

Abstract
Purpose
As corneal stromal cells (keratocytes) become activated prior to transition to the fibroblastic repair phenotype in response to injury (in situ) or serum (in culture), the corneal crystallins, transketolase (TKT) and aldehyde dehydrogenase (ALDH1A1), are lost. We previously showed that the serum cytokine platelet-derived growth factor-BB (PDGF), but not transforming growth factor beta2 (TGF-beta2), stimulates TKT loss. Our goal in this study was to further define molecular mechanisms for PDGF-stimulated loss of crystallins, in order to elucidate the pathway for keratocyte activation.
Methods
Freshly isolated rabbit corneal keratocytes (RCK) were plated in serum-free medium with or without PDGF and/or specific inhibitors of the PDGF-relevant signal pathway components PDGF-receptor, PI3K/AKT, or ras-initiated MAPK proteins. Intracellular TKT protein levels were quantified by immunoblotting. Ubiquinated-TKT levels were assessed by immunoprecipitation and TKT mRNA levels were quantified by quantitative RT-PCR.
Results
PDGF treatment at the same time as inhibition of PDGF-receptor, Akt, JNK and ubiquitin-proteasome pathway (UPP) prevented PDGF-induced TKT protein loss. In contrast, treatment with PDGF did not affect TKT mRNA levels.
Conclusions
The results suggest that PDGF-stimulated TKT loss is mediated via cross talk between PI3K-independent Akt and JNK. This signaling pathway leads to degradation of existing TKT protein, but does not compromise the accumulation of TKT mRNA. Therefore, cells retain the potential to reaccumulate TKT protein that is enabled by PDGF removal. These findings suggest that targeting PDGF signaling could improve repair outcomes following surgical procedures in the cornea.
Keywords: corneal keratocytes, platelet-derived growth factor-BB, aldehyde dehydrogenase, transketolase, signal transduction pathway
Introduction
Injury to connective tissues stimulates a general repair response that shares common features across organ systems including the skin, lung, or cornea [1, 2]. As part of this response, cells at the wound periphery become activated to proliferate and deposit a disorganized, fibrotic extracellular matrix (ECM) that seals off the damaged area. Fibrotic repair tissue can never reproduce the functions of the original tissue that it replaces, however, under some conditions the repair response can take on a more regenerative character. Understanding the molecular pathways controlling fibrotic repair in its many manifestations may enable the design of strategies to improve its regenerative character [3-6].
In the cornea, repair cells appear to be derived, at least in part, from resident keratocytes of the corneal stroma [7-9]. The cornea provides an ideal model for examining activating events that lead to repair phenotypes because corneal stromal cells can be isolated from other cell types and cultured without exposure to serum [4, 10]. We, and others, have characterized fibrotic progression in the cornea as a multi-step process that begins when resident keratocytes of the corneal stroma acquire “activation competency” and assume a “fibroblast” phenotype, which can progress to a more fibrotic form of repair. When corneal stromal cells are freshly isolated from the cornea and plated in culture without serum, they remain quiescent, which is characterized by being non-proliferative, having a dendritic morphology [11] and maintaining expression of the proteoglycans characteristic of a transparent cornea [12]. When exposed to serum however, they take on a phenotype over a period of three days that is very similar to that of fibroblasts seen in repair tissue [10, 13-16]. The transition is characterized by (1) a change in cell shape from dendritic to spindle-shaped associated with reorganization of the actin cytoskeleton to form stress fibers [16], (2) induction of α5-integrin expression and its heterodimerization with β1-integrin to form the fibronectin receptor [11, 16], (3) deposition of a repair-type extracellular matrix (ECM) through induced expression of new molecule such as fibronectin and SPARC [16, 17], and (4) competence to activate an IL-1α autocrine feedback loop essential for the control of collagenase expression and subsequent repair tissue remodeling [18-20].
In cornea, the early repair transition from keratocyte to fibroblast is also associated with loss of specific markers. These include secreted keratan sulfate proteoglycans (KSPGs) such as prostaglandin D synthase [21, 22] as well as members of a group of highly prevalent intracellular proteins called “corneal crystallins” by analogy to the crystallins of the eye's lens. The corneal crystallins provide protection against oxidative damage [23-25] and may also help confer transparency to the cells [26, 27]. The members of this family vary greatly among different species [26, 27]. Two proteins classified as corneal crystallins in the rabbit are the metabolic enzymes transketolase (TKT) and aldehyde dehydrogenase 1A1 (ALDH1A1) [26].
Using a model of freshly isolated rabbit corneal keratocytes (RCK), we previously determined that the serum cytokine platelet derived growth factor-BB (PDGF) stimulates loss of TKT [28]. We sought here to further define the molecular mechanisms for PDGF-stimulated keratocyte loss of corneal crystallins that serves to further understand conversion of keratocytes into repair cells. Together our findings suggest that pharmacologic agents that target PDGF signaling might be used to prevent TKT loss or stimulate TKT re-accumulation following corneal injury, thus improving the regenerative character of corneal repair.
