TNFα. (F) Cumulative data showing significant loss of β2AR phosphorylation due to knock down of GRK2 by siRNA (right panel), (n=3–4), *p< 0.001 versus Vehicle (Veh), # p<0.005 versus ctrl siRNA -TNFα or ISO.
Agonist independent βAR desensitization by TNFα
It is well known that GRK2 mediates βAR desensitization in an agonist-dependent manner.5 To test whether TNFα-mediated βAR desensitization is agonist-dependent or -independent, cells were pre-treated with β-blocker (propranolol) followed by TNFα or ISO to assess β2AR phosphorylation by confocal microscopy. Significant β2AR phosphorylation was visualized with ISO (green) (Fig. 4A, panel g & i) that was abolished with propranolol pre-treatment (Fig. 4A, panels j & l). Despite propranolol pre-treatment, β2AR phosphorylation was observed with TNFα (Fig. 4A, panels p & r) suggesting that TNFα mediates β2AR phosphorylation in an agonist-independent manner. Consistently, immunoblotting showed significant β2AR phosphorylation with TNFα in presence or absence of propranolol (Fig. 4B, upper panel and bar graph).
Figure 4.

TNFα mediated βAR desensitization is agonist independent. (A) FLAG-β2AR phosphorylation was visualized by confocal microscopy using anti-phospho-β2AR antibody (green) following ISO stimulation or TNFα treatment (60 minutes) in the presence and absence of β-blocker propranolol. Nucleus was visualized by DAPI (blue) staining. Scale bar: 10 μm. (B) Immunoblots of phospho-β2AR and FLAG-β2AR following TNFα treatment of HEK-FLAG-β2AR cells in the presence or absence of propranolol. Densitometric analysis of the same is shown on the lower panel. (n=4), *p< 0.001 versus Veh, #p< 0.001 versus Veh. (C) β-Arrestin (green) recruitment to the plasma membrane was visualized by confocal microscopy using double stable cells expressing GFP-β-Arrestin and HA-β2AR following ISO or TNFα in the presence or absence of β-blocker propranolol. Scale bar: 10 μm
Since β2AR phosphorylation by GRK2 recruits β-arrestin,5 we tested whether TNFα recruits β-arrestin to β2ARs. HEK 293 cells stably overexpressing β2AR and β-arrestin-2-GFP were treated with ISO or TNFα in presence or absence of propranolol. ISO treatment resulted in significant recruitment of β-arrestin to the β2ARs (Fig. 4C, panel a) that was markedly reduced in presence of propranolol (Fig. 4C, panel b). Interestingly, we observed β-arrestin recruitment upon TNFα in presence or absence of propranolol (Fig. 4C, panel c & d) showing that TNFα mediates agonist-independent β2AR desensitization through GRK2 and β-arrestin dependent mechanisms.
GRK2 recruitment in response to TNFα is PI3K dependent
GRK2 recruitment to the βAR complex can be mediated by either Gβγ subunits of the G-protein or through phosphoinositides.22, 23 To test whether phosphoinositides generated by PI3K could play a role in GRK2 recruitment, HEK-FLAG-β2AR cells were treated with TNFα in the presence or absence of LY294002 (selective PI3K inhibitor). Significant increase in β2AR phosphorylation and GRK2 recruitment was observed on the plasma membranes following TNFα, which was abolished in the presence of LY294002 (Fig. 5A). Confocal microscopy on HEK-FLAG-β2AR cells showed significant β2AR phosphorylation upon TNFα treatment (green) (Fig. 5B, panels a & c) which was markedly reduced in the presence of LY294002 (Fig. 5B, panels d & f). Since PI3K inhibition reduces GRK2 recruitment and β2AR phosphorylation, we assessed whether PI3Kγ is activated in Myo-Tg mice and in cells treated with TNFα. Significant PI3Kγ activation and upregulation was observed in Myo-Tg mice from 12 weeks that overlaps with βAR desensitization (Supplementary Fig. 4A & B). Similarly, PI3Kγ was also upregulated in TNFα-Tg (Supplementary Fig. 4C). Marked up-regulation of PI3Kγ was observed in HEK-FLAG-β2AR cells (Fig. 5C) and significant PI3Kγ activation was observed in HL-1 cardiac-myoblasts following TNFα (Supplementary Fig. 4D). Together these studies show that phosphoinositides generated by PI3Kγ regulates GRK2 recruitment to the βAR complex upon TNFα.
Figure 5.

