

Abstract
The trifluoromethylation of aryl/heteroaryl iodides has been demonstrated using a flow system, enabling a rapid rate of reaction. A broad spectrum of trifluoromethylated compounds were prepared in good to excellent yields using CF3CO2K as the trifluoromethyl source.
Keywords: trifluoromethylation, cross-coupling, flow chemistry, synthetic methods, copper
The development of synthetic methods to introduce trifluoromethyl groups into aromatic and heteroaromatic compounds is of considerable interest as CF3-substituted molecules often possess improved physical and chemical properties.[1] As a result, a variety of synthetic methods have been developed to accomplish this task.[2,3] MacMillan[4] and Baran[5] reported elegant examples of (hetero)aromatic trifluoromethylation reactions via CF3 radical processes. These have the advantage of being simple and high yielding though a lack of regiocontrol is observed with certain classes of substrates. Although palladium and copper systems have been developed for the trifluoromethylation of (hetero)aromatic precursors, the scope of heterocyclic substrates handled by these methods is limited.[3] In addition, they usually necessitate the use of an excess of expensive or gaseous CF3 reagents (the latter which are more difficult to handle in normal laboratory settings) and have reaction times ranging from hours to days. Thus, it would be desirable to devise a rapid and efficient method that can be applied to a broad range of substrates and that employs a low-cost CF3 source.
Since the pioneering work by Burton,[6] a great deal of effort has been expended to devise convenient means of generating a [CuCF3] species, which is the presumed key intermediate in many copper-mediated trifluoromethylation processes.[3,7–10] In 2008, Vivic disclosed that CuCF3 could be stabilized with NHC ligands,[11] and the resulting complexes were applied to the room temperature transformation of aryl iodides to aryl trifluoromethyl compounds. Amii then reported the first copper-catalyzed aromatic trifluoromethylation process utilizing Et3SiCF3.[3b] The subsequent use of isolated [(phen)CuCF3] reagent by Hartwig allowed the successful transformation of a variety of aryl halides and several heteroaryl iodides into trifluoromethyl (hetero)arenes.[3g]
Among the various trifluoromethylating reagents, fluoroform and trifluoroacetate salts are stable and low price, and thus represent attractive CF3 sources for many applications. Seminal work by Grushin demonstrated the viability of fluoroform in trifluoromethylation processes.[3h,12] While a major step forward, the need to manipulate gaseous CF3H is inconvenient for some applications. Due to the ease of handling and low cost, trifluoroacetate salts have been used by Matsui and others in the trifluoromethylation of aryl halides.[8–10] Only a limited number of heterocycles have been successfully furnished under batch conditions. In those processes, up to 10 equiv. of sodium trifluoroacetate were used, and mixtures of Ar-CF3, Ar-C2F5 and Ar-H were usually obtained during reaction times of several hours. The separation of Ar-CF3 from these side products is usually difficult due to their similar physical properties. Given our experience in both flow and trifluoromethylation chemistry,[3a,13] we wondered if a method combing the two would be advantageous. Herein, we report an efficient aromatic trifluoromethylation process conducted under flow conditions. With this operationally simple method, a broad spectrum of trifluoromethylated aromatic and heteroaromatic compounds can be prepared from the corresponding aryl iodides in a protocol requiring only minutes of residence time, using CF3CO2K as the CF3 source.