Materials and Methods
Cell Culture
Rabbit corneal stromal keratocytes (RCK) were isolated from corneas of New Zealand White rabbits (Pel Freez Co., Rogers, AR) by collagenase digestion modified from a described method [18]. Previously, freshly-isolated keratocytes were exposed to serum overnight to aid cell attachment [29]. Because serum has been shown to reduce TKT levels [28] and serum-induced effects would impact PDGF-induced effects on TKT levels, we modified the technique to maintain RCK in serum-free Dulbecco's modified Eagle's medium (DMEM) [Invitrogen-GIBCO, Carlsbad, CA] throughout the isolation. With this modified technique, PDGF-induced TKT reduction was noted on both a per cell basis (as previously shown [29]) and when assessing equivalent protein levels (Fig. 1). Briefly, epithelium was removed and then RCK were freed from the stroma by incubation in 5 mg/ml bacterial collagenase for 1 hour at 37°C. RCK were pooled from 25 stroma, plated and maintained throughout the culture period in serum-free DMEM supplemented with 50 U/ml penicillin, 50 μg/ml streptomycin and 0.5 μg/ml fungizone amphotericin B at 37°C under 95% humidity and 5% CO2. RCK were rinsed after one day in culture and the following cell treatment reagents were added simultaneously to these original cultures and maintained with no change for four additional culture days unless otherwise indicated: 100 ng/ml Platelet-Derived Growth Factor-BB (PDGF) (R&D Systems, Minneapolis, MN); with or without 10 μM Gleevec (also known as ST1571, CGP57148B) (Novartis, Basel, Switzerland) or various cell-permeable inhibitors obtained from EMD4Biosciences (San Diego, CA): 10 μM Triciribine (Akt Inhibitor V) [does not inhibit PI3K], 2μM SP600125 (JNK Inhibitor II), 10 μM LY294002 (PI3K inhibitor), 50 μM PD98059 (MEK1/ERK Inhibitor), 10 μM SB203580 (p38 MAPK inhibitor) [does not inhibit JNK], 5 μM clasto-lactacystin beta-lactone (UPP inhibitor). Cultures were plated at 5000 cells/cm2 and were approximately 50% confluent at the end of the culture period.
Figure 1.
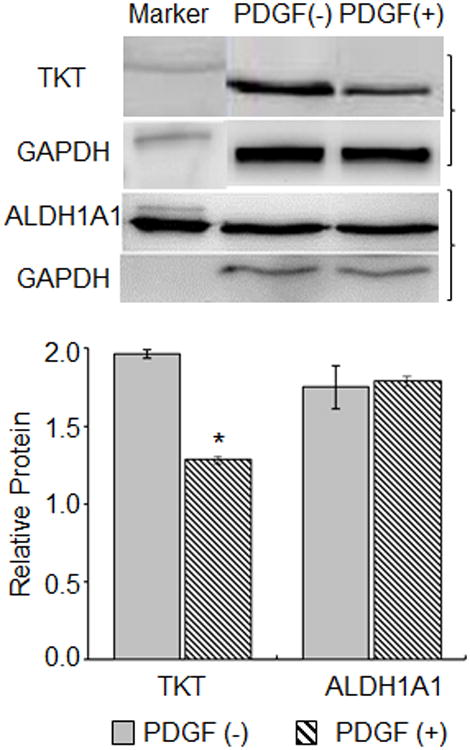
ALDH1A1 loss in RCK is not induced by PDGF. RCK plated at a density of 5 × 104 cells/cm2 were cultured for 4 days in serum-free medium without [(-), solid bars] or with [(+), striped bar] 100 ng/ml PDGF-BB (PDGF). Representative immunoblot of cytoplasmic lysates probed for TKT or ALDH1A1 and GAPDH (top panel). GADPH was used to allow better separation from the ALDH1A1. Brackets indicate immunoblot of protein of interest associated with its control. Marker indicates 90kD molecular marker on TKT blot and 300 ng purified ALDH1A1 on ALDH1A1 blot. Graph of immunoblot data (bottom panel). Relative TKT or ALDH1A1 protein, a ratio of the TKT or ALDH1A1 to GAPDH band densities for each immunoblot (n=3), expressed as average ± standard deviation. TKT, ALDH1A1and GAPDH bands are ∼70 kD, ∼55 kD and ∼37 kD, respectively based on molecular size markers (not shown). Asterisk indicates p < 0.05, PDGF (+) vs PDGF (-).
Immunoblotting
For analysis of TKT and ALDH1A1 protein levels, cell lysates were prepared in lysis buffer (50mM Tris, 1% Triton-X100, 0.1% SDS, 0.5% deoxycholate, 150mM sodium chloride, 10 μg/ml aprotinin (Roche, Indianapolis, IN), leupeptin (Roche) and 5mM sodium orthovanadate (Sigma, St. Louis, MO)). Lysates were cleared by centrifugation and protein concentrations were determined using a protein assay (BioRad) according to manufacturer's instructions. Samples, 1-5 μg total protein, were subjected to SDS-polyacrylamide gel electrophoresis on 10% Tris-glycine gels (BioRad) and electroblotted onto PVDF membranes. The membranes were blocked with 5% dry milk in TBS/Tween20. Membranes probed with 1) 1:5000 of rabbit anti-human ALDH1A1 antibody or purified ALDH1A1 protein for control [30] in 1% dry milk, followed by incubation with horseradish peroxidase (HRP)-linked anti-chicken antibody to avoid cross reacting with rabbit (Jackson ImmunoResearch, WestGrove, PA); or 2) 1:1000 of rabbit anti-mouse TKT antibody (a generous gift from Dr. Joram Piatigorsky, National Eye Institute) in 1% dry milk, followed by incubation with HRP-linked anti-rabbit antibody (Jackson ImmunoResearch) and 3) with 1:2000 of mouse anti-beta-actin or anti-GAPDH (Sigma) in 1% dry milk, followed by incubation with HRP-linked anti-mouse antibody (Jackson ImmunoResearch). HRP was visualized by enhanced chemiluminescence (Sigma). Blots were stripped (Pierce, Rockford, IL) of one set of antibodies, tested to ensure detection reagents were removed and then reprobed with the next set of antibodies. Where indicated, the ∼37 kD GAPDH was used as an internal control, rather than the ∼42 kD beta-actin, to allow better separation between the band of interest and the control band or because the treatment has been shown to alter actin [31]. The specific band for the protein of interest was identified by its relative electrophoretic mobility with respect to the size standard. Specific binding was quantified by densitometry using Quantity One imaging software, (BioRad). The protein levels were normalized to internal beta-actin or GAPDH. For immunoprecipitation, RCK lysate was mixed overnight with anti-TKT antibody, precipitated with Protein-G-Agarose (Roche) as per manufacturers' instructions and supernatants were probed as above with mouse anti-bovine ubiquitin (Millipore-Chemicon, Billerica, MA).
