

Abstract
Transcription antiterminator RfaH alternates between closed (inactive) and open (activated) conformation. In this issue of Cell, Burmann et al. show that opening is accompanied by dramatic all-α to all-β refolding of its C-terminal domain. Each of the folds has a distinct function: all-α-fold acts as a specificity determinant, directing RfaH to a small subset of operons, whereas the all-β-fold recruits ribosome, thereby coupling RfaH-stimulated transcription to translation.
In bacterial cells, RNA polymerases (RNAPs) and ribosomes populate the same space, accessible by simple diffusion, which allows occupation of the same mRNA by both transcription and translation machineries. Cotranscriptional translation (or transcription-translation coupling) was known to play a role in such regulatory mechanisms as transcription attenuation and operon polarity; more recently, trailing ribosomes were shown to affect the rate of transcription by suppressing RNAP backtracking, harmonizing the rates of mRNA and protein synthesis (Proshkin et al., 2010). A direct physical link between the RNAP elongation complex and the trailing ribosome was discovered, wherein the general transcription factor NusG engaged the RNAP with its N-terminal domain (NTD) while interacting with ribosomal protein S10(= NusE) via the C-terminal domain (CTD) (Burmann et al., 2010). RNAP-NusG-S10 bridge complements ribosome binding to mRNA through the engagement of the start codon and Shine-Dalgarno sequence (SDS). In this issue of Cell, Artsimovitch, Rösch, and colleagues report an alternate mechanism of ribosome recruitment to horizontally transferred genes (Burmann et al., 2012).
The recruitment of ribosomes to the RNAP-bound RNAs is not trivial. In rapidly dividing bacterial cells, most of the RNAPs are transcribing nontranslated rRNA genes, leading to a nonrandom segregation of ribosomes and RNAPs. In fact, high-resolution imaging of Escherichia coli cells demonstrated that <10% of all RNAPs colocalized with ribosomes (Bakshi et al., 2012). Although mRNA can diffuse to the areas occupied by ribosomes, impediments to cotranscriptional translation are known to cause premature termination of transcription, making recruitment of ribosomes essential for efficient gene expression. In this light, expression of long operons coding for E. coli virulence factors and other extracytoplasmic components looks particularly problematic. Not only do they often feature suboptimal start codons and SDSs, but RNAPs transcribing these operons are bound not by NusG but by its paralog, RfaH, which is not known to recruit ribosomes (Belogurov et al., 2009).
Antiterminator factor RfaH is recruited to elongation complexes through interactions with a specific sequence, ops (operon polarity suppressor), when it is exposed as a single-stranded nontemplate DNA in the transcription bubble. RfaH shares two-domain architecture with its paralog NusG, but although their NTDs have similar folds, CTDs structures differ dramatically. In crystallographic and NMR studies, NusG CTD was shown to take on a β-barrel-like fold, which made no contacts with the NTD in keeping with the open conformation (Burmann et al., 2011). In contrast, crystallographic structure of RfaH showed a so-called closed conformation of the factor, with the CTD folded into an α-helical hairpin tightly packed against the NTD, the DNA- and RNAP-interacting domain (Belogurov et al., 2007) (Figure 1A). Artsimovitch and colleagues, responsible for the wealth of mechanistic and structural information regarding this rather enigmatic factor, speculated that the β-to-α fold switch occurred in RfaH evolution as the means to convert a NusG-like general transcription factor into a pathway-specific one (Belogurov et al., 2009). Always in open conformation, NusG domains are poised for interactions with RNAP (NTD) and ribosome (CTD). In the case of RfaH, the CTD changed the fold, redeploying hydrophobic amino acids from the interior of the ancestral β barrel onto the surface of the α hairpin, thus allowing it to bind NTD and mask its RNAP interaction surface until engagement of ops DNA would lead to domain dissociation. The hypothetical scenario of β-to-α conversion listed potential CTD interaction with the ribosome as the driving force of RfaH fold evolution (Belogurov et al., 2009).
Figure 1. Conformational Switch that Really Matters.
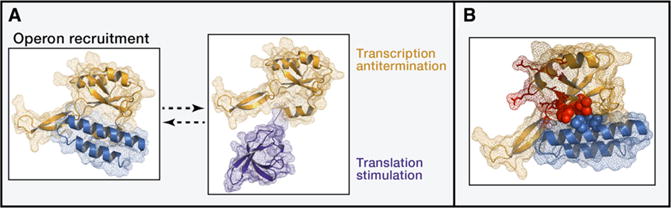
(A) Closed and open conformations of E. coli RfaH. (Left) Closed conformation (2oug,a; Belogurov et al., 2007). N-terminal (yellow) and C-terminal (blue) domains are mesh and cartoon. (Right) Homology model (adding 2lcl as a template) of the open conformation (Burmann et al., 2012). N- and C-terminal domains are mesh and cartoon.
(B) Part of the DNA-binding patch of the RfaH N-terminal domain is obscured by interactions with C-terminal domain in the closed conformation. E. coli RfaH (2oug,a) N- and C-terminal domains are mesh and cartoon; DNA-binding patch residues are red sticks (Tyr8, Cys9, Lys10, Gly12, Arg16, Pro52, Asn53, Thr72, and Val75) or red spheres (Leu6, Tyr54, and Val79). The last three are packed against C-terminal domain residues Leu143 and Ile146 (blue spheres).
