

Abstract
Chromosomes are folded in intricate ways inside cells and their spatial organization is intimately related to regulation of gene expression. Expression of genes can be controlled by regulatory elements that are located at large genomic distances from their target genes (in cis), or even on different chromosomes (in trans). Regulatory elements can act at large genomic distances by engaging in direct physical interactions with their target genes resulting in the formation of chromatin loops. Thus, genes and their regulatory elements come in close spatial proximity irrespective of their relative genomic positions. Analysis of interactions between genes and elements will reveal which elements regulate each gene, and will provide fundamental insights into the spatial organization of chromosomes in general.
Long-range cis- and trans- interactions can be studied at high resolution using the Chromosome Conformation Capture (3C) technology. 3C employs formaldehyde crosslinking to trap physical interactions between loci located throughout the genome. Crosslinked cells are then solubilized and chromatin is digested by a restriction enzyme. After digestion the chromatin is subjected to ligation under very dilute DNA concentrations. These conditions favor intramolecular ligation over intermolecular ligation, and thus result in selective ligation of interacting (and crosslinked) genomic elements. The crosslinks are reversed, the DNA is purified, and interaction frequencies between specific chromosomal loci can be determined by quantifying the amounts of corresponding ligation product that is formed using PCR. This chapter describes detailed protocols for 3C analysis of yeast Saccharomyces cerevisiae and mammalian chromosomes.
Keywords: DNA, chromatin looping, long-range gene regulation, trans-regulation, formaldehyde crosslinking, spatial organization
1. Introduction
The spatial organization of a genome plays an important role in genome regulation. For example, a distant regulatory element may come in direct physical contact with target genes or other regulatory elements even when separated by large genomic distances. Such long-range interactions result in the formation of chromatin loops. Long-range interactions between genes and enhancers have been directly implicated in gene regulation as disruption of these looping interactions abolishes expression (1, 2). The three-dimensional organization of the genome and the mechanisms of long-range gene regulation have been difficult to directly address, mainly due to a lack of suitable assays. The PCR-based technique described here, Chromosome Conformation Capture (3C) has proven to be a powerful method to study physical interactions between genomic elements, e.g. between promoters and enhancers, and to determine the general spatial organization of chromosomes and sub-chromosomal domains (3,4).
3C has been successfully utilized in many applications. Most studies have used 3C to detect interactions between genes and regulatory elements located within chromosomal sub-domains. For instance, analyses of the human and murine beta-globin loci have revealed long-range interactions between globin genes and several regulatory elements (5,6,7). Other 3C studies revealed looping interactions involving insulator elements and imprinting centers (8,9). Thus, it appears that many different types of regulatory elements can act over large genomic distance by engaging in physical interactions with genes and other elements.
In addition to detecting intrachromosomal contacts, 3C has also been used to detect and study functional interactions between elements located on different chromosomes. In yeast, 3C was used to detect such trans-interactions between centromeres of different chromosomes, as well as interactions between homologous chromosomes during meiosis (3). More recently, 3C studies on murine chromosomes have reported functional and highly specific trans-interactions between loci located on different chromosomes. These interactions appear to play important roles in coordinating expression of multiple genes located throughout the genome (10, 11). Together these studies suggest that cis- and trans-interactions occur frequently throughout the genome, which leads us to propose that genomes are organized as three-dimensional networks of interacting genes and regulatory elements (12). Analysis of this network of interactions, using 3C technology, promises to reveal new insights into gene expression in particular and genome regulation in general.
The 3C technology uses formaldehyde crosslinking to covalently link interacting chromatin segments in living cells (Fig. 1). Crosslinked chromatin is then digested with a restriction enzyme, followed by a ligation under dilute DNA concentrations. The crosslinks are then reversed and the DNA is purified. The resulting template, termed the 3C template, represents a library of ligation products. The abundance of a particular ligation product of two chromosomal fragments reflects how frequently these two sites interact inside of the nucleus.
Figure 1.

Schematic representation of the chromosome conformation capture (3C) technology. Formaldehyde is used to treat either yeast or human cells resulting in covalent crosslinks between interacting DNA fragments via protein interactions (indicated by gray square and black circle). The crosslinked cells are then digested, followed by ligation under dilute DNA concentrations. The crosslinks are then reversed and the DNA is purified. The PCR products are detected (PCR primers indicated by arrows) and can then be quantified by gel electrophoresis.
Upon acquisition of a 3C template, quantitative PCR is performed to determine the relative abundance of various ligation products. A control template must also be generated which contains equal amounts of every possible ligation product and serves as an excellent control of primer efficiencies. The ratio of the amount of ligation product of the 3C template and the amount of ligation product of the control template is then determined. This ratio is a quantitative measure for the frequency with which the two corresponding DNA fragments interacted inside the nucleus. Upon determination of a set of interaction frequencies among a number of loci throughout a chromatin domain, the spatial organization of that chromosomal region can be inferred, including the presence of specific looping interactions between genomic elements (Fig 2).
Figure 2.
Titration and gel quantification of yeast 3C and control templates. The left (control template) and right panels (3C template) represent a titration and quantification using two primer pair combinations (detecting interactions between loci separated by 10 kb and 80 kb respectively). The amount of PCR product is plotted versus the template DNA concentration in micrograms. For both primer pair combinations the control template yields similar amounts of PCR product while the 3C template yields significantly more PCR product for the primer pair that detect interactions between loci separated by 10 kb. The linear range for PCR amplification is found to the left of the gray line. The template DNA concentration chosen should be enough to yield sufficient PCR product to allow visualization and quantification on a gel, but should be within the linear range of PCR detection. The same template concentration must be used for all subsequent PCR reactions.
