

Abstract
Huntington’s disease (HD) is an autosomal-dominant neurodegenerative disorder caused by polyglutamine expansion in the amino-terminus of huntingtin (HTT). HD offers unique opportunities for promising RNA-based therapeutic approaches aimed at reducing mutant HTT expression, since the HD mutation is considered to be a “gain-of-function” mutation. Allele-specific strategies that preserve expression from the wild-type allele and reduce the levels of mutant protein would be of particular interest. Here, we have conducted proof-of-concept studies to demonstrate that spliceosome-mediated trans-splicing is a viable molecular strategy to specifically repair the HTT allele. We employed a dual plasmid transfection system consisting of a pre-mRNA trans-splicing module (PTM) containing HTT exon 1 and a HTT minigene to demonstrate that HTT exon 1 can be replaced in trans. We detected the presence of the trans-spliced RNA in which PTM exon 1 was correctly joined to minigene exons 2 and 3. Furthermore, exon 1 from the PTM was trans-spliced to the endogenous HTT pre-mRNA in cultured cells as well as disease-relevant models, including HD patient fibroblasts and primary neurons from a previously described HD mouse model. These results suggest that the repeat expansion of HTT can be repaired successfully not only in the context of synthetic minigenes but also within the context of HD neurons. Therefore, pre-mRNA trans-splicing may be a promising approach for the treatment of HD and other dominant genetic disorders.
Electronic supplementary material
The online version of this article (doi:10.1007/s00018-012-1083-5) contains supplementary material, which is available to authorized users.
Keywords: Neurodegeneration, Huntington’s disease, RNA-based therapeutics, Spliceosome-mediated trans-splicing
Introduction
Huntington’s disease (HD) is an autosomal-dominant neurodegenerative disorder caused by polyglutamine expansion in the amino-terminus of huntingtin (HTT). It is characterized by irrepressible abnormal movements termed chorea which intensify progressively. Furthermore, cognitive function deteriorates and leads to dementia, and death usually occurs within 20 years of disease onset. The pathology of HD is marked by selective and progressive neuronal cell loss in the striatum, specifically caudate and putamen, which is often accompanied by loss of cells in the cerebral cortex and widespread brain atrophy [1].
HTT is a protein of approximately 350 kD which is encoded by 67 exons. It appears to be unique since it has no sequence homology with other proteins, and HTT knockout in mice produces a lethal phenotype as early as embryonic day 9, suggesting a lack of functional compensation by other proteins [2–4]. Except for the extreme amino-terminus, containing the polyglutamine region and proline-rich segments, the entire protein is predicted to be composed of 36 α-helical HEAT repeats. These repeats fold into a spiral structure and may serve as docking sites for other proteins [5, 6]. HTT is expressed ubiquitously in humans and rodents with the highest levels found in CNS neurons and testes [7, 8]. Intracellularly, HTT is associated with various organelles, including the nucleus, endoplasmic reticulum and Golgi complex [9–11]. It is therefore likely that HTT performs multiple functions according to its subcellular context.
The genetic defect causing HD resides in exon 1 of the HTT gene. Exon 1 of the wild-type gene contains a polymorphic stretch of uninterrupted CAG trinucleotide repeats, which is translated into a series of consecutive glutamine residues, the polyglutamine tract. The normal repeat length ranges up to 35. Between 36 and 41 repeats, penetrance is variable. Above 41 repeats, penetrance is complete, and there is a strong inverse correlation between length and age of disease onset [12, 13]. The repeat expansion is generally believed to cause a toxic gain-of-function affecting multiple cellular functions including transcription [14, 15], apoptosis [15, 16], vesicular trafficking [17], cholesterol metabolism [18], and endoplasmic reticulum function [19]. Some experiments also demonstrate a possible loss-of-function due to decreased levels of wild-type protein [20, 21]. For example, wild-type HTT stimulates the production of brain-derived neurotrophic factor (BDNF), a neuronal survival factor, and its decrease in HD may be directly relevant to striatal neuron death [21]. Therefore, an ideal therapy would decrease the levels of mutant HTT while at the same time increasing the amount of wild-type protein.