PI3K regulates TNFα-mediated GRK2 recruitment to the receptor complex. (A) Effect of PI3K inhibition by LY294002 on β2AR phosphorylation and GRK2 recruitment to the plasma membrane following TNFα (n=5). (B) FLAG-β2AR phosphorylation was visualized by confocal microscopy using anti-phospho-β2AR antibody (green) following TNFα in the presence or absence of PI3K inhibitor LY294002. (C) Lysates from HEK-FLAG-β2AR cells following TNFα or vehicle treatment were immunoblotted for PI3Kγ, (n=4).
Cardiac overexpression of TNFα results in βAR desensitization
To test whether TNFα mediates β2AR desensitization in vivo we used transgenic mice with myocyte-specific overexpression of TNFα (TNFα-Tg).13 TNFα-Tg mice have normal cardiac function at 6 weeks that progressively deteriorates to heart failure by 20 weeks13. Therefore, cardiac lysates from TNFα-Tg and Wt (6 & 20 weeks) were immunoblotted for GRK 2, 3, 5 or 6. Selective and significant upregulation of GRK2 was observed in the TNFα-Tg mice at both 6 and 20 weeks compared to Wt (Fig. 6A, Supplementary Fig. 3A) with no appreciable differences in GRK 3 or 6 (Fig. 6A). Interestingly, GRK5 expression was significantly reduced in TNFα-Tg mice compared to Wt (Fig. 6A). Together these data show that GRK2 is selectively upregulated with TNFα in vivo and may play a role in TNFα-induced βAR dysfunction.
Figure 6.

Cardiac dysfunction in TNFα-Tg mice is associated with βAR desensitization (A) Cardiac lysates from Wt and TNFα-Tg mice 6 or 20 weeks of age were immunoblotted for GRK 2, 3, 5, 6, phospho-β2AR and adenylyl cyclase V/VI (n=6). (B) In vitro ISO (I) (closed bars) stimulated cardiac adenylyl cyclase activity compared to vehicle (V) (open bars) in the Wt and TNFα-Tg mice of 6 or 20 weeks (n=6), *p< 0.005 versus respective I Wt samples (6 or 20 weeks). (C) βAR density on the plasma membranes isolated from the hearts of Wt and TNFα-Tg mice (n=6) of 6 or 20 weeks, *p< 0.001 versus 20 weeks Wt. (D) Plasma epinephrine (left panel) and norepinephrine (right panel) levels in Wt and TNFα-Tg mice at 6 weeks (n=6).
To determine whether β2ARs are phosphorylated, immunoblotting was performed on cardiac lysates from TNFα-Tg. Significant β2AR phosphorylation was observed at 6 and 20 weeks in TNFα-Tg compared to Wt (Fig. 6A). Analysis of βAR function in TNFα-Tg mice showed significant loss in adenylyl cyclase activity at 6 and 20 weeks (Fig. 6B). Furthermore, adenylate cyclase expression was not altered in the TNFα-Tg mice compared to Wt (Fig. 6A). Despite significant βAR desensitization by 6 weeks in TNFα-Tg, we observed no alterations in βAR density at the plasma membranes of TNFα-Tg or Wt (Fig. 6C) similar to our observation in Myo-Tg mice (Supplementary Fig. 1B). Furthermore, no differences in epinephrine or norepinephrine levels were observed in TNFα-Tg or Wt (Fig. 6D) showing that βAR desensitization observed in the TNFα-Tg is independent of sympathetic overdrive.
Gβγ independent recruitment of GRK2 mediates TNFα induced-βAR desensitization
GRK2 recruitment to the βAR complex is known to be mediated by Gβγ subunits.5 To determine the mechanism of TNFα-induced GRK2 recruitment to the β2AR complex, transgenic mice with cardiac-specific overexpression of βARK-ct (βARK-ct-Tg) were administered TNFα or ISO. Measurement of cardiac function by echocardiography showed significant loss of % fractional shortening in βARK-ct-Tg and Wt following TNFα treatment (Fig. 7A & B). Since significant cardiac dysfunction was observed in βARK-ct-Tg following TNFα, βAR function was assessed by adenylyl cyclase activity. Significant reduction in adenylyl cyclase activity was observed in both βARK-ct-Tg and Wt following TNFα treatment (Fig. 7C). In contrast, ISO-mediated βAR desensitization was rescued in the βARK-ct-Tg (Fig. 7C). Consistently, ISO-mediated β2AR phosphorylation was reduced in βARK-ct-Tg (Fig. 7D). However, TNFα-mediated β2AR phosphorylation was not reversed in βARK-ct-Tg (Fig. 7D) suggesting that GRK2 recruitment to β2AR complex is Gβγ independent.
Figure 7.