Quantitative Reverse Transcription Polymerase Chain Reaction (qRT-PCR)
Total RNA was extracted from the cells with a total aurum kit as per manufacturer's instructions (BioRad, Hercules, CA). Total RNA was quantified spectrophotometrically at 260 nm and 10ng/μl RNA was reverse transcribed using the iScript cDNA Synthesis Kit (BioRad). One-tenth of the cDNA was subjected to qRT-PCR using iQ SYBR Supermix (BioRad) and primers for 18S rRNA (Invitrogen) and primers for TKT on an iCycler (BioRad). We developed the TKT primers (TKT-F, 5′- ATCATTGCCAAGACCTTCAA -3′; TKT-R, 5′- GATGATCTGCTCAGCCATGT -3′) against mouse TKT (Accession: BC055336; gi:33244004) base pair 782-883 that is shared by human (BC002433, gi:33876691). Reduction in TKT mRNA levels using these primers was confirmed with RCK treated for 4 days with serum (data not shown). Melt curves indicate the primer set produced one product. TKT levels expressed as threshold cycles were normalized to an internal control, 18S. Relative TKT mRNA was calculated as 18S threshold cycles subtracted from TKT threshold cycles to the using iCycler iQ Optical System Software, version 3.0a (BioRad).
Data presentation and analysis
Immunoblot data presented graphically represent immunoblots performed on samples from different experiments (n = 2 or 3, indicated in figure legends). Band densities within each experiment were standardized to an arbitrary band density and band densities for standardized immunoblots were compiled and expressed as averages (± high/low when n=2 and ± standard deviation when n=3). Statistical analysis, ANOVA, was performed with Origin Software (Origin Lab).
Results
The continuous presence of serum in cultures of freshly isolated RCKs leads to a reduction in levels of the intracellular corneal crystallin proteins TKT and ALDH1A1 [32]. Previously, we showed that the serum cytokine PDGF has the capacity to stimulate TKT loss from these cultures [28]. For a more comprehensive context, we sought to determine whether PDGF also stimulates loss of ALDH1A1, a putative corneal crystallin in the rabbit model [33]. RCKs were isolated, plated, washed after one day in culture and then were left untreated or treated in serum-free media spiked with 100 ng/ml PDGF for an additional four days. As determined by immunoblotting, PDGF treatment selectively reduced the TKT protein level as previously shown [28]; in contrast, a new finding is that the ALDH1A1 protein level was unaffected (Fig. 1).
We next sought to identify the signal transduction pathways controlled by PDGF that leads to loss of TKT in corneal keratocytes. To begin, we assessed how antagonizing the PDGF receptor alters PDGF control over TKT in vitro. The pharmaceutical Gleevec (STI571, imatinib) is a protein-tyrosine kinase inhibitor that interferes with signaling through the PDGF-receptor [34]. RCKs in serum-free conditions were cultured in the absence or presence of 100 ng/ml PDGF and 1 μM Gleevec. As determined by immunoblotting, TKT levels were reduced in RCKs treated with PDGF, but not when the RCKs were exposed to Gleevec (Fig. 2), consistent with regulation through the protein tyrosine kinase function of the PDGF receptor.
Figure 2.
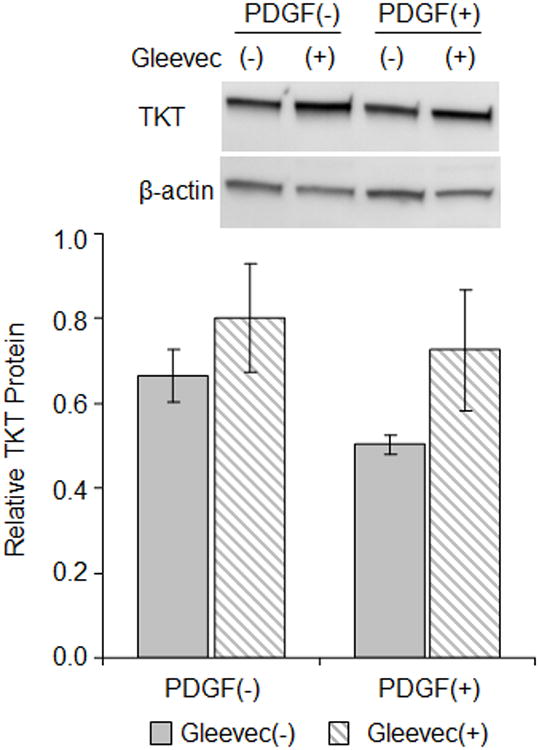
Antagonizing PDGF-receptor alters PDGF-induced loss of TKT. RCK cultured for 4 days in serum-free medium without [PDGF(-)] or with 100 ng/ml PDGF-BB [PDGF(+)] in conjunction without [(-), solid bars] or with [(+), striped bar] 10 μM ST1571 (Gleevec). Representative immunoblot of cytoplasmic lysates probed for TKT and beta-actin (top panel). Graph of immunoblot data (bottom panel). Relative TKT protein, a ratio of the TKT to the beta-actin band densities for each immunoblot (n=2), expressed as average ± high/low. TKT bands are ∼70 kD and beta-actin bands are ∼42 kD based on molecular size markers (not shown).
Following activation of PDGF tyrosine kinase, PDGF's two main pathways for signaling have been described: the PI3K-activated Akt pathways and the ras-activated MAPK [35]. We treated RCKs without or with PDGF in the presence of known cell-permeable inhibitors of signal pathway proteins relevant for PDGF signaling: PI3K (LY294002) and Akt (Triciribine); and three subfamilies of extracellular signal regulated MAPKs, p38 (SB203580), ERK (PD98059), and JNK (SP600125). As determined by immunoblotting, TKT protein levels when compared as a percent of control treatment (no PDGF) were reduced in RCKs exposed to PDGF, even in the presence of inhibitors of PI3K, ERK and p38 (Fig. 3). In contrast, TKT protein levels were unaffected (retaining the same levels as no PDGF control treatments) when cells were treated with PDGF in the presence of inhibitors to Akt and JNK. These data indicate that PDGF-stimulated loss of TKT involves activation of JNK and a PI3K-independent Akt pathway.
Figure 3.