Huntington’s disease is caused by a defined, single mutation, i.e., the CAG repeat expansion. Thus, it is particularly well suited for gene therapy approaches. In particular, allele-specific strategies which preserve expression from the wild-type allele and reduce the levels of mutant protein would be especially advantageous. We therefore explored the possibility of replacing exon 1 of HTT with a corrected, non-pathogenic exon 1 sequence using spliceosome-mediated pre-mRNA trans-splicing. Most splicing reactions occur in cis where both 5′ and 3′ splice sites are located within one RNA molecule. Trans-splicing, however, occurs between two separate RNAs: the endogenous pre-mRNA and the “corrected” PTM RNA. The mature mRNA contains exons from both primary transcripts. Both cis- and trans-splicing follow similar mechanisms and are directed by the spliceosome complexes. Several forms of trans-splicing have been reported in different species from lower to higher eukaryotes including rodents and humans [22–31]. A recent study showed that trans-splicing occurs naturally in normal human cells where 5′ exons of the JAZF1 pre-mRNA are joined to 3′ exons of the JJAZ1/SUZ12 pre-mRNA. This chimeric RNA is translated into JAZF1-JJAZ1, a functional protein with anti-apoptotic activity [32]. Similarly, the ETS fusion protein SLC45A3-ELK4 which is prominent in a subset of prostate cancers commonly occurs in the absence of chromosomal rearrangements and has been ascribed to trans-splicing [33]. Thus, trans-splicing is a naturally occurring process used for the generation of alternative transcripts.
We present proof-of-principle experiments demonstrating that 5′ exon replacement of HTT by spliceosome-mediated pre-mRNA trans-splicing can be achieved in cultured cells using a binary transfection system consisting of a HTT minigene and a pre-mRNA trans-splicing module (PTM). Furthermore, we demonstrate the feasibility of trans-splicing exon 1 from the PTM to the endogenous HTT pre-mRNA. These results suggest that trans-splicing can be used to repair the pathological expansion of HTT exon 1 without impeding the function of the normal allele.
Materials and methods
Plasmid constructs
The HTT minigene contained exon 1 with 42 CAG repeats and exons 2 to 3 separated by intervening sequences. The sequences were based on Genbank accession number NT_006051. The two introns were shortened to 860 and 109 bp, respectively, to make the construct amenable to plasmid cloning. The PTM construct consisted of three portions: (1) the replacement exon 1 of HTT with 21 CAG repeats, (2) the splicing domain with an U1 snRNP binding site at the 3′ end of exon 1 and a triplet repeat of an intronic splice enhancer, and (3) the tether which binds to intron 1 by antisense base pairing. The constructs were generated by custom gene synthesis (Geneart). The minigene was subcloned into pCI-neo (Promega) where its expression was driven by the cytomegalovirus (CMV) promoter/enhancer. The PTM was inserted behind the CMV promoter into pMU1 [34]. pMU1 also contained an eGFP expression module expressed from a separate promoter. For viral delivery, the PTM was inserted into the lentiviral vector pSIN18.
Cell culture and transfection
HEK293 cells, U2OS cells, and HD patient fibroblasts were cultured in Dulbecco’s Modified Eagle’s Medium (DMEM; Invitrogen) containing high glucose and supplemented with 10 % fetal bovine serum (Hyclone) and 100 U penicillin/100 μg streptomycin (Invitrogen) per mL. DBTRG cells were cultured in RPMI 1640 supplemented with 10 % fetal bovine serum and 100 U penicillin/100 μg streptomycin per mL. Cells were transiently transfected when they had reached approximately 90 % confluency using PEI or lipofectamine 2000 (Invitrogen) according to the manufacturer’s recommendations. Minigene and PTM plasmids were co-transfected at the ratios indicated in the text. Sonicated salmon sperm DNA was used to equalize the total amount of DNA input where necessary. Alternatively, the PTM plasmid was transfected by itself in experiments designed for trans-splicing of endogenous HTT pre-mRNA. Cells were harvested 24–48 h post-transfection.
Isolation of primary cortical neurons
All animal experiments were carried out in accordance with the University of Missouri Animal Care and Use Committee. Cerebral cortices from 17-day-old embryos of YAC128 HD transgenic mice (developed by M. Hayden, Jackson Labs stock number 4938) were removed, placed into cold DMEM, and the meninges were discarded. The tissue was suspended in 2 mL of 0.25 % (w/v) trypsin at 37 °C for 40 min. Tissues were washed three times in 10–15 mL DMEM containing 10 % (v/v) FBS, 100 IU/mL penicillin, 100 mg/mL streptomycin, and 7.5 mg/mL fungizone. Tissues were triturated with a glass-fired Pasteur pipette 20 times or until homogeneous. The homogenate was diluted with complete culture medium to the desired concentration and seeded on amine coated six-well plates (Becton–Dickinson). Every 3 days thereafter, half the medium was replaced with B27-AO neurobasal medium [100 IU/mL penicillin, 100 mg/mL streptomycin, 7.5 mg/mL fungizone, 10 ml of B27-AO and neurobasal medium (Gibco-BRL) to 500 mL]. The neurons were used for experiments after 7 days in culture (DIV7).