βAR desensitization by TNFα is Gβγ independent. (A) Representative echocardiography images from Wt or βARK-ct-Tg pre- and post-TNFα treatment for 2 weeks. (B) % fractional shortening (% FS) from Wt or βARK-ct-Tg mice with or without TNFα treatment. (n=6), *p< 0.005 versus Vehicle (both Wt and βARK-ct-Tg). (C) In vitro ISO (I) (closed bars) stimulated cardiac adenylyl cyclase activity compared to vehicle (V) (open bars) in the hearts of Wt and βARK-ct-Tg mice following 2 weeks of ISO or TNFα3 treatment. (n=6), *p< 0.05 versus in vitro ISO (I) stimulated 2 weeks vehicle treated Wt or βARK-ct-Tg or 2 weeks ISO treated βARK-ct-Tg cardiac membranes. (D) Upper panel: Cardiac lysates from Wt or βARK-ct-Tg mice given a bolus of Vehicle (AA-Ascorbic Acid or Sal-saline), TNFα or ISO were immunoblotted for phospho-β2AR. Lower panel: The blot was stripped and re-probed for β-actin.
Cardiac-specific ablation of GRK2 rescues myocyte contractility
Adult cardiac myocytes were isolated from cardiac-specific GRK2 knock out (GRK2 del) mice and their littermate controls (GRK2 f/f).8 Following isolation, the myocytes were pre-incubated with ISO or TNFα and contractility was assessed in presence of ISO. Marked contractility was observed upon ISO in myocytes from GRK2 f/f and GRK2 del mice in the absence of ISO or TNFα pre-treatment (Fig. 8A, B and C, black bars, positive control). Myocyte contractility was significantly reduced in GRK2 f/f following pre-treatment with ISO or TNFα (Fig. 8A, B and C, light & dark grey bars). In contrast, myocyte contractility was rescued in GRK2 del mice despite pre-treatment with ISO or TNFα (Fig. 8A, B and C, light & dark grey bars) indicating that TNFα-mediated β2AR dysfunction is GRK2 dependent.
Figure 8.

βAR desensitization by TNFα is GRK2 dependent (A) Representative tracings of isolated myocytes from GRK2 floxed mice (GRK2 f/f) or GRK2 knockout mice (GRK2 del) following pre-treatment with ISO or TNFα. (B & C) Cell-contractility measurements upon ISO in myocytes from GRK2 f/f or GRK2 del mice pre-treated with Veh, ISO or TNFα. *p < 0.01 versus ISO + ISO GRK2 f/f; #p < 0.01 versus TNFα + ISO GRK2 f/f (n =5, ~30 cells/experiment). (D) Plasma membrane from mouse aortic endothelial cells of Wt, TNFR1 or TNFR2 knock out mice (TNFR1−/− or TNFR2−/−) treated with Veh or TNFα were immunoblotted for phospho-β2AR. The blots were stripped and re-probed for GRK2 and β-actin. (E) TNFR1 was immunoprecipitated from cardiac lysates of Wt or TNFR2−/− mice and TNFR2 was immunoprecipitated from Wt or TNFR1−/− mice and immunoblotted for co-immunoprecipitating GRK2 (n=6). (F) Illustration depicting mechanism of TNFα-mediated desensitization of βAR through TNFR2 and GRK2.
Differential βAR phosphorylation by TNFα receptors (TNFRs)
TNFα can mediate its responses through TNFα receptors TNFR1 and TNFR2.24 To determine the contribution of these receptor subtypes in βAR desensitization, we used aortic endothelial cells which have good representation of βARs from TNFR1 or TNFR2 knockout mice.25 Significant β2AR phosphorylation and GRK2 recruitment to the plasma membrane was observed in endothelial cells from Wt and TNFR1 knockout mice (Fig. 8D, upper and middle panel) that was markedly reduced in TNFR2 knockout mice (Fig. 8D, upper and middle panel) following TNFα. To investigate whether GRK2 is recruited to the TNFα receptor, cardiac TNFR1 or TNFR2 was immunoprecipitated and immunoblotted for co-immunoprecipitating GRK2. Significant levels of GRK2 co-immunoprecipitated with TNFR2 in the Wt or TNFR1 knockout mice (Fig. 8E). While in contrast, we observed reduced co-immunoprecipitation of GRK2 with TNFR1 from Wt or TNFR2 knockout mice (Fig. 8E). These studies show that TNFR2 may preferentially recruit GRK2 to the plasma membranes. Together our data show that TNFαrecruits GRK2 to the plasma membranes in a Gβγ independent manner.