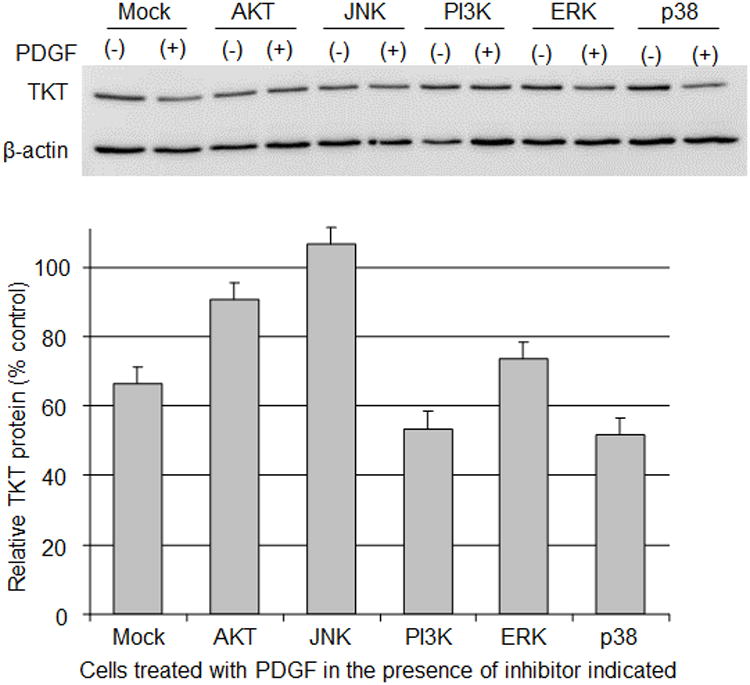
PI3K-independent Akt and JNK mediates PDGF-induced loss of TKT. RCK cultured for 4 days in serum-free medium with DMSO (mock), 10 μM Triciribine (AKT), 2μM SP600125 (JNK), 10 μM LY294002 (PI3K), 50 μM PD98059 (ERK) or10 μM SB203580 (p38) in conjunction without [PDGF(-)] or with 100 ng/ml PDGF-BB [PDGF(+)]. Representative immunoblot of cytoplasmic lysates probed for TKT and beta-actin (top panel). Graph of immunoblot data (bottom panel). Relative TKT protein, a ratio of the TKT to the beta-actin band densities, assessed for comparison as percentage of RCK with inhibitor, but without PDGF (% control). Data expressed as average ± high/low. TKT bands are ∼70 kD and beta-actin bands are ∼42 kD based on molecular size markers (not shown).
Previously, we reported that TKT loss during exposure to serum is associated with activation of the UPP [32]. In our next experiments, we directly assessed the role of the UPP in PDGF stimulated TKT loss. RCKs were cultured without or with PDGF and clasto-lactacystin β-lactone, a cell-permeable proteasome inhibitor. As determined by immunoblotting, PDGF-stimulated TKT protein loss was inhibited in the presence of clasto-lactacystin β-lactone (Fig. 4A). In addition, RCKs treated without or with PDGF were assessed for TKT ubiquitination by immunoprecipitation with TKT and blotting for ubiquitin. The levels of ubiquinated TKT were increased in RCKs treated with PDGF (Fig. 4B). These results directly support the hypothesis that PDGF-stimulated loss of TKT occurs by degradation via the UPP.
Figure 4.
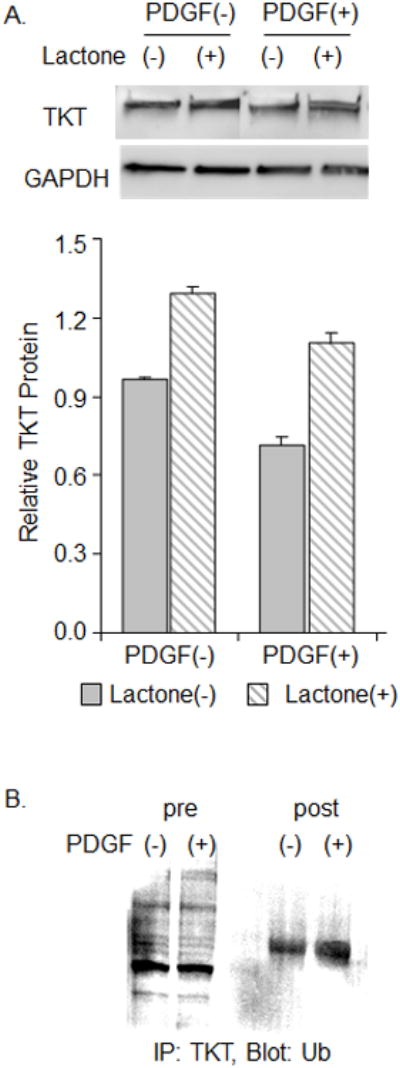
Inhibition of the UPP abrogates PDGF-induced loss of TKT as PDGF leads to increased TKT ubiquination. (A) RCK cultured for 4 days in serum-free medium without [PDGF(-)] or with 100 ng/ml PDGF-BB [PDGF(+)] in conjunction without [ (-), solid bars] or with [(+), striped bar] 5 μM clasto-lactacystin beta-lactone, a UPP inhibitor (lactone). Representative immunoblot of lysates probed for TKT and GAPDH (top panel). Graph of immunoblot data (bottom panel). Relative TKT protein, a ratio of the TKT to GAPDH band densities for each immunoblot (n=2), expressed as average ± high/low. GADPH was used here instead of beta-actin as actin responds to lactone. (B) RCK were cultured for 4 days in serum-free medium without [PDGF(-)] or with 100 ng/ml PDGF-BB [PDGF(+)]. RCK lysate was not (pre) or was (post) immunoprecipitated with TKT antibody (IP:TKT) and probed for ubiquitin (Blot:Ub).
While PDGF clearly affects the levels of TKT protein accumulated within RCKs, it might affect TKT gene transcription and accumulation of TKT mRNA very differently. In the next set of experiments, we investigated the effects of PDGF on the accumulated levels of TKT mRNA. RCKs cultured under serum-free conditions were left untreated or treated with PDGF for 4 days. Total RNA was isolated and qRT-PCR was used to quantify mRNA levels for TKT, normalized to 18S ribosomal RNA levels. In striking contrast to results when protein was assayed, PDGF treatment of RCK did not appreciably alter TKT mRNA levels (Fig. 5A).