Generation of PTM lentivirus and transduction of cultured cells
The PTM under the control of the CMV promoter was cloned into the unique EcoRV restriction site of pSIN18 [35], thereby retaining the GFP expression cassette of this vector. Virus was produced by triple transfection of HEK293 FT cells with pSIN18-PTM, the helper plasmid psPAX2 (originally developed by D. Trono and obtained from Addgene) and the envelope plasmid pVSV-G for pseudotyping. After 48 h, cell culture supernatant was collected and filtered through a 0.45-μm PES membrane, followed by centrifugation at 53,000g for 90 min to pellet viral particles. Pellets were resuspended in phosphate-buffered saline (PBS) and stored at 4 °C until use. HEK293 T cells, HD patient fibroblasts or primary HD neurons from YAC128 mice were transduced with varying amounts of virus preparations in the presence of 8 μg/ml polybrene. Cells were harvested 48–72 h later for analyses.
RNA isolation and RT-PCR
RNA was isolated using Tri-Reagent (Sigma) following the manufacturer’s instructions. RNA was resuspended in 10 mM Tris–HCl pH 8.2, 1 mM EDTA, and concentrations were measured using a Nanodrop (Thermo Fisher). cDNA was synthesized using 1 μg of RNA and random primers following the SuperScript III protocol (Invitrogen). PCR was performed using two different procedures. For amplifications outside the CAG repeat and the adjacent GC-rich region, Pfu enzyme (prepared in-house) with Thermopol buffer (New England Biolabs) was used. This method was not successful for amplification across the CAG repeat, and we therefore used Taq PCRx with 2× enhancer solution (Invitrogen) for these reactions. PCR products were visualized by agarose gel electrophoresis. Selected PCR products were cloned into the TOPO pCR2.1 vector (Invitrogen) and sequenced by the University of Missouri DNA Core Facility. Primer sequences were: GAPDH fw 5′ TCCGCGCAGCCGAGCCA; GAPDH rev 5′ ACGCCAGTGGACTCCACG; F1 5′ GCAGAAGTTGGTCGTGAGGC; F2 5′ CCGGCCATCTAGGCCAAGC; R1 5′ CACACGGTCTTTCTTGGTAGCTG; R2 5′ CTGACAGACTGTGCCACTATG; R3 5′ ACTCTGCGTCATCACTGCACAGC; R4 5′ AGGCATTCGTCAGCCACCATCC; R5 5′ GATAACTTTGTTGAGGCATTCG; Ex1.1 5′ CTGCTGGAAGGACTTGAGGG; Ex1.2 5′ GGCGGCTGAGGAAGCTGAGGA; HD53 5′ GGTTCTGCTTTTACCTGCGGC. The efficiency of the trans-splicing reaction was determined by RT-PCR with primer pair HD53 and R1, in which HD53 was fluorescently labeled. The PCR product was purified by phenol/chloroform extraction and Sephadex gel filtration followed by restriction digestion with PstI. The products were separated on an 8 % polyacrylamide gel and visualized and quantitated (Typhoon FLA 9000).
Immunofluorescence
Cortical neurons were cultured in eight-chamber slides. Cells were fixed with methanol at 4 °C for 30 min and air dried. After blocking for 1 h in PBS with 5 % normal goat serum and 0.1 % Triton X-100, wells were washed 3 × 5 min with PBS followed by incubation with mouse anti-NeuN antibody (1:50 dilution; Chemicon) in PBS with 0.1 % Triton X-100 overnight at 4 °C. After three washes, secondary antibody (TRITC-conjugated goat anti-mouse IgG, 1:200 dilution; Jackson Immunoresearch) was added for 2 h at room temperature. After three washes, the specimens were coverslipped with Vectashield and DAPI (Vector Laboratories) and examined using a Leica 5500 compound microscope.