2. Materials
2.1 Preparation of 3C Template From Mammalian Cells
Mammalian cells growing in appropriate culture medium
37% formaldehyde (see Note 1) (Mallindkrodt)
2.5 M glycine
Lysing buffer I: 10 mM Tris-Cl, pH 8.0, 10 mM NaCl, 0.2% Igepal (NP-40). Make fresh on the day of the experiment.
Protease inhibitor cocktail for use with mammalian cells
Restriction enzyme and corresponding 10x restriction enzyme buffer.
1% sodium dodecyl sulfate (SDS)
10% sodium dodecyl sulfate (SDS)
10% Triton X-100
Ligation buffer (10x): 500 mM Tris-Cl, pH 7.5, 100 mM MgCl2, 100mM dithiothreitol. Store in 1 mL aliquots for up to 1 year at −80°C.
10 mg/mL bovine serum albumin (BSA)
100 mM adenosine triphosphate (ATP)
T4 DNA ligase (Invitrogen)
10 mg/mL proteinase K in TE buffer, pH 8.0
Phenol
1:1 phenol/chloroform
3M sodium acetate, pH 5.2
100% ethanol
70% ethanol
TE buffer, pH 8.0
Chloroform
10 mg/ml DNase-free RNase A (Sigma)
2.2 Preparation of Control Template from Mammalian DNA
Bacterial artificial chromosome clones
Ethidium bromide
Agarose
TBE (10x): 108 g Tris base, 55 g boric acid, 40 mL 0.5 M EDTA, pH 8.0
DNA loading buffer (see Note 2)
TE buffer, pH 8.0
Restriction enzyme and corresponding 10x restriction enzyme buffer
10 mg/mL bovine serum albumin (BSA)
1:1 phenol/chloroform
3M sodium acetate, pH 5.2
70% ethanol
100% ethanol
Ligation buffer (10x): 500 mM Tris-Cl, pH 7.5, 100 mM MgCl2, 100mM dithiothreitol. Store in 1 mL aliquots for up to 1 year at −80°C.
100 mM adenosine triphosphate (ATP)
T4 DNA ligase (Invitrogen)
Chloroform
10 mg/ml DNase-free RNase A (Sigma)
2.3 Analysis of Mammalian 3C and Control Template by Quantitative PCR
3C and Control Templates
DNA Molecular weight standard of known concentration
Ethidium bromide
Agarose
TBE (10x): 108 g Tris base, 55 g boric acid, 40 mL 0.5 M EDTA, pH 8.0
DNA loading buffer (see Note 2)
PCR buffer for mammalian templates (10x): 600 mM Tris, adjust to pH 8.9 with H2SO4, 180 mM (NH4)2SO4. Store up to 1 year at −80°C.
100 mM dNTPs (Invitrogen)
Primers designed for specific region of interest
Taq DNA polymerase (NEB)
50 mM MgSO4
2.4 Preparation of 3C Template from Yeast Cells
-
1
Saccharomyces cerevisiae cells of interest
-
2
Spheroplasting buffer I: 0.4 M sorbitol, 0.4 M KCl, 40 mM sodium phosphate buffer, pH 7.2, 0.5 mM MgCl2. Store up to 6 months at 4°C.
-
3
20 mg/mL zymolyase 100-T solution: 20 mg/mL zymolyase 100-T, 2% glucose, 50 mM Tris-Cl, pH 7.5. Make solution at least one day prior to experiment. This solution can be stored up to one month at 4°C.
-
4
MES wash buffer: 0.1 M MES, 1.2 M sorbitol, 1 mM EDTA, pH 8.0, 0.5 M MgCl2, adjust to pH 6.4 with NaOH. Store up to 6 months at 4°C.
-
5
37% formaldehyde (see Note 1) (Mallindkrodt)
-
6
2.5 M glycine
-
7
Restriction enzyme and corresponding 10x restriction enzyme buffer
-
8
1% sodium dodecyl sulfate (SDS)
-
9
10% sodium dodecyl sulfate (SDS)
-
10
10% Triton X-100
-
11
Ligation buffer (10x): 500 mM Tris-Cl, pH 7.5, 100 mM MgCl2, 100mM dithiothreitol. Store in 1 mL aliquots for up to 1 year at −80°C.
-
12
10 mg/mL bovine serum albumin (BSA)
-
13
100 mM adenosine triphosphate (ATP)
-
14
T4 DNA ligase (Invitrogen)
-
15
10 mg/mL proteinase K in TE buffer, pH 8.0
-
16
1:1 phenol/chloroform
-
17
3 M sodium acetate, pH 5.2
-
18
100% ethanol
-
19
70% ethanol
-
19
TE buffer, pH 8.0
-
20
10 mg/mL DNase-free RNase A (Sigma)
2.5 Preparation of Control Template from Yeast Genomic DNA
Saccharomyces cerevisiae cells of interest
Spheroplasting buffer II: 10 mM sodium phosphate buffer, pH 7.2, 10 mM EDTA, pH 8.0, 1% 2-mercaptoethanol, 100 μg/mL zymolyase 100-T. Store up to 6 months at 4°C.