Early in our work, in studies carried out under batch conditions, we found that the rate of the decarboxylation of CF3CO2K could be substantially accelerated by increasing the reaction temperature from 160 °C to 200 °C in the presence of copper iodide.[14] In principle, this rapid decarboxylation process could facilitate the generation of the desired [CuCF3] intermediate. While the high temperature and the increased pressure that occurs during decarboxylation[15] can present problems under batch conditions, especially on a larger scale, flow processes are able to readily manage reaction parameters such as temperature, pressure and residence times.[16–18]
The handling of solids under flow conditions can be problematic. Considering the low solubility of CuI in NMP, we first examined a number of ligands, such as 1,10-phenanthroline, 2,2’-bipyridine, TMEDA, DMEDA and pyridine, to establish suitable reaction conditions. Pyridine proved to be the optimal ligand, furnishing a homogenous solution in combination with CuI. The CuI (2.0 equiv)/pyridine (2.4 equiv) combination was applied to a mixture of CF3CO2K (2.0 equiv) and 4-iodobipenyl (1.0 equiv) at 200 °C for 15 minutes to provide an excellent yield of 4-trifluoromethylbiphenyl (95 % as determined by 19F NMR). Only trace amounts of reduced product (biphenyl) or double insertion product, 4-pentafluoroethyl biphenyl, were detected (<2% as judged by GC analysis). Interestingly, when CF3CO2K was replaced with CF3CO2Na under otherwise identical conditions, only a 40 % yield of the desired product was realized.[19]
We next adapted our process to flow conditions. A solution of CF3CO2K, CuI and pyridine in NMP was mixed with a solution of 4-trifluoromethylbiphenyl in NMP and introduced into a stainless steel tube reactor submerged in a preheated 200 °C bath using an optimized 16 minute residence time (Figure 1). At this point the mixture was diluted with a stream of ethyl acetate (controlled with back-pressure regulators). Finally, the resulting mixture was collected and subsequently purified in a standard fashion by column chromatography to afford 2a in 87 % isolated yield (Table 1).
Figure 1.

Flow setup for the aromatic trifluoromethylation. See the Supporting Information for details.
Table 1.
Substrate scope of trifluoromethylation of aromatic compounds under flow conditions.[a]
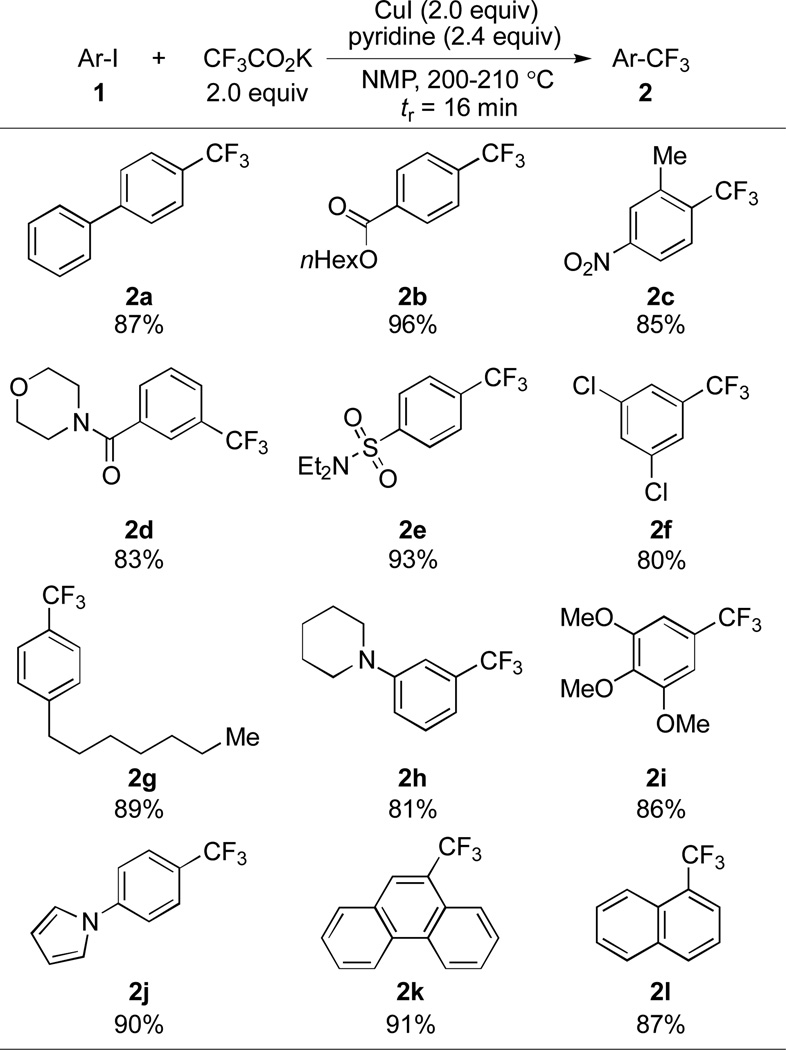 |
Yields of isolated products (average of two runs) based on 1 mmol scale are shown. See the supporting information for details.