Here, Artsimovitch, Rösch, and colleagues used time-resolved NMR to demonstrate that RfaH CTD undergoes the all-α to all-β fold switch—thought to have required a long evolutionary transition—during the lifetime of the protein upon dissociation from, or a proteolytic removal of, the NTD (Burmann et al., 2012) (Figure 1A). Using an RfaH mutant with destabilized interdomain interactions (E48S), the authors discovered that α and β forms of the CTD exist at equimolar equilibrium in solution, indicating that these drastically different folds probably have similar energies and are separated by a rather low-energy barrier. Although the exceptions to it continue to accumulate, the classic notion of the protein's tertiary structure being uniquely determined by its sequence and representing the global free energy minimum still dominates the ways that protein folding and structure are viewed today. Unprecedented in its scale, refolding of RfaH CTD, driven by functionally relevant interactions, has profound implications for structural and structure-based analysis of proteins—not only the well-known metamorphic ones, such as prions, but also those not yet thought to change folds (Bryan and Orban, 2010).
Even more remarkable is the finding that both CTD forms are fully functional. Whereas α form was shown to act as a determinant of pathway specificity, the β form was found to retain not only the fold, but also at least one of the functions of the ancestral (NusG) CTD: in an array of experiments, including mass spectrometry, ChIP-chip, and in vivo reporter assays, RfaH CTD activated translation via recruitment of the S10(NusE) component of the ribosome (Burmann et al., 2012). CTD-dependent stimulation of translation by RfaH was particularly prominent when mRNA lacked efficient means of ribosome recruitment, characteristic of horizontally transferred operons under its control. This poses an interesting problem from an evolutionary standpoint: whereas paralogs are thought to evolve through duplication, divergence, and functionalization, RfaH appeared to have never lost the ribosome recruitment function of its ancestor, and the new function was added to the ancestral one via evolved CTD metamorphism.
Activation of translation by RfaH has another important implication for gene expression: it reduces susceptibility of the nascent transcript to Rho-dependent termination. Together with other consequences of RfaH action—exclusion of Rho cofactor NusG from elongation complexes and direct antitermination effect on elongation—ribosome recruitment by RfaH results in 400-fold reversal of Rho-dependent operon polarity in vivo (Burmann et al., 2012; Sevostyanova et al., 2011).
The exact mechanism by which RfaH domain dissociation is triggered by binding to ops remains unknown. Previously, Artsimovitch and colleagues identified five RfaH residues (Lys10, Arg16, His20, Thr72, and Arg73) that are likely to bind DNA; all of them are located on the surface of the NTD, where ops binding would not compete with the CTD-NTD interaction (Belogurov et al., 2010). Because RfaH lacks a discernible DNA-binding motif, we extracted NTD from the published structure of RfaH and subjected it to computational analysis by Patchfinder (http://patchfinder.tau.ac.il). With a significant score of 0.6065, this algorithm predicted a potential DNA-binding patch comprising 13 amino acids (including all residues that were previously implicated in ops binding) (Figure 1B). Notably, three of these residues, Leu6, Tyr54, and Val79, are located at the interface with the CTD, packed against Leu143 and Ile146, providing a potential basis for competition between DNA-binding and NTD-CTD interactions. Molecular details governing RfaH function, including the allosteric effect on RNAP processivity, domain dissociation, and ops recognition, still await their full elucidation. This metamorphic transcription-translation factor also provides an excellent platform for studies of protein fold transition in evolution and in real time, domain structure-function relationships, and evolutionary strategies of virulence/horizontally transferred operons.
Acknowledgments
This work was supported by a grant from the NIH R01 GM58750 (E.N.).
References
- Bakshi S, Siryaporn A, Goulian M, Weisshaar JC. Mol Microbiol. 2012;85:21–38. doi: 10.1111/j.1365-2958.2012.08081.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belogurov GA, Vassylyeva MN, Svetlov V, Klyuyev S, Grishin NV, Vassylyev DG, Artsimovitch I. Mol Cell. 2007;26:117–129. doi: 10.1016/j.molcel.2007.02.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belogurov GA, Mooney RA, Svetlov V, Landick R, Artsimovitch I. EMBO J. 2009;28:112–122. doi: 10.1038/emboj.2008.268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belogurov GA, Sevostyanova A, Svetlov V, Artsimovitch I. Mol Microbiol. 2010;76:286–301. doi: 10.1111/j.1365-2958.2010.07056.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bryan PN, Orban J. Curr Opin Struct Biol. 2010;20:482–488. doi: 10.1016/j.sbi.2010.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burmann BM, Schweimer K, Luo X, Wahl MC, Stitt BL, Gottesman ME, Rösch P. Science. 2010;328:501–504. doi: 10.1126/science.1184953. [DOI] [PubMed] [Google Scholar]
- Burmann BM, Scheckenhofer U, Schweimer K, Rösch P. Biochem J. 2011;435:783–789. doi: 10.1042/BJ20101679. [DOI] [PubMed] [Google Scholar]
- Burmann BM, Knauer SH, Sevostyanova A, Schweimer K, Mooney RA, Landick R, Artsimovitch I, Rosch P. Cell. 2012;150:291–303. doi: 10.1016/j.cell.2012.05.042. this issue. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Proshkin S, Rahmouni AR, Mironov A, Nudler E. Science. 2010;328:504–508. doi: 10.1126/science.1184939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sevostyanova A, Belogurov GA, Mooney RA, Landick R, Artsimovitch I. Mol Cell. 2011;43:253–262. doi: 10.1016/j.molcel.2011.05.026. [DOI] [PMC free article] [PubMed] [Google Scholar]