Lysing buffer II: 0.25 M EDTA, pH 8.0, 0.5 M Tris base, 2.5% SDS. Make solution fresh on day of experiment.
20 mg/mL proteinase K in TE buffer, pH 8.0
5 M potassium acetate
80% ethanol
100% ethanol
TE buffer, pH 8.0 containing 10 μg/mL DNase-free Rnase A
1:1 phenol/chloroform
100% isopropanol
1 mg/mL bovine serum albumin (BSA)
10 mM adenosine triphosphate (ATP)
T4 DNA ligase (Invitrogen)
0.5 M EDTA, pH 8.0
10 mg/ml DNase-free RNase A (Sigma)
2.6 Analysis of Yeast 3C and Control Template by Quantitative PCR
Yeast 3C and Control Templates
DNA Molecular weight standard of known concentration
Ethidium bromide
Agarose
TBE (10x): 108 g Tris base, 55 g boric acid, 40 mL 0.5 M EDTA, pH 8.0
DNA loading buffer (see Note 2)
PCR buffer for mammalian templates (10x): 600 mM Tris, adjust to pH 8.9 with H2SO4, 180 mM (NH4)2SO4. Store up to 1 year at −80°C.
100 mM dNTPs (Invitrogen)
Primers designed for specific region of interest
Taq DNA polymerase (NEB)
3. Methods
3.1 Initial Preparation of 3C Experiments for Mammalian and Yeast Cells
3.2 Preparation of 3C Template From Mammalian Cells
Acquire 1 × 108 (see Note 5) cells grown in the appropriate culture medium for the cell type (see Note 6). Centrifuge cells at 450 × g for 10 minutes, remove the supernatant and dissolve the cell pellet in 45 mL fresh culture medium.
Crosslink cells by adding 1.35 mL of 37% formaldehyde (final concentration 1%) (see Note 1), mix well by pipetting up and down, and incubate at room temperature for 10 minutes.
To quench the reaction, add 2.5 mL of 2.5 M glycine. Mix well by pipetting up and down and incubate at room temperature for 5 minutes.
Store on ice for at least 15 minutes, centrifuge cells for 10 minutes at 800 × g and resuspend the pellet in 1 mL of ice-cold lysing buffer I containing 0.1 mL protease inhibitor cocktail. Incubate on ice for 15 minutes.
Dounce homogenize the cells on ice with pestle B by stroking 15 times, incubating on ice for 1 minute, and then stroking an additional 15 times.
Transfer cells to a microcentrifuge tube and centrifuge cells for 5 minutes at 2500 × g. Wash pelleted cells with 0.5 mL of 1x of the appropriate restriction enzyme buffer for the restriction enzyme chosen. Centrifuge washed cells under the same conditions and resuspend in 0.5 mL of 1 x restriction enzyme buffer.
Distribute cells over 20 individual microcentrifuge tubes (25 μL per tube), centrifuge 5 minutes at 2500 × g, resuspend each pellet in 362 μL of 1 x restriction enzyme buffer, add 38 μL of 1% SDS to each tube and incubate for 10 minutes at 65°C.
Add 44 μL of 10% Triton X-100 to each tube and mix gently by pipetting up and down. Add 400 U of restriction enzyme per tube, mix well, and incubate reactions overnight at the appropriate conditions corresponding to the restriction enzyme as recommended by the manufacturer.
Add 86 μL of 10% SDS per tube and incubate at 30 minutes at 65°C to inactivate the enzyme.
Add 745 μL of 10% Triton X-100, 745 of 10x ligation buffer, 80 μL of 10 mg/ml BSA, 80 μL of 10 mg/ml BSA, 5960 μL of distilled water, and 4000 cohesive end units T4 DNA ligase and incubate for 2 hours at 16°C (see Note 7).
Add 50 μL of 10 mg/mL proteinase K in TE buffer, pH 8.0, and incubate overnight at 65°C to reverse the crosslinks.
Add 50 μL of 10 mg/mL proteinase K in TE buffer, pH 8.0 and incubate for 2 hours at 42°C.
Extract DNA by adding an equal amount of phenol to each of the tubes, vortex for 30 seconds, and centrifuge for 5 minutes at 2460 × g. Collect the upper, aqueous layer and repeat the phenol extraction.
Collect the upper, aqueous layer and add an equal volume of 1:1 phenol/chloroform, vortex for 30 seconds, and centrifuge for 5 minutes at 2460 × g.
Pool the upper, aqueous layers from the 20 samples and add 1/10 volume of 3 M sodium acetate, pH 5.2, vortex briefly and add 2.5 volume of ice-cold 100% ethanol. Incubate for at least 30 minutes at −80°C and centrifuge for 20 minutes at 12,000 × g at 4°C. Decant the supernatant and resuspend the pellets in a total volume of 1 mL TE buffer, pH 8.0 and transfer to two fresh microcentrifuge tube each containing 500 μL of DNA solution.
Phenol extract by addition of equal amount of phenol, vortex for 30 seconds, and centrifuge for 5 minutes at 2460 × g. Collect the upper, aqueous layer and extract DNA by addition of an equal volume of 1:1 phenol/chloroform, vortex for 30 seconds, and centrifuge for 5 minutes at 2460 × g. Repeat the phenol/chloroform extraction.
Add an equal volume of chloroform to each tube containing the upper, aqueous layer, vortex for 30 seconds, and centrifuge for 5 minutes at 1100 × g. Collect the upper, aqueous layer.