Having established efficient flow conditions, the substrate scope was investigated (Table 1) using the flow setup shown in Figure 1. All of the substrates underwent complete conversion using a 16 minutes residence time, affording products in good to excellent isolated yields and in over 95% purity. The high selectivity for trifluoromethylation further enhances the efficiency of this method, since the separation of Ar-H, a typical side product of trifluoromethylation processes, from Ar-CF3 can often be difficult. The aromatic trifluoromethylation reaction could be successfully carried out with para-, meta-, and ortho-substituted aryl substrates. In addition, electron-deficient, electron-neutral and electron-rich iodoarenes are all suitable substrates. As illustrated in Scheme 2, the flow conditions tolerate ester- (2b), nitro- (2c), amide- (2d), sulfonamide- (2e) and chloro-substituted (2f) aryl compounds as well.
Scheme 2.

Trifluoromethylation of ethyl 4-iodobenzoate on 10 mmol scale. See the Supporting Information for details.
In light of the above results, we next focused on expanding the scope of this method to heteroaryl substrates (Table 2).[8b] Nitrogen-containing heterocycles are known to coordinate with copper species, and thus represent a challenging class of substrates for copper-catalyzed processes. As shown in Scheme 3, a variety of heterocyclic compounds including pyridines (2m, 2n and 2o), indole (2p), pyrimidine (2q), pyrazine (2r), quinoline (2s), isoquinoline (2t), imidazopyridine (2u), and pyrrolopyridine (2v) substrates could be efficiently coupled with the trifluoromethyl group under the flow conditions. Trifluoromethylpyrazole is the core unit of many pharmaceuticals, agrochemicals and potential candidates (e.g., Celecoxib, Mavacoxib, Razaxaban, SC-560),[20] but these five-membered heterocycles are poor substrates for Pd-catalyzed processes.[21] Conversely, using this flow method, both 2- and 3-trifluoromethyl substituted pyrazoles (2w and 2x) could be prepared in good yields.
Table 2.
Substrate scope of trifluoromethylation of heteroaromatic compounds under flow conditions.[a]
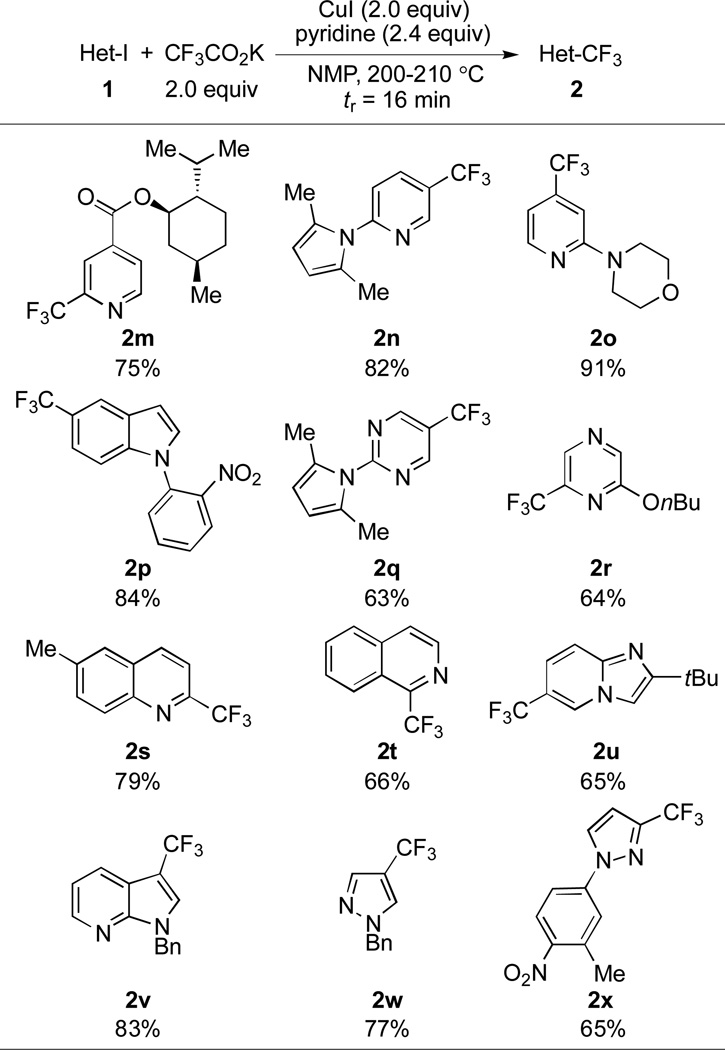 |
Yields of isolated products (average of two runs) based on 1 mmol scale are shown. See the supporting information for details.