Figure 5.
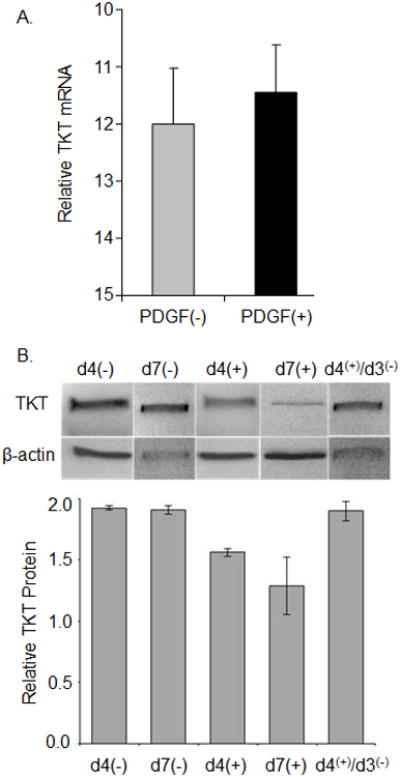
PDGF does not affect the TKT mRNA level and RCK retain the potential to re-accumulate TKT. (A) RCK cultured for 4 days in serum-free medium without [PDGF(-)] or with 100 ng/ml PDGF-BB [PDGF(+)]. Total RNA was assessed by qRT-PCR. Relative TKT mRNA calculated as threshold cycles for 18S subtracted from TKT. (B) RCK cultured under serum-free conditions were left untreated (-) or treated (+) with PDGF for 4 (d4) or 7 (d7) days; or after 4 days, PDGF was removed and RCK were cultured for an additional 3 days [d4(+)/d3(-)]. Representative immunoblot of cytoplasmic lysates probed with TKT and beta-actin antibody (top panel). Graph of immunoblot data (bottom panel). Relative TKT protein, a ratio of the TKT to the beta-actin band densities for each immunoblot (n=2) expressed as average ± high/low.
Taken together, the preceding data indicate that PDGF mediates TKT protein loss without affecting mRNA levels. This suggests that RCKs might be able to re-accumulate TKT protein once PDGF is withdrawn. To test this hypothesis, RCKs cultured under serum-free conditions were left untreated or treated with PDGF for 4 or 7 days. In a subset of the cultures treated with PDGF for 4 days, PDGF was removed and RCKs were cultured for an additional 3 days. Total protein was isolated and immunoblotting was used to quantify protein levels for TKT, normalized to β-actin levels. We found that in 7 day cultures from which PDGF was removed on the 4th day [d4(+)/d3(-)], TKT protein levels were comparable to those in cultures left untreated for 4 or 7 days. In addition PDGF-induced loss of TKT is accentuated after 7 days of PDGF treatment when compared to 4 days of PDGF treatment (Fig. 5B).
Discussion
In the cornea, fibrosis creates opacity and its contraction alters corneal shape and vision [reviewed in [36]]. Repair cells appear to be derived, at least in part, from resident keratocytes of the corneal stroma [7-9]. The specific goal of this study was to understand more about the molecular mechanisms mediating keratocyte transition to a “fibroblast” phenotype. We used a rabbit cell culture model characterized in our lab, and focused on proteins called corneal crystallins, whose loss serves as a marker for the transition from keratocyte to fibroblast. The corneal crystallins accumulate to high levels in keratocytes of the quiescent cornea, but most of this accumulated protein is lost when keratocytes are activated in vitro by exposure to serum.
In the rabbit, the metabolic enzymes transketolase (TKT) and aldehyde dehydrogenase 1A1 (ALDH1A1) [26] are two of the major corneal crystallins. Previously we showed that TKT loss in rabbit corneal keratocytes (RCK) is stimulated by treatment of RCKs with PDGF or bFGF and this loss is uncoupled from fibrotic markers whose expression is selectively stimulated by TGF-beta [26]. We showed here that treatment of RCKs with PDGF does not cause loss of the corneal crystallin ALDH1A1 [33]. This result is consistent with reports that ALDH expression is controlled by TGF-beta regulated pathways [37]. The finding provides additional support for the concept that the repair phenotype can be modulated to a more regenerative character by selectively interfering with specific regulatory pathways.
PDGF signals [reviewed in [38]] are mediated from PDGF-receptors through PI3K/Akt [39] and MAPKs subfamilies, e.g. p38, ERK and JNK [40, 41]. We presented results indicating that PDGF stimulates TKT protein loss through the PDGF-receptor because TKT loss did not occur in the presence of a PDGF-receptor inhibitor, Gleevec. Additionally, PDGF-induced TKT loss occurred in the presence of a PI3K, ERK and p38 inhibitor, but was not noted in the presence of a specific inhibitor of Akt that did not impact PI3K and of JNK that did not impact p38. These results indicate that PDGF induced loss of TKT via a signal pathway involving PI3K-independent Akt and JNK and are consistent with reports of a PI3K-independent Akt activation pathway [42] and of Akt crosstalk with JNK [43, 44].
There is an ever growing interest in an Akt pathway that is activated by mTOR, rather than PI3K, because deregulation of mTOR is an emerging theme in diverse human diseases [reviewed in [45]]. Drugs that target mTOR, such as rapamycin, already have therapeutic uses as immunosuppressants. We have preliminary data that inhibition of mTOR abrogates PDGF-induced loss of TKT (A.J. LaGier and M.E. Fini, unpublished data). However, unlike other pathway inhibitors, rapamycin treatment led to an observable loss of RCK numbers, assumedly because rapamycin leads to irreversible cell cycle arrest [46]. We have previously noted that there is an association between TKT loss and proliferation, but concluded based on cell-density studies that cell division is not sufficient to explain TKT loss [28].