To the upper layer, add 1/10 volume of 3M sodium acetate, pH 5.2, and vortex briefly. Precipitate DNA by adding 2.5 volumes of ice-cold 100% ethanol, mix gently, incubate at −20°C and centrifuge for 20 minutes at 18,000 × g at 4°C.
Wash the DNA pellet 5 times with 70% ethanol.
Let the DNA pellets dry completely before dissolving in a total volume of 1 mL TE buffer, pH 8.0. The obtained DNA sample is termed the 3C template.
Remove RNA by adding 2 μL of 10 mg/mL of DNase-free RNase A followed by incubation for 15 minutes at 37°C.
3.3 Preparation of Control Template from Mammalian DNA
-
1
Acquire BAC clones (see Note 8) spanning the entire region of interest (see Note 9), purify the BAC DNA by alkaline lysis with SDS (Qiagen) and estimate the DNA concentration by running 1 μL of DNA on a 0.8% agarose/0.5x TBE gel side by side with a molecular weight standard of known concentration (see Note 10).
-
2
Digest ~20 μg of BAC DNA (see Note 11,12) in 10x restriction enzyme buffer, and 10 mg/mL BSA if recommended by the manufacturer. Add 40 U/μL of restriction enzyme stock so that the amount of enzyme corresponds to 8.75% of the final digestion volume. Incubate overnight at the appropriate conditions recommended by the manufacturer.
-
3
Extract DNA by adding an equal volume of 1:1 phenol/chloroform, vortex for 30 seconds, and centrifuge for 5 minutes at 18,000 × g. Transfer the upper, aqueous layer to a fresh microcentrifuge tube.
-
4
Precipitate the DNA by adding 1/10 volume of 3M sodium acetate, pH 5.2, vortex briefly, and add 2.5 volumes of ice-cold 100% ethanol, mix gently, incubate for at least 15 minutes at −20°C, and centrifuge for 20 minutes at 18,000 × g at 4°C.
-
16
Remove the supernatant promptly, wash the DNA pellet with 1 mL of 70% ethanol, and centrifuge for 15 minutes at 18.000 × g at 4°C. Let the pellet air dry briefly (see Note 13) and resuspend in 161 μL of water. Incubate at 37°C to dissolve the DNA completely.
-
17
Combine 157 μL of digested BAC DNA, 20 μL of 10x ligation buffer, 2 μL of 10 mg/mL BSA, 2 μL of 100 mM ATP, and 7600 cohesive end units T4 DNA ligase and adjust to 200 μL final volume (see Note 7). Incubate overnight at 16°C.
-
18
Inactivate the ligase by incubating the solution for 15 minutes at 65°C.
-
19
Purify DNA by adding 200 μL of 1:1 phenol/chloroform, vortex for 30 seconds, and centrifuge for 5 minutes at 18,000 × g. Transfer the upper, aqueous layer to a microcentrifuge tube and repeat the phenol/chloroform extraction.
-
20
To the upper, aqueous layer add 200 μL of chloroform, vortex for 30 seconds, and centrifuge for 5 minutes at 18,000 × g. Transfer the upper, aqueous layer to a microcentrifuge tube.
-
21
Add 1/10 volume of 3M sodium acetate, pH 5.2, vortex briefly, and add 2.5 volumes of ice-cold 100% ethanol. Mix gently, incubate for 15 minutes at −20°C, and centrifuge for 20 minutes at 18,000 × g.
-
22
Carefully decant the supernatant, air dry the pellet slightly, and resuspend in TE buffer, pH 8.0 to obtain a DNA solution with the final concentration of ~100 ng/μL. The obtained DNA sample is termed the control template.
-
23
Remove RNA by adding 20 μL of 10 mg/mL of DNase-free RNase A followed by incubation for 15 minutes at 37°C.
3.4 Analysis of Mammalian 3C and Control Template by Quantitative PCR
Determine the DNA concentration of the obtained templates by agarose gel electrophoresis by running in parallel with a known molecular weight standard (see Note 10).
Make a series of 8–10 two-fold dilutions of template DNA starting with the highest DNA amount at 500 ng and ending with a water control.
Set up PCR reactions for each dilution in a 25 μL reaction containing 2 μL each template dilution (equaling 500 ng of DNA), 1x PCR buffer for mammalian templates, 4 mM MgSO4, 0.2 mM dNTPs, 0.4 μM of each primer, and 1 U Taq DNA polymerase (see Note 14). For quantitative results use the following hot start PCR parameters to program the thermal cycler: 1 cycle for 1 minute at 95°C; 34 cycles 1 minute at 95°C followed by 45 seconds at 65°C followed by 2 minute at 72°C: 1 cycle for 1 minute at 95°C followed by 45 seconds at 65°C followed by 8 minutes at 72°C.
Analyze the PCR products by agarose gel electrophoresis using a 1.5% agarose gel containing 0.5 TBE and 0.5 μg/mL ethidium bromide (see Note 2). Quantify PCR products using a gel documentation setup and determine the amount of PCR product obtained for each concentration of 3C and control template (see Note 15 and Fig 3). Determine the range of template concentrations that are within the linear range of PCR detection as described in the legend of Figure 3.
Using the DNA concentration determined by the PCR titration set up three reactions for each primer pair of interest for both the 3C and control template following the same PCR conditions and program as described above. Analyze and quantify the PCR products using the same conditions as described above (see Note 16).