Results
The possibility of correcting exon 1 of HTT was explored in cell culture using transient expression plasmids. To this end, we adopted a dual plasmid strategy consisting of a novel HTT minigene and a trans-splicing plasmid, the PTM. The splicing competent HTT minigene consisted of a subgenomic segment of the HTT gene and expresses exon 1, exon 2 and exon 3 of human HTT and shortened intervening sequences (Fig. 1). The expanded allele in humans has on average 42 CAG repeats. We therefore designed exon 1 of the minigene to serve as a molecular model of disease by expressing 42 consecutive CAG repeats. This is substantially longer than the 21 CAG repeats of the PTM (described below), making size discrimination feasible. Exon 2 and exon 3 of the minigene were identical to the human HTT sequence. Intron 1 and intron 2 of the human gene are both larger than 10 kb and therefore are not well suited for standard mammalian expression vectors. Therefore, the 5′ and 3′ regions of each intron were fused to generate shorter intervening sequences: 860 bp for intron 1 and 109 bp for intron 2. The construct is splicing competent and produces the expected fully spliced mRNA after transfection (data not shown). The SV40 polyadenylation signal was located at the 3′ end of the construct. Expression was driven by the CMV promoter/enhancer.
Fig. 1.

Schematic of the minigene and the PTM trans-splicing constructs. The minigene contains exons 1 to 3 from the human HTT gene and shortened introns 1 and 2 as well as a polyadenylation signal (pA). Exon 1 was designed to harbor 42 consecutive CAG repeats. The trans-splicing construct (termed PTM) contains exon 1 of human HTT with 21 CAG repeats, followed by an engineered intron. Features include an optimized U1 snRNP binding site at the 5′ splice site, a triplet repeat of intronic splice enhancers (ISE), a branch point (BP), and a tether region complementary to the 5′ region of intron 1 which serves to bring both molecules into close proximity and facilitating the trans-splicing reaction (thick arrow). The resulting chimeric RNA contains trans-exon 1 spliced to cis-exons 2 and 3. The constructs are not drawn to scale
The HTT trans-splicing donor plasmid (termed PTM) was designed to contain the complete HTT exon 1 with 21 CAG repeats which represents a non-pathological number of triplets (Fig. 1). Directly downstream, we placed a U1 snRNP binding site, followed by a spacer and three repeats of the intronic splice enhancer (ISE) sequence, TGCATG, each separated by a short spacer sequence. A branch point (BP) sequence was inserted further downstream. Finally, 100 bp of sequence complementary to the 5′ end of HTT intron 1 was added as the “tether”. This tether serves to bind the PTM RNA to the minigene pre-mRNA, thus bringing the two molecules into close proximity. The trans-splicing reaction is indicated by the thick arrow (Fig. 1).
In order to specifically detect and discriminate between the RNAs derived from the minigene, the PTM, and the expected trans-spliced product, we designed primers for RT-PCR. The specificity of these primers was determined in preliminary experiments which demonstrated that amplification of the individual cDNAs occurred only with the appropriate primer combinations (Fig. 2). Briefly, primers F1, and R1 to R2 are specific for the minigene, whereas F2 is specific for the PTM. Therefore, a PCR product obtained with forward primer F2 and the reverse primer R1 or R2 is indicative of trans-splicing between PTM exon 1 and minigene exon 2. Such a product was not obtained when amplifying from the minigene (Fig. 2a) or the PTM (Fig. 2b) alone, demonstrating the fidelity of detection. Amplification with primers Ex1.1 and Ex1.2 which anneal to exon 1, in conjunction with forward primer F2, resulted in the generation of the expected PCR products from the PTM (Fig. 2b).
Fig. 2.

Primer specificity. Primers were designed to specifically discriminate between the cDNAs generated from the minigene and PTM plasmids, as well as the presumptive trans-splicing product. To this end, we took advantage of the sequence divergence in the 5′-UTRs of the two cDNAs. Primer F1 binds uniquely to the minigene, but not to the PTM, whereas primer F2 specifically recognizes the PTM but not the minigene. Reverse primers R1 and R2 are located in exon 2 (minigene only), whereas reverse primers Ex1.1 and Ex1.2 are complementary to both constructs. a PCR with the minigene and F1 (lanes 1 and 2) or F2 (lanes 3 and 4) and the indicated reverse primer only generates a specific product with the F1 primer. b PCR with the PTM and F1 (lanes 1 and 2) or F2 (lanes 3–6) and the indicated reverse primer only generates a specific product with F2 and Ex1.1 or Ex1.2 (lanes 5 and 6). Identical specificities were observed when cDNA from HEK293 cells co-transfected with these two plasmids was used as PCR input (data not shown)
To test the capacity of the PTM RNA to trans-splice to the minigene pre-mRNA, the two plasmids were co-transfected into HEK293 cells. Total RNA was isolated and RT-PCR was performed using the PTM-specific primer F2 and minigene-specific reverse primers in exon 2 (R1 and R2) and exon 3 (R3–R5). Specific bands of the expected sizes were observed, suggesting that trans-splicing occurred between the two RNAs (Fig. 3). The F2–R4 PCR product was cloned, and sequence analysis identified the correct exon junctions, demonstrating that trans-exon 1 was correctly spliced to exons 2–3 of the minigene (Suppl. Fig. 1). Taken together, the detection of products using F2 and reverse primers R1–R5 is indicative of the successful joining (trans-splicing) of PTM exon 1 to minigene exons 2 and 3.