Discussion
In this study, we show that βAR dysfunction is independent of sympathetic overdrive in conditions of inflammation. Furthermore, the observed βAR dysfunction is associated with selective upregulation of GRK2 in two pro-inflammatory mouse models of heart failure (Myo-Tg and TNFα-Tg). Our in vitro and in vivo studies show that TNFα alone is sufficient to induce βAR dysfunction. TNFα treatment of HEK-FLAG-β2AR cells or cardiac overexpression of TNFα resulted in marked upregulation of GRK2. Interestingly, TNFα-induced βAR desensitization is agonist-independent as βAR phosphorylation is observed despite the presence of βAR antagonist propranolol. Importantly, TNFα-mediated βAR desensitization is independent of Gβγ subunits as cardiac-overexpression of βARK-ct peptide did not rescue βAR dysfunction in response to TNFα. Inhibition of PI3K significantly reduced GRK2 recruitment and β2AR phosphorylation upon TNFα. Cardiac ablation of GRK2 (GRK2 del) was able to normalize the reduction in myocyte contractility following pre-treatment with TNFα. Furthermore, studies from TNFR1 or TNFR2 knockout mice show that TNFR2 preferentially recruits GRK2 mediating β2AR phosphorylation. Therefore, our study has identified a cross-talk between TNFα and βAR function that accounts for the reduced cardiac contractility observed in conditions of inflammation.
Elevated inflammatory cytokines are an underlying cause for cardiac dysfunction in patients with congestive heart failure, myocarditis, or sepsis-associated cardiac dysfunction.1, 26, 27 Accumulating evidence from previous studies28–30 and our current study together suggests the presence of a direct cross-talk between the βAR desensitization machinery and upregulated cytokines. Indeed studies have shown that treatment of neonatal rat myocytes with supernatants from activated immune cells diminished contractility28 and have identified the cardiac suppressive components to be TNFα and IL-128. Studies in neonatal myocytes28 and human airway smooth muscle cells31 have shown that TNFα pre-disposes βARs towards decreased G-protein coupling. Although our studies have been limited to TNFα-mediated β2AR dysfunction, it is possible that TNFα could similarly mediate β1AR desensitization. Consistently, studies have shown that chronic TNFα infusion in rat results in ventricular dysfunction associated with reduced in vitro cardiomyocyte contractility32. In this context, we have observed significant βAR desensitization in Myo-Tg or TNFα-Tg mice with cytokine upregulation which is independent of catecholamine overdrive. Correspondingly, our cellular studies show that TNFα alone is sufficient to mediate βAR desensitization. Despite βAR desensitization and cardiac dysfunction, there is no downregulation of plasma membrane receptors in Myo-Tg or TNFα-Tg mice. This is in contrast to catecholamine-mediated βAR dysfunction wherein the βAR desensitization is accompanied with loss of βARs from the plasma membrane.10 We speculate that cytokine-mediated βAR desensitization may not robustly recruit subsequent components to dynamically drive receptor internalization suggesting the presence of different mechanisms yet contributing to cardiac dysfunction.
Despite the knowledge that TNFα could desensitize βARs, little is known about the mechanisms proximal to TNFα-mediated βAR desensitization. Studies using GTPγS loading have shown that TNFα-mediated receptor desensitization is upstream of G-protein coupling28. Interestingly, our studies show that TNFα selectively upregulates GRK2 without altering other ubiquitous GRKs (Fig. 6A). Data from our studies suggest that TNFα mediates βAR dysfunction upstream of G-protein coupling via GRK2 as supported by GRK2 siRNA and conditional cardiac knockout studies. Despite significant increase in GRK2 in response to TNFα, the recruitment of GRK2 to the βAR complex may not be as robust as catecholamine-mediated recruitment. Thus, both sympathetic overdrive and pro-inflammatory cytokines mediate elevation of GRK2 expression and recruitment to the βAR complex driving phosphorylation and desensitization. Importantly, in vitro and in vivo studies show that TNFα mediates upregulation of GRK2, a molecule proximal to βARs mediating receptor dysfunction.
Our studies show that TNFα-mediated βAR desensitization is agonist independent as βAR antagonist propranolol did not inhibit β2AR phosphorylation or β-arrestin recruitment to the β2AR complex upon TNFα. This observation suggests that GRK2 recruitment to the βAR complex following TNFα is independent of G-protein activation. Studies in βARK-ct-Tg mice show that cardiac-specific overexpression of βARK-ct could not prevent TNFα-mediated βAR desensitization or cardiac dysfunction strengthening the idea that GRK2 recruitment to the βARs occurs in a Gβγ independent manner. Studies have shown that GRK2 recruitment to βARs requires both Gβγ subunits and phospholipids 22, 23. Moreover, generated phospholipids are known to regulate GRK2 activity22 as mutation of the phospholipid binding site on GRK2 results in loss of βAR phosphorylation despite the presence of Gβγ binding sites22, 33. Consistently, inhibition of PI3K resulted in significant loss of β2AR phosphorylation and GRK2 recruitment following TNFα indicating that PI3K activity is required for GRK2 recruitment. Indeed, we have observed increased expression of PI3Kγ in both Myo-Tg (Supplementary Fig. 4B) and TNFα-Tg mice (Supplementary Fig. 4C). We have also observed significant increase in PI3Kγ activity in Myo-Tg mice as well as HL-1 cardiac-myoblasts further supporting the role of PI3Kγ in TNFα-βAR cross-talk. Together these data suggest that GRK2 recruitment to the β2AR complex can occur through phospholipid-dependent mechanisms independent of classical Gβγ subunits of G-proteins.