To determine the interaction frequency for each primer pair divide the amount of PCR product obtained while using the 3C template (the average of 3 PCR reactions) by the amount of PCR product obtained by using the control template (the average of 3 PCR reactions) (see Note 17).
Combine a number of interaction frequencies from which the spatial organization of the genomic region of interest can be analyzed (see Notes 18,19). A hypothetical 3C experiment is shown in Figure 2.
Figure 3.

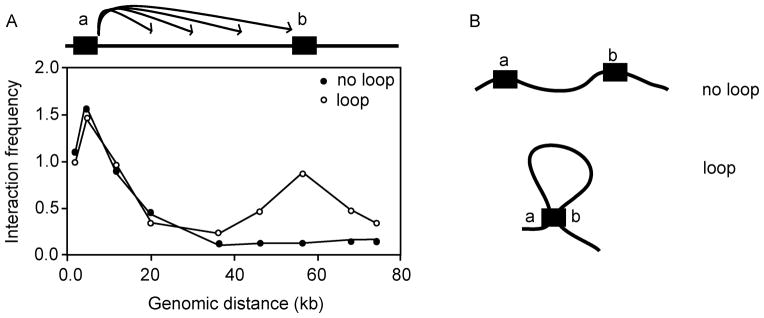
Hypothetical results for a 3C looping experiment. (A) Analysis of a looping interactions between two elements (a and b) under two conditions: The open circles indicate the expected pattern of interactions when a and b engage in a specific looping interaction. The black circles indicate the expected pattern of interactions when and b do not engage in a specific interactions. For both datasets a number of interactions were determined between element a and element b as well as a set of interactions between element a and loci located in between elements a and b and loci located downstream of b. In both cases, a interacts highly with sites very close to it and this interaction frequency decreases as the genomic distance between loci increases. However, in the case of the looping interaction, a local peak of high interaction frequencies is observed ~55 kb which is indicative of a looping interaction at the site of the sequence element b. As well as determining whether a looping interaction is present, information regarding the flexibility and the level of compaction can be inferred from the graphs (3,4). (B) A hypothetical model inferred from the datasets in (A).
3.5 Preparation of 3C Template From Yeast Cells
Acquire a 200 mL culture of the Saccharomyces cerevisiae cells for analysis at OD600 = 1.
Centrifuge cells for 10 minutes at 1250 × g, remove the supernatant promptly, and resuspend in 10 mL spheroplasting buffer I.
Add 50 μL of 20 mg/mL of zymolyase 100-T solution (see Note 20). Mix tube gently and incubate in roller drum for 40 minutes at 30°C (see Note 21).
Centrifuge cells (now spheroplasts) for 5 minutes at 2460 × g at room temperature. Wash cells two times in 10 mL MES wash buffer. Dissolve washed spheroplasts in 10 mL MES wash buffer.
Add 263 μL of 37% formaldehyde (final concentration 1%) (see Note 1), mix thoroughly, and incubate for 10 minutes at room temperature.
Quench reaction by addition of 0.5 mL of 2.5 M glycine and incubate for 5 minutes at room temperature (see Note 22).
Add 50 μL of crosslinked spheroplasts to each of 40 individual microcentrifuge tubes (see Note 23). Wash spheroplasts three times with 100 μL of 1x restriction enzyme buffer corresponding to the restriction enzyme chosen per tube. Between each wash, mix by pipetting up and down and centrifuge for 3 minutes at 18,000 × g. Resuspend washed pellet in 36.2 μL of 1x restriction enzyme buffer.
Add 3.8 μL of 1% SDS per tube, and incubate for 10 minutes at 65°C (see Note 24).
Add 4.4 μL of 10% Triton X-100 per tube and mix gently by pipetting up and down.
Add 60 U of restriction enzyme per tube, mix well, and incubate reactions overnight at the appropriate conditions recommended by the manufacturer.
Add 8.6 μL of 10% SDS per tube and incubate for 20 minutes at 65°C to inactivate the enzyme.
Add 74.5 μL of 10% Triton X-100, 74.5 μL 10x ligation buffer, 8 μL of 10 mg/ml BSA, 8 μL 100 mM ATP, 596 μL distilled water, and 800 cohesive end units of T4 DNA ligase per tube (see Note 7). Incubate for 2 hrs at 16°C.
Add 5 μL of 10 mg/mL proteinase K in TE buffer, pH 8.0, and incubate overnight at 65°C to reverse the crosslinks.
Add an additional 5 μL of 10 mg/mL proteinase K in TE buffer and incubate for 2 hours at 42°C.
Combine ten reactions to end up with 4 larger pooled reactions in 50 mL centrifuge tubes (see Note 25).
Add an equal volume of 1:1 phenol/chloroform to each of the reactions, vortex for 30 seconds, and centrifuge for 5 minutes at 2460 × g in a table top centrifuge. Remove the upper, aqueous layer promptly taking care not to include the interface layer. Repeat the phenol/chloroform extraction until the aqueous layer is clear.
Transfer clear aqueous solution to a 30 mL screw cap centrifuge tube. Add 1/10 volume of 3 M sodium acetate, pH 5.2, to the clear aqueous layer and vortex briefly. Precipitate the DNA by adding 2.5 volumes of cold 100% ethanol and mixing gently. Incubate for 15 minutes at −80°C and centrifuge for 20 minutes at 10,000 × g at 4°C.