Fig. 3.

Detection of trans-splicing of exon 1 of the PTM to exons 2 and 3 of the minigene. HEK293 cells were co-transfected with the minigene and PTM constructs and RNA was isolated 48 h later. RT-PCR was performed with PTM-specific F2 forward primer and one of the minigene-specific reverse primers, R1–R5. A specific trans-spliced product of the expected size was detected using reverse primers for exon 2 (R1, R2) and exon 3 (R3–R5)
Since both plasmids contain exon 1, i.e., a stretch of homologous sequence, it is possible that the two plasmids could recombine, thereby generating a DNA molecule containing PTM exon 1 and minigene exon 2 and exon 3. This recombined DNA could then give rise to a RNA species that is indistinguishable from the trans-spliced RNA species. To address this issue, the CMV promoter driving the expression of the PTM was deleted; therefore, the trans-splicing could not occur. The minigene plasmid was co-transfected into HEK293 cells with identical amounts of PTM plasmid with or without the CMV promoter, RNA was isolated and RT-PCR was performed. Expression of the PTM was predictably essentially undetectable without the CMV promoter, and the putative trans-splicing product was absent (Fig. 4, left side). Recombination should have occurred at a similar rate irrespective of the presence of the promoter since identical amounts of DNA were used. Therefore, this strongly suggests that the HTT mRNA species are indeed products of trans-splicing. As an additional control, the tether within the PTM was made into the reverse complement. The tether consists of a sequence complementary to the coding strand. It allows for base-pairing with the pre-mRNA, or potentially with the coding strand of the gene. To further confirm that the observed PCR product originates from an interaction of RNAs, we tested a PTM in which the tether was made into the reverse complement (sense) sequence. The PTM with either the sense or the antisense tether was co-transfected with the minigene into HEK293 cells and RT-PCR was performed on isolated RNA to detect trans-splicing products. As expected, the efficacy of the sense tether was drastically reduced, suggesting again that the generation of the chimeric HTT mRNA species is a result of bona fide trans-splicing (Fig. 4, right side).
Fig. 4.

The trans-splicing product is generated by interaction of RNA molecules, not via plasmid DNA recombination. a HEK293 cells were co-transfected with identical amounts of the minigene plasmid and a PTM construct either with (left lanes, labeled “+”) or without (right lanes, labeled “−”) the CMV promoter. After RNA isolation and RT-PCR using appropriate primer pairs (Fig. 3), mRNA from the minigene (top panel) or the PTM (second panel) was detected. As expected, minigene expression levels were comparable with or without the CMV promoter in the PTM construct. However, in the absence of the CMV promoter in the PTM, only a very small amount of PTM mRNA was detected. Residual levels of PTM RNA which can be detected after prolonged PCR (data not shown) are likely due to the presence of AAV ITRs in the PTM plasmid which have weak intrinsic promoter activity. However, no trans-splicing product was detected using reverse primers for either exon 2 (third panel, F2–R1) or exon 3 (fourth panel, F2–R4) when the CMV promoter was absent in the PTM. GAPDH was used as internal control. Duplicate reactions are shown. b A PTM plasmid in which the 100-bp tether domain sequence (antisense) was replaced by its reverse complement (sense) was co-transfected with the minigene construct into HEK293 cells. With this sense construct, the trans-splicing reaction was much less effective
To determine whether trans-splicing was dose-dependent, a dose–response experiment was performed in which a constant level of minigene target was co-transfected into HEK293 cells with increasing quantities of the PTM plasmid (Fig. 5a). Results indicated that the amount of trans-spliced product increased with the level of PTM expression over nearly a log-fold change in the PTM concentration (Fig. 5a). As expected, trans-splicing was not detected in the absence of the PTM, whereas trans-splicing was detected even at the lowest concentration of the PTM tested. The PTM dose-dependent trans-splicing of the minigene occurred similarly in U2OS osteosarcoma and DBRTG glioblastoma cells (Fig. 5b, c). This suggests that trans-splicing parameters are amenable to optimization and that different cell types are capable of repairing HTT exon 1 via trans-splicing.