Although it is known from previous studies, ISO mediated deleterious cardiac remodeling is ameliorated by the presence TNFR2,34 it is not known which of the TNFα receptors mediate βAR desensitization. Interestingly, our studies using TNFR1 or TNFR2 knockout mice show that TNFR2 selectively recruits GRK2 in response to TNFα treatment (Fig. 8 D & E). Such an observation is intriguing given the beneficial role of TNFR2 in cardiac remodeling. We speculate that the beneficial effects of TNFR2 signaling in presence of sympathetic overdrive could be through preferential TNFR2-mediated recruitment of GRK2 to mediate βAR desensitization reducing deleterious cardiac signaling and remodeling. Thus, our studies show that TNFR2 may be a key player in regulating the TNFα-GRK2-βAR axis and its effects on cardiac function.
Identification of the TNFα-βAR cross-talk in our current studies has significant implications in obesity, diabetes, dyslipidemia and hypertension35, 36 which are all cardiovascular risk factors with elevated levels of TNFα. Indeed, TNFα is upregulated in all models of obesity and type II diabetes37 and therefore may lead to upregulation of GRK2 pre-disposing the heart towards cardiac dysfunction via βAR desensitization. In addition to implications in cardiovascular axis consistent with our studies, treatment of lung epithelial cells with Dexamethasone (an anti-inflammatory agent) resulted in significant inhibition of GRK2 expression38. Since asthma is associated with significant inflammatory response accompanied by marked β2AR dysfunction, our study suggests that TNFα-GRK2 cross-talk may underlie β2AR dysfunction in a pro-inflammatory milieu. Therefore, our studies are exciting as they establish a direct signaling pathway linking pro-inflammatory cytokine TNFα to βAR desensitization via GRK2 (Fig. 8F) providing insights on elevated inflammatory cytokines being secondary to initiation of cardiac dysfunction and progression of heart failure39.
Supplementary Material
Clinical Perspective.
It is well known that pro-inflammatory cytokine tumor necrosis factor-α (TNFα) is elevated in congestive heart failure and contributes to pathological left ventricular remodeling. The surprising failure of clinical trials on TNF blockade indicates that more needs to be understood about the role of TNFα in cardiac signaling/function to develop better therapeutic approaches. Although TNFα mediates negative inotropy potentially through β-adrenergic receptors (βARs), mechanisms underlying this process are not well understood. Our current studies show the presence of a direct cross-talk between TNFα receptor signaling and βAR function. TNFα treatment results in non-classical recruitment of G-protein coupled receptor kinase 2 (GRK2) to βARs that results in βAR phosphorylation inhibiting βAR function. Most surprisingly, TNFα mediates βAR desensitization in a βAR agonist/antagonist independent manner contrary to the current paradigm of βAR activation and signaling. This finding has significant implications as it suggests that just the presence of TNFα is sufficient to pre-dispose βARs towards dysfunction independent of the sympathetic inputs from epinephrine/norepinephrine. Importantly, TNFα may reduce the number of responsive βARs accounting for reduced myocyte contractility and deleterious remodeling. Therefore, our findings suggest that novel strategies for targeting βARs may be required to overcome the TNFα-mediated βAR dysfunction as TNFα is elevated in co-morbid conditions like hypertension, dyslipidemia, diabetes and obesity that may underlie deleterious cardiac remodeling and heart failure.
Acknowledgments
We would like to thank Dr. Robert J. Lefkowitz for FLAG-β2AR cells, Dr. Walter J. Koch for βARK-ct-Tg mice and Dr. Mark. G. Caron for HA-β2AR/GFP-β-arrestin2 cells. We would like to thank Dr. Sadashiva Karnik for constant feedback and insightful thoughts during the development and progression of the project. We would like to thank Dr. Sadashiva Karnik and Dr. Edward F. Plow for critically reading the manuscript.
Funding Sources: This work is supported in part by NIH grants HL89473 and HL89473-02S1 (S.V. Naga Prasad), NIH HL47794 (S. Sen), NIH HL29582 (P. E. DiCorleto), R01 HL087871 (G.W. Dorn II), PO1 HL091799 (A.M. Feldman), AHA post-doctoral fellowship (N.T.V and M.K.G).