In this regard, the preexisting TKT protein could passively be diluted as the cells divide in response to PDGF. For this to occur, PDGF induction of the Akt and JNK signal pathways would activate transcription factors, e.g. FKHR, CREB, AP-1, that simply downregulate TKT gene transcription. Alternatively, RCK cultured in serum-free conditions undergo little cell division [11] and would therefore, maintain preexisting TKT protein levels. Data presented here indicate that PDGF does not affect the TKT mRNA level indicating that PDGF does not simply turn ‘off’ TKT production. In addition, analysis of the TKT promoter (Accession: U90889; gi:2286041) did not reveal the presence of consensus sequences [47] appropriate for the FKHR, CREB or AP-1 transcription factors. Together, this data indicated that PDGF-induced Akt and JNK directly impacted TKT protein rather than affecting TKT gene expression.
Several reports have recently indicated that AKT and JNK play a direct role in regulating protein degradation [48-50] via UPP, which we have previously shown to be involved in serum-induced loss of TKT [32]. We demonstrate here that RCK treated with PDGF in conjunction with clasto-lactacystin beta-lactone, a UPP inhibitor, retain TKT protein levels similar to untreated RCK. In addition, RCK treated with PDGF had enhanced levels of ubiquinated TKT. These results confirm our previous findings with serum that UPP regulates TKT protein. We also noted that lactone treatment sans PDGF led to an increase in TKT protein levels. Since lactones have been shown to inactivate protein kinases such as JNK [51], this ancillary data further supports our findings that TKT loss involves JNK signaling. We suggest here that PDGF stimulates TKT protein loss because it directly compromises TKT protein stability via a PDGF-receptor driven signal pathway involving PI3K-independent Akt and JNK.
The findings that PDGF does not alter the ability of RCKs to produce TKT indicate that the RCKs retain the potential to re-accumulate TKT. Our data suggests that if PDGF is removed after it has effected a TKT loss, i.e. day 4, the TKT protein levels approach TKT protein levels noted in untreated, ‘quiescent’ RCK. We did not observe a return of the ‘quiescent’ morphology in these cells exposed to PDGF, which maintained an overall cell elongation [28] indicating that PDGF-induced changes to RCK phenotype are not reversible simply by removing PDGF.
In summary, we provide in this report a new insight into the molecular mechanisms for PDGF-induced keratocyte loss of the TKT crystallin. We suggest that PDGF-induced TKT loss is mediated via cross-talk between Akt and JNK that directly compromises existing TKT protein stability. However PDGF does not compromise new TKT protein production and therefore, corneal keratocytes retain the potential to reaccumulate TKT. Our studies suggest that targeting of the PDGF receptor could be used to retain TKT within corneal repair cells, and thus modulate repair outcomes following injury or surgical procedures in the cornea.
Acknowledgments
The authors thank Dr. Dolena Ledee for technical assistance with the immunoprecipitation, Dr. Andrius Kazlauskas from Harvard Medical School for helpful discussion about the approach and for review of the manuscript and Witte-Maria Weber from Novartis for the gift of Gleevec. This work was supported by NIH grants EY09828 (MEF), EY014801 (MEF), and EY17963 (VV) and an unrestricted grant to the University of Miami from Research to Prevent Blindness. MEF held the Research to Prevent Blindness Senior Scientific Investigator Award and the Walter G. Ross Chair in Ophthalmic Research when this work was performed.
Supported by NIH grants EY09828, EY014801 and EY17963, and an unrestricted grant to the University of Miami from Research to Prevent Blindness. MEF held the Research to Prevent Blindness Senior Scientific Investigator Award and the Walter G. Ross Chair in Ophthalmic Research.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Desmouliere A, Gabbiani G. The role of the myofibroblast in wound healing and fibrocontractive diseases. In: Clark RAF, editor. The molecular and cellular biology of wound repair. New York: Plenum Press; 1996. pp. 391–423. [Google Scholar]
- 2.Grinnell F. Fibroblasts, myofibroblasts, and wound contraction. J Cell Biol. 1994;124:401–404. doi: 10.1083/jcb.124.4.401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bisgaard HC, Thorgeirsson SS. Hepatic regeneration. The role of regeneration in pathogenesis of chronic liver diseases. Clin Lab Med. 1996;16:325–339. [PubMed] [Google Scholar]
- 4.Fini ME, Stramer BM. How the Cornea Heals: cornea-specific repair mechanisms impacting on surgical outcomes. Cornea. 2005;24(8 suppl):S2–S11. doi: 10.1097/01.ico.0000178743.06340.2c. [DOI] [PubMed] [Google Scholar]
- 5.Gurtner GC, Werner S, Barrandon Y, Longaker MT. Wound repair and regeneration. Nature. 2008;453:314–321. doi: 10.1038/nature07039. [DOI] [PubMed] [Google Scholar]
- 6.McCallion RL, Ferguson MWJ. Fetal Wound Repair. In: Clark RAF, editor. The Molecular and Cellular Biology of Wound Repair. New York: Plenum Press; 1996. pp. 561–600. [Google Scholar]