Fig. 5.

The trans-splicing product titrates with PTM input in different cell lines. a HEK293 cells were co-transfected with a constant amount of minigene plasmid and increasing amounts of the PTM construct. After RNA isolation, expression levels of the trans-splicing product were determined using the F2 and R1 primer pair. The quality of the RNAs was tested by amplification of GAPDH with or without RT reaction (data not shown). b, c Expression levels of the trans-splicing product in U2OS and DBTRG cells was determined with the F2 and R4 primer pair and also correlated with the amount of PTM input
The minigene system is a straightforward means to initially investigate trans-splicing activity; however, the system is intrinsically artificial and the level of “target” transcript produced by the CMV promoter is exceptionally high. To begin to examine HTT trans-splicing in a more complex environment, we next sought to determine if the PTM is also suitable for trans-splicing of endogenous HTT pre-mRNA. HEK293 cells were transfected with the PTM plasmid alone. RT-PCR was performed with the primer combination F2–R1, and a product of the expected size was detected at all concentrations of the PTM plasmid (Fig. 6). This result indicates that exon 1 of endogenous HTT is amenable to replacement by trans-splicing.
Fig. 6.

Trans-splicing of endogenous HTT pre-mRNA. HEK293 cells were transfected with PTM plasmid alone. RT-PCR was performed using the trans-splicing-specific primer pair F2–R1. The expression levels of the trans-spliced product titrated with the amount of PTM plasmid input
For the delivery and long-term expression of therapeutic candidates in the central nervous system, viral agents are frequently employed, and the feasibility and efficacy of delivering regulatory RNAs has been shown using several HD mouse models [36–41]. Therefore, we transferred the CMV-driven PTM into the pSIN18 lentivirus vector [35] and pseudotyped the virus with VSV-G, which confers a broad tropism and allows entry into a large number of cells, including neuronal cells. To initially test the effectiveness of the PTM lentivirus, HEK293 cells were transiently transfected with the 42 CAG minigene, followed by transduction with the PTM lentivirus. The specific trans-spliced mRNA was detected, suggesting that lentiviral vectors can be used successfully (Fig. 7a). Furthermore, we wished to explore if the virally encoded PTM could direct the exchange of endogenous HTT exon 1. To this end, HEK293 cells were transduced in the absence of the minigene, and the trans-spliced product was detected by RT-PCR (Fig. 7b). Decreasing viral multiplicity of infection correlated with reduced synthesis of PTM RNA and diminished abundance of the trans-spliced endogenous HTT pre-mRNA.
Fig. 7.

Viral delivery of the PTM. a HEK293 cells were transiently transfected with the minigene plasmid, followed by transduction with a lentivirus harboring the PTM and an eGFP module (MOI: 25). Identical amounts of virus were administered to all wells. After 48 h, the cells were harvested and RNA was isolated for RT-PCR. Specific primer pairs were used to amplify GAPDH, minigene, and trans-spliced product. Duplicate reactions are shown. b Viral transduction (MOI: 50, 25, 12.5, 6, 2.5) was performed in the absence of the minigene. Trans-splicing to the endogenous HTT pre-mRNA of HEK293 cells was proportional to the amount of virus added. M mock transduction (no virus added)
In order to determine whether the exon 1 replacement strategy was applicable to a more disease-specific context, we examined trans-splicing in primary fibroblasts from HD patients. First, fibroblasts were transiently transfected with the minigene target, followed by transduction with the PTM lentivirus. Cells were harvested 48–72 h after transduction. As observed before in the other cell types, the amount of trans-spliced product titrated with the quantity of virus added to the fibroblasts cultures using constant minigene input (Fig. 8a). Importantly, trans-splicing was not restricted to the minigene as robust trans-splicing of endogenous HTT by the PTM lentivirus in the absence of minigene was detected in fibroblasts from three independent HD patients (Fig. 8a, bottom panel, and b). These results suggest that the trans-splicing approach is successful in an important disease-appropriate context, primary HD cells.
Fig. 8.