Remove the supernatant and allow each of the 4 the DNA pellets to dry completely before resuspending in 100 μL TE buffer, pH 8.0. Pool all samples to obtain a 400 μL DNA solution if starting with 40 microcentrifuge tubes, adjusting if necessary solution.
Add an equal volume of phenol/chloroform to the purified DNA. Vortex for 30 seconds, and centrifuge for 5 minutes at 18.000 × g. Remove the upper, aqueous layer and transfer to a clean 1.7 mL microcentrifuge tube.
To the upper, aqueous layer, add 1/10 volume of 3 M sodium acetate, pH 5.2 and vortex briefly. Precipitate the DNA by adding 2.5 volumes of cold 100% ethanol and mix gently. Centrifuge for 10 minutes at 18,000 × g at 4°C. Remove the supernatant.
Wash the DNA pellet by adding 0.5 mL 70% ethanol. Resuspend the pellet and centrifuge for 5 minutes at 18.000 × g.
Remove the supernatant carefully and allow the DNA pellet to dry completely before resuspending in 100 μL TE buffer, pH 8.0. The obtained DNA sample is termed the 3C template.
Remove RNA by adding 2 μL of 10 mg/mL of DNase-free RNase A followed by incubation for 15 minutes at 37°C.
3.6 Preparation of Control Template from Yeast Genomic DNA
Acquire an overnight, saturated culture of the Saccharomyces cerevisiae strain that was used for generation of the 3C template.
Centrifuge cells for 10 minutes at 1250 × g and remove the supernatant promptly.
Dissolve in 20 mL of spheroplasting buffer II and distribute amongst 40 individual microcentrifuge tubes (0.5 ml each; see Note 26). Incubate at 37°C for 40 minutes.
Add 100 μL lysing buffer I to each tube.
Add 10 μL of 20 mg/mL proteinase K in TE buffer, pH 8.0 to each tube and incubate tubes at 65°C for 30 minutes.
Add 100 μL of 5 M potassium acetate to each tube and incubate in ice water for 10 minutes.
Centrifuge for 20 minutes at 18.000 × g at 4°C.
Transfer supernatant to fresh tubes containing 0.5 mL of ice-cold 100% ethanol, invert tubes 5 times, and spin 10 minutes at 18,000 × g.
Remove the supernatant by suction and allow DNA pellets to dry before dissolving DNA in 500 μL TE buffer, pH 8.0, containing 10 μg/mL DNase-free RNase A. Incubate for 30 minutes or until all DNA is dissolved at 37°C, occasionally tapping tubes.
Extract DNA once by adding an equal volume 1:1 phenol//chloroform, vortex for 30 seconds, and centrifuge for 5 minutes at 1100 × g.
Transfer upper, aqueous layer to a clean microcentrifuge tube, add 0.5 mL isopropanol to precipitate DNA, invert tubes 5 times and centrifuge for 10 minutes at 18,000 × g.
Wash DNA once with 80% ethanol and centrifuge for 10 minutes at 18,000 × g.
Allow DNA pellets to dry and dissolve each pellet in 100 μL TE buffer, pH 8.0. Pool samples and determine the DNA concentration by absorption spectroscopy. This is the genomic DNA to be used in generation of the control template.
Digest 20 tubes each containing 10 μg of genomic DNA isolated in previous steps in a 400 μL reaction using 60 U of the same restriction enzyme and appropriate restriction enzyme buffer as used in preparation of yeast 3C template (see Note 26). Incubate tubes for 3 hours at the appropriate conditions recommended for the enzyme.
Extract DNA once by addition of an equal volume of 1:1 phenol/chloroform, vortex for 30 seconds, and centrifuge for 5 minutes at 1100 × g.
Transfer the upper, aqueous layer to a fresh tube, add 1/10 volume of 3 M sodium acetate, pH 5.2, and vortex briefly. Precipitate DNA by adding 2.5 volume of ice-cold 100% ethanol, mix gently, centrifuge for 10 minutes at 18,000 × g. Carefully remove the supernatant.
Wash pellet by addition of 0.5 mL 80% ethanol, centrifuge for 10 minutes at 18,000 × g, carefully remove the supernatant, and allow the pellet to air dry completely.
-
Dissolve each DNA pellet in 20 μL autoclaved water.
Add 3 μL 10x ligation buffer, 3 μL of 1 mg/mL BSA, 3 μL of 10 mM ATP, and 800 cohesive end units of T4 DNA ligase to each tube (see Note 7) and incubate at 16°C for 1 hour.
Pool ten reactions together to end up with two larger reactions, and stop the reaction by adding 6 μL of 0.5 M EDTA, pH 8.0 to each pool.
Extract DNA once by addition an equal volume of 1:1 phenol/chloroform, vortex for 30 seconds, and centrifuge for 5 minutes at 1100 × g.
Transfer the upper, aqueous layer to a fresh tube. Add 1/10 volume of 3 M sodium acetate, pH 5.2, and vortex briefly. Precipitate DNA by adding 2.5 volumes of ice-cold 100% ethanol, mix gently, centrifuge for 10 minutes at 18.000 × g, carefully remove the supernatant, and allow the pellet to air dry completely.
Wash pellet by addition of 0.5 mL 80% ethanol. Centrifuge for 5 minutes at 18,000 × g and let pellet air dry completely. Dissolve each pellet in 400 μL TE buffer, pH 8.0. Pool all samples to obtain an 800 μL DNA solution. The obtained DNA sample is termed the control template.