One of the major advantages of flow chemistry involves the ease to scale up reactions. To further demonstrate the robustness of our flow system, we successfully performed the trifluoromethylation of ethyl 4-iodobenzoate under flow conditions on a 10 mmol scale (Scheme 2). In a period of 208 minutes, we were able to generate two grams (10 mmol) of product.
To probe the mechanism of this reaction, we performed a Hammett study. Due to the small size of microflow reactors, an advantage of their use is their efficient mass and heat transfer with short reaction times that are difficult to control in a batch reactor.[16] Accordingly, a flow system with the same setup as shown in Figure 1 was used to conduct the kinetic study. All experiments were performed under conditions of 1 min residence time at 200 °C. In competition experiments, an excess of the equimolar mixture of 3- or 4-substituted aryl iodides and iodobenzene relative to CF3CO2K was used. The relative rates of different reactions were extracted from the product distributions after reaching a steady state. As shown in Figure 2, a linear correlation (R2 = 0.98) with a slope of 0.52 was observed using this flow method. The positive ρ value is consistent with the presence of a nucleophilic [CuCF3] species that undergoes oxidative addition with aryl iodides, which slightly favors substrates with electron-withdrawing groups. The ρ value observed here was similar to that obtained from other copper-catalyzed or mediated processes in our group.[22]
Figure 2.

Hammett plots of competition experiments. See the Supporting Information for details.
In conclusion, we have developed a simple, scalable and efficient technique to introduce a trifluoromethyl group into aromatic and heteroaromatic compounds via copper-mediated cross-coupling reactions. This method represents the first example of aromatic trifluoromethylation conducted in flow, using stable, cost-effective and easy-to-handle CF3CO2K as the CF3 source. Of importance, very short reaction times (minutes) are required to achieve full conversion of (hetero)aryl starting materials in this process. To increase the practicality of the protocol, all components used to assemble our reactors are available from commercial sources. Moreover, we have also successfully performed a Hammett study using a 1 min residence time at 200 °C employing flow techniques, providing a reliable method of studying the reaction under “hot and fast” and synthetically relevant conditions.
Experimental Section
General procedure
A solution of aryl iodide (0.6 M) in NMP was loaded into a stainless steel syringe and a solution of CuI (0.8 M), CF3CO2K (0.8 M) and pyridine (0.96 M) in NMP was loaded into a second stainless steel syringe. The flow rates of two solutions were 40 uL/min and 60 uL/min separately delivering with two Harvard Apparatus syringe pumps. After mixing with a T-mixer, the resulting mixture was delivered into the stainless steel reactor (1/16 inch OD, 0.046 inch ID, 1.6 mL volume) at 200–210 °C (Caution: Hot! A suitable bath oil should be used[23]). Upon exiting the reactor, the reaction was quenched with EtOAc (100 uL/min). Next, after reaching steady state, the sample was collected in order to obtain exactly 1 mmol of product. Further safety information as well as details of the flow setup and workup procedures can be found in the supporting information.
Sample analysis
NMR and IR spectroscopies were used to identify the products.
Supplementary Material
Scheme 1.

Aromatic trifluoromethylation under batch and flow conditions.