- 7.Cintron C, Kublin CL. Regeneration of corneal tissue. Dev Biol. 1977;61:346–357. doi: 10.1016/0012-1606(77)90304-9. [DOI] [PubMed] [Google Scholar]
- 8.Matsuda H, Smelser GK. Electron microscopy of corneal wound healing. Exp Eye Res. 1973;16:427–442. doi: 10.1016/0014-4835(73)90100-0. [DOI] [PubMed] [Google Scholar]
- 9.Weimar V. Healing processes in the cornea. Oxford, UK: Blackwell Scientific Publications; 1960. [Google Scholar]
- 10.Fini ME. Keratocyte and fibroblast phenotypes in the repairing cornea. Prog Retin Eye Res. 1999;18:529–551. doi: 10.1016/s1350-9462(98)00033-0. [DOI] [PubMed] [Google Scholar]
- 11.Jester JV, Barry-Lane PA, Cavanagh HD, Petroll WM. Induction of alpha-smooth muscle actin expression and myofibroblast transformation in cultured corneal keratocytes. Cornea. 1996;15:505–516. [PubMed] [Google Scholar]
- 12.Funderburgh JL, Chandler JW. Proteoglycans of rabbit corneas with nonperforating wounds. Invest Ophthalmol Vis Sci. 1989;30:435–442. [PubMed] [Google Scholar]
- 13.Beales MP, Funderburgh JL, Jester JV, Hassell JR. Proteoglycan synthesis by bovine keratocytes and corneal fibroblasts: maintenance of the keratocyte phenotype in culture. Invest Ophthalmol Vis Sci. 1999;40:1658–1663. [PubMed] [Google Scholar]
- 14.Berryhill BL, Kader R, Kane B, Birk DE, Feng J, Hassell JR. Partial restoration of the keratocyte phenotype to bovine keratocytes made fibroblastic by serum. Invest Ophthalmol Vis Sci. 2002;43:3416–3421. [PubMed] [Google Scholar]
- 15.Garana RM, Petroll WM, Chen WT, Herman IM, Barry P, Andrews P, Cavanagh HD, Jester JV. Radial keratotomy. II. Role of the myofibroblast in corneal wound contraction. Invest Ophthalmol Vis Sci. 1992;33:3271–3282. [PubMed] [Google Scholar]
- 16.Jester JV, Barry PA, Lind GJ, Petroll WM, Garana R, Cavanagh HD. Corneal keratocytes: in situ and in vitro organization of cytoskeletal contractile proteins. Invest Ophthalmol Vis Sci. 1994;35:730–743. [PubMed] [Google Scholar]
- 17.Berryhill BL, Kane B, Stramer BM, Fini ME, Hassell JR. Increased SPARC accumulation during corneal repair. Exp Eye Res. 2003;77:85–92. doi: 10.1016/s0014-4835(03)00060-5. [DOI] [PubMed] [Google Scholar]
- 18.Cook JR, Mody MK, Fini ME. Failure to activate transcription factor NF-kappaB in corneal stromal cells (keratocytes) Invest Ophthalmol Vis Sci. 1999;40:3122–3131. [PubMed] [Google Scholar]
- 19.West-Mays JA, Sadow PM, Tobin TW, Strissel KJ, Cintron C, Fini ME. Repair phenotype in corneal fibroblasts is controlled by an interleukin-1 alpha autocrine feedback loop. Invest Ophthalmol Vis Sci. 1997;38:1367–1379. [PubMed] [Google Scholar]
- 20.West-Mays JA, Strissel KJ, Sadow PM, Fini ME. Competence for collagenase gene expression by tissue fibroblasts requires activation of an interleukin 1 alpha autocrine loop. Proc Natl Acad Sci U S A. 1995;92:6768–6772. doi: 10.1073/pnas.92.15.6768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Berryhill BL, Beales MP, Hassell JR. Production of prostaglandin D synthase as a keratan sulfate proteoglycan by cultured bovine keratocytes. Invest Ophthalmol Vis Sci. 2001;42:1201–1207. [PubMed] [Google Scholar]
- 22.Funderburgh JL, Mann MM, Funderburgh ML. Keratocyte phenotype mediates proteoglycan structure: a role for fibroblasts in corneal fibrosis. J Biol Chem. 2003;278:45629–45637. doi: 10.1074/jbc.M303292200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Estey T, Chen Y, Carpenter JF, Vasiliou V. Structural and functional modifications of corneal crystallin ALDH3A1 by UVB light. PLoS One. 2010;5:e15218. doi: 10.1371/journal.pone.0015218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Estey T, Piatigorsky J, Lassen N, Vasiliou V. ALDH3A1: a corneal crystallin with diverse functions. Exp Eye Res. 2007;84:3–12. doi: 10.1016/j.exer.2006.04.010. [DOI] [PubMed] [Google Scholar]
- 25.Lassen N, Black WJ, Estey T, Vasiliou V. The role of corneal crystallins in the cellular defense mechanisms against oxidative stress. Semin Cell Dev Biol. 2008;19:100–112. doi: 10.1016/j.semcdb.2007.10.004. [DOI] [PubMed] [Google Scholar]
- 26.Jester JV, Moller-Pedersen T, Huang J, Sax CM, Kays WT, Cavangh HD, Petroll WM, Piatigorsky J. The cellular basis of corneal transparency: evidence for ‘corneal crystallins’. J Cell Sci. 1999;112(Pt 5):613–622. doi: 10.1242/jcs.112.5.613. [DOI] [PubMed] [Google Scholar]
- 27.Piatigorsky J. Enigma of the abundant water-soluble cytoplasmic proteins of the cornea: the “refracton” hypothesis. Cornea. 2001;20:853–858. doi: 10.1097/00003226-200111000-00015. [DOI] [PubMed] [Google Scholar]
- 28.Stramer BM, Fini ME. Uncoupling keratocyte loss of corneal crystallin from markers of fibrotic repair. Invest Ophthalmol Vis Sci. 2004;45:4010–4015. doi: 10.1167/iovs.03-1057. [DOI] [PubMed] [Google Scholar]
- 29.Stramer BM, Zieske JD, Jung JC, Austin JS, Fini ME. Molecular mechanisms controlling the fibrotic repair phenotype in cornea: implications for surgical outcomes. Invest Ophthalmol Vis Sci. 2003;44:4237–4246. doi: 10.1167/iovs.02-1188. [DOI] [PubMed] [Google Scholar]
- 30.Pappa A, Sophos NA, Vasiliou V. Corneal and stomach expression of aldehyde dehydrogenases: from fish to mammals. Chem Biol Interact. 2001;130-132:181–191. doi: 10.1016/s0009-2797(00)00233-7. [DOI] [PubMed] [Google Scholar]