Add 2 μL of 10 mg/mL DNase-free RNase and incubate for 15 minutes at 37°C.
3.6 Analysis of Yeast 3C and Control Template by Quantitative PCR
Determine the DNA concentration of the yeast 3C and control templates by agarose gel electrophoresis by running in parallel with a known molecular weight standard (see Note 10).
Make a series of 8–10 two-fold dilutions of template DNA starting with the highest amount at 2 μg and ending with a water control.
Set up PCR reactions for each dilution in a 50 μL reaction containing 2 μL each template dilution, 1x PCR buffer for yeast templates, 0.5 mM dNTPs, 0.4 μM of each primer, and 2.5 U Taq DNA polymerase (see Note 14). For quantitative results use the following hot start PCR parameters to program the thermal cycler: 1 cycle for 1 minute at 95°C; 32 cycles 1 minute at 95°C followed by 45 seconds at 60°C followed by 2 minute at 72°C: 1 cycle for 1 minute at 95°C followed by 45 seconds at 60°C followed by 8 minutes at 72°C.
Analyze the PCR products by agarose gel electrophoresis using a 1.5% agarose gel containing 0.5 TBE and 0.5 μg/mL ethidium bromide (see Note 2). Quantify PCR products using a gel documentation setup and determine the amount of PCR product per individual reaction (see Note 15 and Fig 3).
Using the DNA concentration determined by the PCR titration set up three reactions for each primer pair of interest for both the 3C and Control template following the same PCR conditions and program as described above. Analyze and quantify the PCR products using the same conditions as described above (see Note 16).
To determine the interaction frequency for each primer pair divide the amount of PCR product obtained while using the 3C template (in triplicate) by the amount of PCR product obtained by using the control template (in triplicate) (Note 17).
Combine a number of interaction frequencies from which the spatial organization of the genomic region of interest can be analyzed (see Notes 18,19). A hypothetical 3C experiment is shown in Figure 2.
Footnotes
The formaldehyde used for all 3C experiments should not be more than 1 year old as the effective concentration of formaldehyde gradually decreases, which will reduce the efficiency of crosslinking.
The loading buffer used should not contain bromophenol blue as this dye will run at the same position as the PCR products which will interfere with gel quantification. The use of Ficoll is encouraged.
Cut sites for the enzyme should be evenly distributed throughout the genomic region of interest and if analyzing a potential looping interaction the putative looping elements should be contained within restriction fragments that are no bigger than 10 kb and no less than 1 kb as this may result in slightly higher or slightly lower interaction frequencies, respectively. Several to many restriction fragments should be located between the two potential looping elements to add more confidence to the results and to obtain a high resolution looped structure.
The primers should be designed with the following criteria: Primers are designed unidirectionally ~80–150 basepairs 5′ of the restriction cut site so as to result in a PCR product between 160 and 300 basepairs in size. Primers are designed to be around 28 bp in length and have a GC content of ~50%. Primer combinations should be tested with the control templates of either the yeast or mammalian control template. Primers that behave aberrantly i.e. do not amplify the correct product, amplify multiple products, or produce high levels of primer dimers should be discarded and redesigned. Each primer pair should yield roughly the same amount of PCR product when using the control template. If this is not the case then discard the primer. Primers should not be designed on fragments that are greater than 10 kb or less than 1 kb in size.
The amount of cells used in this assay can be altered as necessary depending on the amount of interactions one wishes to quantify with the 3C template. The procedure described here is enough to measure ~1000 interactions.
The quality of the starting material plays an important factor in the template quality. Therefore, cells should be exponentially growing in the appropriate growth conditions and display healthy growth curves appropriate to its cell type.
When setting up ligation reactions due to the large number of tubes, a master mix of all components is typically made and distributed.
Because of the complexity of the mammalian genome, a detectable level of ligation product will not occur in a control template generated from whole genomic DNA. Therefore, a control template is generated from a bacterial artificial chromosome (BAC) clone or set of clones, which only contains the genomic region of interest.
If more than one BAC clone is needed, the experiment must be designed so that there is minimal overlap and minimal gaps between the clones as these regions will be over represented in the case of the overlap and absent in the case of the gaps.
The templates should run as a DNA species of high molecular weight; however, the appearance may differ between samples and may be smear-like. A template titration should be assembled regardless of template appearance.
If more than one BAC clone is needed to span the entire region of interest, the clones must be mixed in equimolar ratios before digestion. Therefore, the amount of DNA in the BAC preparations should be quantified. Molar quantities of BAC clones can be determined by real-time PCR using a set of universal primers that amplify a small region within the common vector backbone.
The amount of total BAC DNA should equal 20 μg if more than one BAC clone is required.
Do not let DNA pellet dry completely as this will compromise its ability to dissolve.
Two primer pairs should be chosen while preparing a template titration. The primers chosen should be of high quality and differ in genomic size separation, (i.e. one primer pair should amplify a ligation product representing an interaction between restriction fragments that are close in location, such as 10 kb, while the other should amplify a ligation product representing an interaction between restriction fragments that are far apart, such as 80 kb).
The amount of template chosen for PCR analysis and all subsequent reactions used with the template obtained should be in the linear range of amplication. If a linear range of amplication cannot be determined the cause is most likely due to high concentrations of salt. If this occurs, the template should be repurified by phenol/chloroform, ethanol precipitated, and washed with 70% ethanol as described in the above protocols.