Footnotes
Conflict of Interest Disclosures: None.
References
- 1.Prabhu SD. Cytokine-induced modulation of cardiac function. Circ Res. 2004;95:1140–1153. doi: 10.1161/01.RES.0000150734.79804.92. [DOI] [PubMed] [Google Scholar]
- 2.Amadou A, Nawrocki A, Best-Belpomme M, Pavoine C, Pecker F. Arachidonic acid mediates dual effect of tnf-alpha on ca2+ transients and contraction of adult rat cardiomyocytes. Am J Physiol Cell Physiol. 2002;282:C1339–1347. doi: 10.1152/ajpcell.00471.2001. [DOI] [PubMed] [Google Scholar]
- 3.Grandel U, Fink L, Blum A, Heep M, Buerke M, Kraemer HJ, Mayer K, Bohle RM, Seeger W, Grimminger F, Sibelius U. Endotoxin-induced myocardial tumor necrosis factor-alpha synthesis depresses contractility of isolated rat hearts: Evidence for a role of sphingosine and cyclooxygenase-2-derived thromboxane production. Circulation. 2000;102:2758–2764. doi: 10.1161/01.cir.102.22.2758. [DOI] [PubMed] [Google Scholar]
- 4.Stein B, Frank P, Schmitz W, Scholz H, Thoenes M. Endotoxin and cytokines induce direct cardiodepressive effects in mammalian cardiomyocytes via induction of nitric oxide synthase. J Mol Cell Cardiol. 1996;28:1631–1639. doi: 10.1006/jmcc.1996.0153. [DOI] [PubMed] [Google Scholar]
- 5.Rockman HA, Koch WJ, Lefkowitz RJ. Seven-transmembrane-spanning receptors and heart function. Nature. 2002;415:206–212. doi: 10.1038/415206a. [DOI] [PubMed] [Google Scholar]
- 6.Iaccarino G, Barbato E, Cipolletta E, De Amicis V, Margulies KB, Leosco D, Trimarco B, Koch WJ. Elevated myocardial and lymphocyte grk2 expression and activity in human heart failure. Eur Heart J. 2005;26:1752–1758. doi: 10.1093/eurheartj/ehi429. [DOI] [PubMed] [Google Scholar]
- 7.Vinge LE, Raake PW, Koch WJ. Gene therapy in heart failure. Circ Res. 2008;102:1458–1470. doi: 10.1161/CIRCRESAHA.108.173195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Matkovich SJ, Diwan A, Klanke JL, Hammer DJ, Marreez Y, Odley AM, Brunskill EW, Koch WJ, Schwartz RJ, Dorn GW., 2nd Cardiac-specific ablation of g-protein receptor kinase 2 redefines its roles in heart development and beta-adrenergic signaling. Circ Res. 2006;99:996–1003. doi: 10.1161/01.RES.0000247932.71270.2c. [DOI] [PubMed] [Google Scholar]
- 9.Naga Prasad SV, Barak LS, Rapacciuolo A, Caron MG, Rockman HA. Agonist-dependent recruitment of phosphoinositide 3-kinase to the membrane by beta-adrenergic receptor kinase 1. A role in receptor sequestration. J Biol Chem. 2001;276:18953–18959. doi: 10.1074/jbc.M102376200. [DOI] [PubMed] [Google Scholar]
- 10.Nienaber JJ, Tachibana H, Naga Prasad SV, Esposito G, Wu D, Mao L, Rockman HA. Inhibition of receptor-localized pi3k preserves cardiac beta-adrenergic receptor function and ameliorates pressure overload heart failure. J Clin Invest. 2003;112:1067–1079. doi: 10.1172/JCI18213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Perrino C, Schroder JN, Lima B, Villamizar N, Nienaber JJ, Milano CA, Naga Prasad SV. Dynamic regulation of phosphoinositide 3-kinase-gamma activity and beta-adrenergic receptor trafficking in end-stage human heart failure. Circulation. 2007;116:2571–2579. doi: 10.1161/CIRCULATIONAHA.107.706515. [DOI] [PubMed] [Google Scholar]
- 12.Sarkar S, Leaman DW, Gupta S, Sil P, Young D, Morehead A, Mukherjee D, Ratliff N, Sun Y, Rayborn M, Hollyfield J, Sen S. Cardiac overexpression of myotrophin triggers myocardial hypertrophy and heart failure in transgenic mice. J Biol Chem. 2004;279:20422–20434. doi: 10.1074/jbc.M308488200. [DOI] [PubMed] [Google Scholar]
- 13.Kubota T, McTiernan CF, Frye CS, Slawson SE, Lemster BH, Koretsky AP, Demetris AJ, Feldman AM. Dilated cardiomyopathy in transgenic mice with cardiac-specific overexpression of tumor necrosis factor-alpha. Circ Res. 1997;81:627–635. doi: 10.1161/01.res.81.4.627. [DOI] [PubMed] [Google Scholar]