a Primary fibroblasts from HD patients were transfected with identical amounts of minigene plasmid followed by transduction with varying amounts of the PTM lentivirus (top panels) (MOI: 126, 63, 31.5, 15). After 72 h, cells were harvested, RNA was isolated and RT-PCR was performed using the specific primer pair F2–R1. Bottom panel HD patient fibroblasts were transduced with the PTM lentivirus in the absence of minigene and endogenous trans-splicing was detected using the specific primer pair F2–R1. b Two additional HD patient fibroblast lines with different CAG repeat lengths (GM09197 and GM00305) were transduced with the PTM lentivirus (MOI 126 and 31.5, respectively) and trans-splicing to endogenous HTT pre-mRNA was detected. c The relative efficiency of the trans-splicing reaction was determined by performing PCR with a primer pair common to both the cis- and trans-spliced RNAs. The 5′ primer was labeled fluorescently (asterisk), and the products of the PstI digestion were separated on a 8 % non-denaturing polyacrylamide gel. The band intensities were quantified using a Typhoon FLA 9000, and the relative fraction of the 90-bp band is indicated below the scan. The (cis + trans) band varies in size between cell lines due to different CAG tract lengths. HEK293 have 16 and 17 CAG repeats (data not shown), the normal allele of the patient fibroblasts has 17 repeats (Corriell data), and the minigene has 42 repeats
The trans-spliced product is detected using specific primer pairs that differ from those used to detect the minigene mRNA. To quantify the relative amount of trans-spliced product generated, i.e., the efficiency of the trans-splicing versus the cis-splicing reaction, a unique PstI restriction site was introduced in the 5′-UTR of exon 1 of the PTM (Fig. 8c). A common primer pair was then used to amplify both cis- and trans-spliced RNAs. The 5′ primer was labeled fluorescently to allow quantitation. After PstI digestion of the PCR product, the intensity of the 90-bp band is indicative of the relative fraction of trans-spliced RNA. In HEK293 cells, approximately 3 and 5 % trans-splicing was achieved when using endogenous HTT pre-mRNA, or the minigene as target (Fig. 8c). Similarly, patient fibroblasts transduced with the PTM lentivirus showed a slightly lower 1.6 % trans-splicing efficiency.
Since the primary defect in HD manifests itself in the brain, we were interested in examining the possibility of HTT exon 1 replacement in neurons. To this end, primary cortical neuron cultures were prepared from embryonic day 16.5–17.5 brains of YAC128 transgenic mice. Importantly, these mice carry a complete human HTT gene with 128 CAG repeats in exon 1, including the intronic sequence which is targeted by the PTM’s tether [42]. To ascertain the purity of the neuronal cultures, cells were plated in eight-well chamber slides and processed for immunohistochemistry. Staining with an antibody against NeuN, a neuron-specific marker, showed that the vast majority of the cultured cells were neurons (Fig. 9, top panels). We then transduced the neurons with the PTM lentivirus and isolated RNA after 72 h. As expected, PCR products were not generated when we used wild-type neurons from mice not expressing the human HTT gene (data not shown). In contrast, the specific trans-splicing product was successfully detected after RT-PCR with the specific primer set in HD neurons from YAC128 mice expressing the human HTT gene (Fig. 9, bottom panel), demonstrating that the pathogenic expanded exon 1 region can be successfully replaced by spliceosome-mediated pre-mRNA trans-splicing in this important disease context.
Fig. 9.
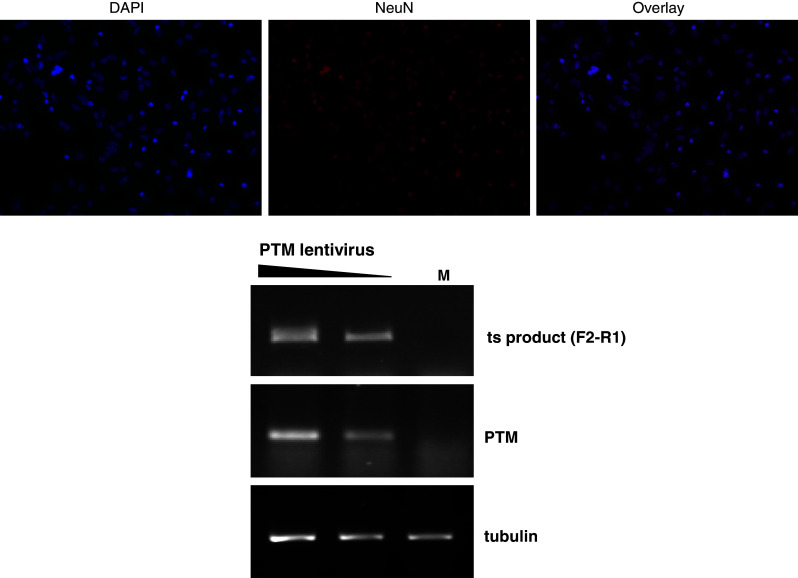
Primary cortical neurons were isolated from YAC128 HD transgenic mice and cultured until DIV7. Top panels Cells were fixed and stained with the neuronal nuclear marker, NeuN, and counterstained with the nuclear stain DAPI to assess the relative purity of the preparations. Bottom panel Cells were transduced with the PTM lentivirus and harvested 72 h later. After RNA isolation and RT-PCR, the trans-splicing product of transgenic HTT pre-mRNA was detected using the specific primer pair F2–R1
Discussion
Targeting RNA for the treatment of inherited disorders is an alternative to conventional gene replacement therapy. RNA-based approaches have a number of potential advantages [43]. For example, since targeted sequences are generally relatively short, they can easily be corrected with current delivery systems, such as adeno-associated virus. In addition, insertional mutagenesis and adverse effects due to genomic integration can be avoided. Importantly, the modification of the disease-related RNA takes place within the framework of a normal regulatory environment where the spatial and temporal expression of the underlying gene is controlled by its intrinsic regulatory mechanisms, and optimal expression is ultimately controlled via the endogenous promoter.