PCR analysis of the control template and 3C template for each primer pair should be performed during the same PCR run. Further, the products obtained should be run side by side on the same gel as there is gel to gel variation.
All PCR reactions are performed in triplicate, and thus for each primer pair three interaction frequencies should be obtained. The average of these three values is the final interaction frequency. The standard error of the mean of the three interaction frequencies found should not be more than 15%. If this is the case, additional PCR reactions may be necessary to reduce the error.
If analyzing a putative looping interaction, it is imperative to test interactions with the elements and sites in between the two putative looping elements. These interactions should be less frequent than the interaction between the two looping elements (12).
If comparing interaction frequencies between two different cell types, it is necessary to first normalize the two datasets to each other to correct for differences in DNA concentrations and other experimental differences between the templates. This is done by analyzing interactions (usually 10–20) throughout a region other than the particular region of interest and that is expected to remain the same between the two cell types at hand. For each interaction the log ratio between the two datasets is then determined and the average of all log ratios will serve as the factor to normalize the 3C datasets (4, 12).
Twenty mg/mL Zymolyase 100-T will not go into solution completely and should be mixed well before addition to cells.
The efficiency of cell wall digestion should be tested by cell lysis which can be done by observation under a microscope. After addition of water to a small amount of cells ~80% of cells should burst open and exhibit hypotonic lysis within 1–2 minutes.
Cells can now be stored for up to a year at −80°C in 1 mL aliquots if desired.
Reactions should not be pooled as this will compromise the quality of the template. Tubes containing 50 μL of cells yield the best results; however, the amount of tubes may vary depending on need. The procedure described here is enough to measure ~350 reactions.
This step is essential in template generation and care should be taken to ensure proper incubation temperature.
Do not pool more than ten reactions as this will compromise the quality of the template.
Reactions should not be pooled.
References
- 1.Drissen R, Palstra R, Gillemano N, Splinter E, Grosveld F, Philipsen S, de Laat W. The active spatial organization of the β-globin locus requires the transcription factor EKLF. Genes & Dev. 2004;18:2485–2490. doi: 10.1101/gad.317004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Vakoc C, Letting DL, Gheldof N, Sawado T, Bender MA, Groudine M, Weiss MJ, Dekker J, Blobel GA. Proximity among distant regulatory elements at the beta-globin locus requires GATA-1 and FOG-1. Mol Cell. 2005;17:453–462. doi: 10.1016/j.molcel.2004.12.028. [DOI] [PubMed] [Google Scholar]
- 3.Dekker J. Capturing chromosome conformation. Science. 2002;295:1306–1311. doi: 10.1126/science.1067799. [DOI] [PubMed] [Google Scholar]
- 4.Gheldof N, Tabuchi TM, Dekker J. The active FMR1 promoter is associated with a large domain of altered chromatin conformation with embedded local histone modifications. Proc Natl Acad Sci USA. 2006;103:12463–12468. doi: 10.1073/pnas.0605343103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Toluis B, Palstra RJ, Splinter E, Grosveld F, de Laat W. Looping interaction between hypersensitive sites in the active beta-globin locus. Mol Cell. 2002;10:1435–1465. doi: 10.1016/s1097-2765(02)00781-5. [DOI] [PubMed] [Google Scholar]
- 6.Palstra RJ, Tolhuis B, Splinter E, Nijmeijer R, Grosveld F, de Laat W. The β-globin nuclear compartment in development and erythroid differentiation. Nat Genet. 2003;25:190–194. doi: 10.1038/ng1244. [DOI] [PubMed] [Google Scholar]
- 7.Dostie J, Richmond RA, Arnaout RA, Selzer RR, Lee WL, Honan A, Rubio ED, Krumm A, Lamb J, Nusbaum C, Green RD, Dekker J. Chromosome Conformation Capture Carbon Copy (5C): A massively parallel solution for mapping interactions between genomic elements. Genome Research. 2006;16:1299–1309. doi: 10.1101/gr.5571506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Liu Z, Garrard WT. Long-range interactions between three transcriptional enhancers, active Vkappa gene promoters, and a 3′ boundary sequence spanning 46 kilobases. Mol Cell Biol. 2005;25:3220–3231. doi: 10.1128/MCB.25.8.3220-3231.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Murrell A, Heeson S, Reik W. Interaction between differentially methylated regions partitions the imprinted genes Igf2 and H19 into parent-specific chromatin loops. Nat Genet. 2004;36:889–893. doi: 10.1038/ng1402. [DOI] [PubMed] [Google Scholar]
- 10.Spilianakis CG, Lalioti MD, Town T, Lee GR, Flavell RA. Interchromosomal associations between alternatively expressed loci. Nature. 2005;435:637–645. doi: 10.1038/nature03574. [DOI] [PubMed] [Google Scholar]
- 11.Lomvardas S, Barnea G, Pisapia DJ, Mendelsohn M, Kirkland J, Axel R. Interchromosomal interactions and olfactory receptor choice. Cell. 2006;126:403–413. doi: 10.1016/j.cell.2006.06.035. [DOI] [PubMed] [Google Scholar]
- 12.Dekker J. The three C’s of chromosome conformation capture: controls, controls, controls. Nat Methods. 2006;3:17–21. doi: 10.1038/nmeth823. [DOI] [PubMed] [Google Scholar]