- 14.Rockman HA, Chien KR, Choi DJ, Iaccarino G, Hunter JJ, Ross J, Jr, Lefkowitz RJ, Koch WJ. Expression of a beta-adrenergic receptor kinase 1 inhibitor prevents the development of myocardial failure in gene-targeted mice. Proc Natl Acad Sci U S A. 1998;95:7000–7005. doi: 10.1073/pnas.95.12.7000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chandrasekharan UM, Siemionow M, Unsal M, Yang L, Poptic E, Bohn J, Ozer K, Zhou Z, Howe PH, Penn M, DiCorleto PE. Tumor necrosis factor alpha (tnf-alpha) receptor-ii is required for tnf-alpha-induced leukocyte-endothelial interaction in vivo. Blood. 2007;109:1938–1944. doi: 10.1182/blood-2006-05-020875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Vasudevan NT, Mohan ML, Gupta MK, Hussain AK, Naga Prasad SV. Inhibition of protein phosphatase 2a activity by pi3kgamma regulates beta-adrenergic receptor function. Mol Cell. 2011;41:636–648. doi: 10.1016/j.molcel.2011.02.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cavadini G, Petrzilka S, Kohler P, Jud C, Tobler I, Birchler T, Fontana A. Tnf-alpha suppresses the expression of clock genes by interfering with e-box-mediated transcription. Proc Natl Acad Sci U S A. 2007;104:12843–12848. doi: 10.1073/pnas.0701466104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Perrino C, Naga Prasad SV, Mao L, Noma T, Yan Z, Kim HS, Smithies O, Rockman HA. Intermittent pressure overload triggers hypertrophy-independent cardiac dysfunction and vascular rarefaction. J Clin Invest. 2006;116:1547–1560. doi: 10.1172/JCI25397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Rengo G, Lymperopoulos A, Leosco D, Koch WJ. Grk2 as a novel gene therapy target in heart failure. J Mol Cell Cardiol. 2011;50:785–792. doi: 10.1016/j.yjmcc.2010.08.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Petrofski JA, Hoopes CW, Bashore TM, Russell SD, Milano CA. Mechanical ventricular support lowers pulmonary vascular resistance in a patient with congential heart disease. Ann Thorac Surg. 2003;75:1005–1007. doi: 10.1016/s0003-4975(02)04372-2. [DOI] [PubMed] [Google Scholar]
- 21.Tran TM, Friedman J, Qunaibi E, Baameur F, Moore RH, Clark RB. Characterization of agonist stimulation of camp-dependent protein kinase and g protein-coupled receptor kinase phosphorylation of the beta2-adrenergic receptor using phosphoserine-specific antibodies. Mol Pharmacol. 2004;65:196–206. doi: 10.1124/mol.65.1.196. [DOI] [PubMed] [Google Scholar]
- 22.DebBurman SK, Ptasienski J, Benovic JL, Hosey MM. G protein-coupled receptor kinase grk2 is a phospholipid-dependent enzyme that can be conditionally activated by g protein betagamma subunits. J Biol Chem. 1996;271:22552–22562. doi: 10.1074/jbc.271.37.22552. [DOI] [PubMed] [Google Scholar]
- 23.DebBurman SK, Ptasienski J, Boetticher E, Lomasney JW, Benovic JL, Hosey MM. Lipid-mediated regulation of g protein-coupled receptor kinases 2 and 3. J Biol Chem. 1995;270:5742–5747. doi: 10.1074/jbc.270.11.5742. [DOI] [PubMed] [Google Scholar]
- 24.Defer N, Azroyan A, Pecker F, Pavoine C. Tnfr1 and tnfr2 signaling interplay in cardiac myocytes. J Biol Chem. 2007;282:35564–35573. doi: 10.1074/jbc.M704003200. [DOI] [PubMed] [Google Scholar]
- 25.Graf K, Grafe M, Dummler U, O’Connor A, Regitz-Zagrosek V, Kunkel G, Auch-Schwelk W, Fleck E. Regulation of beta-adrenergic receptors on endothelial cells in culture. Eur Heart J. 1993;14 (Suppl I):173–176. [PubMed] [Google Scholar]