Footnotes
We thank Novartis International AG and the National Institutes of Health for financial support (GM46059).The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health. We thank Dr. John R. DeBergh and Dr. Christine Nguyen for assistance with the preparation of this manuscript.
Supporting information for this article is available on the WWW under http://www.angewandte.org or from the author.
References
- 1.a) Müller K, Faeh C, Diederich F. Science. 2007;317:1881. doi: 10.1126/science.1131943. [DOI] [PubMed] [Google Scholar]; b) Schlosser M. Angew. Chem. Int. Ed. 2006;45:5432. doi: 10.1002/anie.200600449. [DOI] [PubMed] [Google Scholar]; c) Purser S, Moore PR, Swallow S, Gouverneur V. Chem. Soc. Rev. 2008;37:320. doi: 10.1039/b610213c. [DOI] [PubMed] [Google Scholar]; d) Isanbor C, O’Hagan D. J. Fluorine Chem. 2006;127:303. [Google Scholar]; e) Kirsch P. Modern Fluoroorganic Chemistry: Synthesis Reactivty, Applications. Weinheim: Wiley-VCH; 2004. [Google Scholar]; f) Umemoto T. Chem. Rev. 1996;96:1757. doi: 10.1021/cr941149u. [DOI] [PubMed] [Google Scholar]; g) Prakash SGK, Yudin AK. Chem. Rev. 1997;97:757. doi: 10.1021/cr9408991. [DOI] [PubMed] [Google Scholar]; h) Langlois BR, Billard T. Synthesis. 2003:185. [Google Scholar]; i) McClinton MA, McClinton DA. Tetrahedron. 1992;48:6555. [Google Scholar]; j) Singh RP, Shreeve JM. Tetrahedron. 2000;56:7613. [Google Scholar]; k) Kieltsch I, Eisenberger P, Stanek K, Togni A. Chima. 2008;62:260. [Google Scholar]
- 2.For recent reviews of trifluoromethylation, see: Tomashenko OA, Grushin VV. Chem. Rev. 2011;111:4475. doi: 10.1021/cr1004293. Ma J-A, Cahard D. J. Fluorine Chem. 2007;128:975. Lundgren RJ, Stradiotto M. Angew. Chem. Int. Ed. 2010;49:9322. doi: 10.1002/anie.201004051. Roy S, Gregg BT, Gribble GW, Le V-D, Roy S. Tetrahedron. 2011;67:2161. Wu X-F, Neumann H, Beller M. Chem. Asian J. 2012;7:1744. doi: 10.1002/asia.201200211. Furuya T, Kamlet AS, Ritter T. Nature. 2011;473:470. doi: 10.1038/nature10108.
- 3.For selected recent examples, see: Cho EJ, Senecal TD, Kinzel T, Zhang Y, Watson DA, Buchwald SL. Science. 2010;328:1679. doi: 10.1126/science.1190524. Oishi M, Kondo H, Amii H. Chem. Commun. 2009:1909. doi: 10.1039/b823249k. Ball ND, Gary JB, Ye Y, Sanford MS. J. Am. Chem. Soc. 2011;133:7577. doi: 10.1021/ja201726q. Zhang X-G, Dai H-X, Wasa M, Yu J-Q. J. Am. Chem. Soc. 2012;134:11948. doi: 10.1021/ja305259n. Ye Y, Sanford MS. J. Am. Chem. Soc, 2012;134:9034. doi: 10.1021/ja301553c. Knauber T, Arikan F, Röschenthaler G-V, Gooβen LJ. Chem. Eur. J. 2011;17:2689. doi: 10.1002/chem.201002749. Morimoto H, Tsubogo T, Litvinas ND, Hartwig JF. Angew. Chem. Int. Ed. 2011;50:3793. doi: 10.1002/anie.201100633. Novák P, Lishchynskyi A, Grushin VV. Angew. Chem. Int. Ed. 2012;51:7767. doi: 10.1002/anie.201201613. Kremlev MM, Tyrra W, Mushta AI, Naumann D, Yagupolskii YL. J. Fluorine Chem. 2010;131:212. Zhang CP, Wang ZL, Chen QY, Zhang CT, Gu YC, Xiao JC. Angew. Chem. Int. Ed. 2011;50:1896. doi: 10.1002/anie.201006823. Döbele M, Wiehn MS, Bräse S. Angew. Chem. Int. Ed. 2011;50:11533. doi: 10.1002/anie.201105446. Liu T, Shao X, Wu Y, Shen Q. Angew. Chem. Int. Ed. 2012;51:540. doi: 10.1002/anie.201106673. Chu L, Qing F-L. J. Am. Chem. Soc. 2012;134:1298. doi: 10.1021/ja209992w. Weng Z, Lee R, Jia W, Yuan Y, Wang W, Feng X, Huang KW. Organometallics. 2011;30:3229. Dai J-J, Fang C, Xiao B, Yi J, Xu J, Liu Z-J, Lu X, Liu L, Fu Y. J. Am. Chem. Soc. 2013;135:8436. doi: 10.1021/ja404217t. Danoun G, Bayarmagnai B, Grünberg MF, Gooβen LJ. Angew, Chem. Int. Ed. 2013 doi: 10.1002/anie.201304276.