- 31.Fenteany G, Standaert RF, Reichard GA, Corey EJ, Schreiber SL. A beta-lactone related to lactacystin induces neurite outgrowth in a neuroblastoma cell line and inhibits cell cycle progression in an osteosarcoma cell line. Proc Natl Acad Sci U S A. 1994;91:3358–3362. doi: 10.1073/pnas.91.8.3358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Stramer BM, Cook JR, Fini ME, Taylor A, Obin M. Induction of the ubiquitin-proteasome pathway during the keratocyte transition to the repair fibroblast phenotype. Invest Ophthalmol Vis Sci. 2001;42:1698–1706. [PubMed] [Google Scholar]
- 33.Manzer R, Pappa A, Estey T, Sladek N, Carpenter JF, Vasiliou V. Ultraviolet radiation decreases expression and induces aggregation of corneal ALDH3A1. Chem Biol Interact. 2003;143-144:45–53. doi: 10.1016/s0009-2797(02)00171-0. [DOI] [PubMed] [Google Scholar]
- 34.Buchdunger E, Cioffi CL, Law N, Stover D, Ohno-Jones S, Druker BJ, Lydon NB. Abl protein-tyrosine kinase inhibitor STI571 inhibits in vitro signal transduction mediated by c-kit and platelet-derived growth factor receptors. J Pharmacol Exp Ther. 2000;295:139–145. [PubMed] [Google Scholar]
- 35.Pinzani M. PDGF and signal transduction in hepatic stellate cells. Front Biosci. 2002;7:d1720–1726. doi: 10.2741/A875. [DOI] [PubMed] [Google Scholar]
- 36.Stramer BM, Austin JS, Roberts AB, Elizabeth Fini M. Selective reduction of fibrotic markers in repairing corneas of mice deficient in Smad3. J Cell Physiol. 2005;203:226–232. doi: 10.1002/jcp.20215. [DOI] [PubMed] [Google Scholar]
- 37.Ciuclan L, Ehnert S, Ilkavets I, Weng HL, Gaitantzi H, Tsukamoto H, Ueberham E, Meindl-Beinker NM, Singer MV, Breitkopf K, Dooley S. TGF-beta enhances alcohol dependent hepatocyte damage via down-regulation of alcohol dehydrogenase I. J Hepatol. 2010;(52):407–416. doi: 10.1016/j.jhep.2009.12.003. [DOI] [PubMed] [Google Scholar]
- 38.Tallquist M, Kazlauskas A. PDGF signaling in cells and mice. Cytokine Growth Factor Rev. 2004;15:205–213. doi: 10.1016/j.cytogfr.2004.03.003. [DOI] [PubMed] [Google Scholar]
- 39.Lei H, Velez G, Kazlauskas A. Pathological signaling via platelet-derived growth factor receptor {alpha} involves chronic activation of Akt and suppression of p53. Mol Cell Biol. 2011;31:1788–1799. doi: 10.1128/MCB.01321-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Franke TF, Yang SI, Chan TO, Datta K, Kazlauskas A, Morrison DK, Kaplan DR, Tsichlis PN. The protein kinase encoded by the Akt proto-oncogene is a target of the PDGF-activated phosphatidylinositol 3-kinase. Cell. 1995;81:727–736. doi: 10.1016/0092-8674(95)90534-0. [DOI] [PubMed] [Google Scholar]
- 41.Johnson GL, Lapadat R. Mitogen-activated protein kinase pathways mediated by ERK, JNK, and p38 protein kinases. Science. 2002;298:1911–1912. doi: 10.1126/science.1072682. [DOI] [PubMed] [Google Scholar]
- 42.Chua BT, Gallego-Ortega D, Ramirez de Molina A, Ullrich A, Lacal JC, Downward J. Regulation of Akt(ser473) phosphorylation by choline kinase in breast carcinoma cells. Mol Cancer. 2009;8:131. doi: 10.1186/1476-4598-8-131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Aikin R, Maysinger D, Rosenberg L. Cross-talk between phosphatidylinositol 3-kinase/AKT and c-jun NH2-terminal kinase mediates survival of isolated human islets. Endocrinology. 2004;145:4522–4531. doi: 10.1210/en.2004-0488. [DOI] [PubMed] [Google Scholar]
- 44.Wei L, Liu Y, Kaneto H, Fanburg BL. JNK regulates serotonin-mediated proliferation and migration of pulmonary artery smooth muscle cells. Am J Physiol Lung Cell Mol Physiol. 2010;298:L863–869. doi: 10.1152/ajplung.00281.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Sarbassov DD, Ali SM, Sabatini DM. Growing roles for the mTOR pathway. Curr Opin Cell Biol. 2005;17:596–603. doi: 10.1016/j.ceb.2005.09.009. [DOI] [PubMed] [Google Scholar]
- 46.Heitman J, Movva NR, Hall MN. Targets for cell cycle arrest by the immunosuppressant rapamycin in yeast. Science. 1991;253:905–909. doi: 10.1126/science.1715094. [DOI] [PubMed] [Google Scholar]
- 47.Ghosh D. Status of the transcription factors database (TFD) Nucleic Acids Res. 1993;21:3117–3118. doi: 10.1093/nar/21.13.3117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Bogoyevitch MA, Kobe B. Uses for JNK: the many and varied substrates of the c-Jun N-terminal kinases. Microbiol Mol Biol Rev. 2006;70:1061–1095. doi: 10.1128/MMBR.00025-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Terme JM, Lhermitte L, Asnafi V, Jalinot P. TGF-beta induces degradation of TAL1/SCL by the ubiquitin-proteasome pathway through AKT-mediated phosphorylation. Blood. 2009;113:6695–6698. doi: 10.1182/blood-2008-07-166835. [DOI] [PubMed] [Google Scholar]
- 50.Weidenfeld-Baranboim K, Koren L, Aronheim A. Phosphorylation of JDP2 on threonine-148 by the c-Jun N-terminal kinase targets it for proteosomal degradation. Biochem J. 2011;436:661–669. doi: 10.1042/BJ20101031. [DOI] [PubMed] [Google Scholar]
- 51.Schirmer A, Kennedy J, Murli S, Reid R, Santi DV. Targeted covalent inactivation of protein kinases by resorcylic acid lactone polyketides. Proc Natl Acad Sci. 2006;103:4234–4239. doi: 10.1073/pnas.0600445103. [DOI] [PMC free article] [PubMed] [Google Scholar]