- 26.Birks EJ, Latif N, Owen V, Bowles C, Felkin LE, Mullen AJ, Khaghani A, Barton PJ, Polak JM, Pepper JR, Banner NR, Yacoub MH. Quantitative myocardial cytokine expression and activation of the apoptotic pathway in patients who require left ventricular assist devices. Circulation. 2001;104:I233–240. doi: 10.1161/hc37t1.094872. [DOI] [PubMed] [Google Scholar]
- 27.Ohtsuka T, Hamada M, Hiasa G, Sasaki O, Suzuki M, Hara Y, Shigematsu Y, Hiwada K. Effect of beta-blockers on circulating levels of inflammatory and anti-inflammatory cytokines in patients with dilated cardiomyopathy. J Am Coll Cardiol. 2001;37:412–417. doi: 10.1016/s0735-1097(00)01121-9. [DOI] [PubMed] [Google Scholar]
- 28.Gulick T, Chung MK, Pieper SJ, Lange LG, Schreiner GF. Interleukin 1 and tumor necrosis factor inhibit cardiac myocyte beta-adrenergic responsiveness. Proc Natl Acad Sci U S A. 1989;86:6753–6757. doi: 10.1073/pnas.86.17.6753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Chung MK, Gulick TS, Rotondo RE, Schreiner GF, Lange LG. Mechanism of cytokine inhibition of beta-adrenergic agonist stimulation of cyclic amp in rat cardiac myocytes. Impairment of signal transduction. Circ Res. 1990;67:753–763. doi: 10.1161/01.res.67.3.753. [DOI] [PubMed] [Google Scholar]
- 30.Gulick T, Chung MK, Pieper SJ, Schreiner GF, Lange LG. Immune cytokine inhibition of beta-adrenergic agonist stimulated cyclic amp generation in cardiac myocytes. Biochem Biophys Res Commun. 1988;150:1–9. doi: 10.1016/0006-291x(88)90478-0. [DOI] [PubMed] [Google Scholar]
- 31.Shore SA, Moore PE. Regulation of beta-adrenergic responses in airway smooth muscle. Respir Physiol Neurobiol. 2003;137:179–195. doi: 10.1016/s1569-9048(03)00146-0. [DOI] [PubMed] [Google Scholar]
- 32.Bozkurt B, Kribbs SB, Clubb FJ, Jr, Michael LH, Didenko VV, Hornsby PJ, Seta Y, Oral H, Spinale FG, Mann DL. Pathophysiologically relevant concentrations of tumor necrosis factor-alpha promote progressive left ventricular dysfunction and remodeling in rats. Circulation. 1998;97:1382–1391. doi: 10.1161/01.cir.97.14.1382. [DOI] [PubMed] [Google Scholar]
- 33.Carman CV, Barak LS, Chen C, Liu-Chen LY, Onorato JJ, Kennedy SP, Caron MG, Benovic JL. Mutational analysis of gbetagamma and phospholipid interaction with g protein-coupled receptor kinase 2. J Biol Chem. 2000;275:10443–10452. doi: 10.1074/jbc.275.14.10443. [DOI] [PubMed] [Google Scholar]
- 34.Garlie JB, Hamid T, Gu Y, Ismahil MA, Chandrasekar B, Prabhu SD. Tumor necrosis factor receptor 2 signaling limits beta-adrenergic receptor-mediated cardiac hypertrophy in vivo. Basic Res Cardiol. 2011;106:1193–1205. doi: 10.1007/s00395-011-0196-6. [DOI] [PubMed] [Google Scholar]
- 35.Park EJ, Lee JH, Yu GY, He G, Ali SR, Holzer RG, Osterreicher CH, Takahashi H, Karin M. Dietary and genetic obesity promote liver inflammation and tumorigenesis by enhancing il-6 and tnf expression. Cell. 2010;140:197–208. doi: 10.1016/j.cell.2009.12.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wellen KE, Hotamisligil GS. Inflammation, stress, and diabetes. J Clin Invest. 2005;115:1111–1119. doi: 10.1172/JCI25102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hotamisligil GS. Mechanisms of tnf-alpha-induced insulin resistance. Exp Clin Endocrinol Diabetes. 1999;107:119–125. doi: 10.1055/s-0029-1212086. [DOI] [PubMed] [Google Scholar]
- 38.Vroon A, Heijnen CJ, Kavelaars A. Grks and arrestins: Regulators of migration and inflammation. J Leukoc Biol. 2006;80:1214–1221. doi: 10.1189/jlb.0606373. [DOI] [PubMed] [Google Scholar]
- 39.Mann DL. Inflammatory mediators and the failing heart: Past, present, and the foreseeable future. Circ Res. 2002;91:988–998. doi: 10.1161/01.res.0000043825.01705.1b. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.