- 4.Nagib DA, MacMillan DWC. Nature. 2011;480:224. doi: 10.1038/nature10647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ji Y, Brueckl T, Baxter RD, Fujiwara Y, Seiple IB, Su S, Blackmond DG, Baran PS. Proc. Natl. Acad. Sci. USA. 2011;108:14411. doi: 10.1073/pnas.1109059108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wiemers DM, Burton DJ. J. Am. Chem. Soc. 1986;108:832. [Google Scholar]
- 7.a) Prakash GKS, Krishnamurti R, Olah GA. J. Am. Chem. Soc. 1989;121:393. [Google Scholar]; b) Stahly GP, Bell DR. J. Org. Chem. 1989;54:2873. [Google Scholar]; c) Kruse A, Siegemund G, Schumann DCA, Ruppert I. Ger. Offen. 1988 DE3805534. [Google Scholar]
- 8.For examples of using CF3CO2Na as CF3 sources, see: Matsui K, Tobita E, Ando M, Kondo K. Chem. Lett. 1981:1719. b) This paper investigated the reactions of heteroaryl substrates: Carr GE, Chambers RD, Holmes TF. J. Chem. Soc. Perkin Trans. 1988;1:921. Li Y, Chen T, Wang H, Zhang R, Jin K, Wang X, Duan C. Synlett. 2011:1713.
- 9.For examples using CF3CO2Me, see: Langlois BR, Roques N. J. Fluorine Chem. 2007;128:1318. Schareina T, Wu XF, Zapf A, Cotte A, Gotta M, Beller M. Top. Catal. 2012;55:426.
- 10.For examples of using CF3CO2K as CF3 sources, see Lin RW, Davidson RI. 4808748. U. S. Patent. 1989
- 11.Dubinina GG, Furutachi H, Vicic DA. J. Am. Chem. Soc. 2008;130:8600. doi: 10.1021/ja802946s. [DOI] [PubMed] [Google Scholar]
- 12.Zanardi A, Novikov MA, Martin E, Benet-Buchholz J, Grushin VV. J. Am. Chem. Soc. 2011;133:20901. doi: 10.1021/ja2081026. [DOI] [PubMed] [Google Scholar]
- 13.For selected recent examples, see: DeAngelis A, Wang D-H, Buchwald SL. Angew. Chem. Int. Ed. 2013;52:3434. doi: 10.1002/anie.201208544. Chen M, Buchwald SL. Angew. Chem. Int. Ed. 2013;52:4247. doi: 10.1002/anie.201300615. Chernyak N, Buchwald SL. J. Am Chem. Soc. 2012;134:12466. doi: 10.1021/ja305660a.
- 14.See Figure S-1 in the Supporting Information for details.
- 15.When 10 mmol CF3CO2K is decarboxylated in a 20 mL sealed flask at 200 °C, the pressure increases to about 15 atm according to the ideal gas law.
- 16.For selected reviews in flow, see: Ley SV. Chem. Rec. 2012;12:378. doi: 10.1002/tcr.201100041. Wegner J, Ceylan S, Kirschning A. Chem. Commun. 2011;47:4583. doi: 10.1039/c0cc05060a. Mason BP, Price KE, Steinbacher JL, Bogdan AR, McQuade DT. Chem. Rev. 2007;107:2300. doi: 10.1021/cr050944c. Yoshida J-i, Nagaki A, Yamada T. Chem. Eur. J. 2008;14:7450. doi: 10.1002/chem.200800582. Wiles C, Watts P. Chem. Commun. 2011;47:6512. doi: 10.1039/c1cc00089f. Cukalovic A, Monbaliu JCMR, Stevens CV. Top. Heterocycl. Chem. 2010;23:161. Fukuyama T, Rahman MT, Sata M, Ryu I. Synlett. 2008:151.
- 17.a) Ehrfeld W, Hessel V, Löwe H. Microreactors: New Technology for Modern Chemistry. Weiheim: Wiley-VCH; 2000. [Google Scholar]; b) Seeberger PH, Blume T. New Avenues to Efficient Chemical Synthesis-Emerging Technologies. Berlin: Springer; 2007. [Google Scholar]; c) Wirth T. Microreactors in Organic Synthesis and Catalysis. Weiheim: Wiley-VCH; 2008. [Google Scholar]
- 18.For examples of handling high-temperature conditions in flow, see: Sedelmeier J, Baxendale IR, Ley SV. Green Chem. 2009;11:683. Baxendale IR, Griffiths-Jones CM, Ley SV, Tranmer GK. Chem. Eur. J. 2006;12:4407. doi: 10.1002/chem.200501400. Zaborenko N, Bedore MW, Jamison TF, Jensen KF. Org. Process Res. Dev. 2011;15:131. O’Brien AG, Horváth Z, Lévesque F, Lee JW, Seidel-Morgenstern A, Seeberger PH. Angew. Chem. Int. Ed. 2012;51:7028. doi: 10.1002/anie.201202795. Organ MG, Hanson PR, Rolfe A, Samarakoon TB, Farman U. J. Flow Chem. 2011;1:32. doi: 10.1556/jfchem.2011.00008. Zhang Y, Jamison TF, Patel S, Mainolfi N. Org. Lett. 2011;13:280. doi: 10.1021/ol1026848. Cantillo D, Sheibani H, Kappe CO. J. Org. Chem. 2012;77:2463. doi: 10.1021/jo3001645. Kawanami H, Matsushima K, Sato M, Ikushima Y. Angew. Chem. Int. Ed. 2007;46:5129. doi: 10.1002/anie.200700611. Stevens JG, Bourne RA, Twigg MV, Poliakoff M. Angew. Chem. Int. Ed. 2010;49:8856. doi: 10.1002/anie.201005092. Gutmann B, Roduit J-P, Roberge D, Kappe CO. Angew. Chem. Int. Ed. 2010;49:7101. doi: 10.1002/anie.201003733. Baek J, Allen PM, Bawendi MG, Jensen KF. Angew. Chem. Int. Ed. 2011;50:627. doi: 10.1002/anie.201006412.
- 19.See Table S-1 in the Supporting Information for details.
- 20.a) Lamberth C. Heterocycles. 2007;71:1467. [Google Scholar]; b) Elguero J, Silva AMS, Tomé AC. In: Modern Heterocyclic Chemistry. Alvarez-Builla J, Vaquero JJ, Barluenga J, editors. Weinheim: Wiley-VCH; 2011. pp. 635–725. [Google Scholar]
- 21.Ye Y, Sanford MS. Synlett. 2012;23:2005. doi: 10.1055/s-0032-1316988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Strieter ER, Bhayana B, Buchwald SL. J. Am. Chem. Soc. 2009;131:78. doi: 10.1021/ja0781893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.For example, the silicone oil from Alfa Aesar, catalogue number 45896, with a temperature range of 25–250 °C.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.