Dominantly inherited disorders, such as HD, are particularly well suited for correction by RNA targeting because the alternative therapeutic introduction of a functional gene does not eliminate the underlying toxic gain-of-function caused by expression of the mutant allele. In this case, reducing or inhibiting the expression of the mutant allele would be advantageous. A promising approach to the suppression of HTT expression is the use of small RNAs. Antisense oligonucleotides have been used successfully to reduce HTT mRNA expression in vitro and in cell culture [44, 45]. These approaches targeted regions common to wild-type and mutant HTT and demonstrated the feasibility of suppressing HTT expression. A recent study using peptide nucleic acid and locked nucleic acid chemistry revealed that it is possible to target the CAG repeat region of HTT and ATXN3 [46]. Depending on the specific sequence of the oligonucleotide and the concentration used, mutant protein levels were reduced while the amount of wild-type protein was not strongly affected. It is unclear how much loss of wild-type HTT can be reasonably tolerated in a therapeutic approach. Non-allele-specific knockdowns of HTT suggest that wild-type HTT can be reduced below 50 % of normal levels, at least in the short term, although this was shown to be accompanied by significant changes in gene expression profiles whose consequences need to be explored [38, 47]. On the other hand, HTT gene knockout in the mouse results in embryonic lethality [2–4], and reduced levels lead to developmental brain defects and perinatal lethality [48]. In addition, lack of HTT in the adult mammalian brain has adverse effects [20, 49]. This suggests that a certain level of wild-type HTT is required for appropriate brain function, making allele-selective approaches highly attractive. Significant discrimination between the expanded and the wild-type allele has been successfully demonstrated by targeting SNPs in the mutant allele using RNAi in vitro [50–53]. HD-associated SNPs have been described [54], and a survey of 225 human samples found that about 75 % of expanded alleles were associated with three specific SNPs that were successfully targeted with a cocktail of five siRNAs, suggesting that the majority of HD patients would be amenable to such a therapeutic regimen [53]. Similarly, Hayden and colleagues have recently developed SNP-based antisense oligonucleotides that reduce mutant HTT expression in an allele-selective fashion [55]. This is potentially very powerful approach to the reduction of mutant HTT in the brain, with the possible drawback that the patient population is not homogeneous in the occurrence of SNPs, and different antisense oligonucleotides may have to be tailored towards specific individuals. Importantly, McBride and colleagues demonstrated that inhibitory RNAs incorporated into artificial miRNA vector scaffolds supported efficient expression and silencing while at the same time exhibiting low levels of toxicity in mouse brain [37], and that reduction of HTT expression levels in rhesus monkeys is overall well tolerated [56].
We explored the feasibility of a novel RNA-based approach for the allele-specific suppression of mutant HTT, i.e., spliceosome-mediated pre-mRNA trans-splicing. In general, trans-splicing offers several advantages. Spatial and temporal expression patterns of HTT should remain unchanged, since the gene is driven by its intrinsic regulatory elements. In addition, exon replacement occurs only in cells expressing HTT, and adverse effects due to expression of the PTM in cells that do not express HTT should therefore be minimal.
An important part of Huntington pathology is the accumulation of specific protein aggregates. The expanded HTT protein forms high molecular weight, β-sheet-rich amyloid-like aggregates similar to those seen in other neurodegenerative disorders, such as Alzheimer’s disease and Parkinson’s disease. Similar protein aggregation structures are observed in prion diseases, such as Creutzfeld–Jakob disease, where these amyloid fibrils are responsible for the infection of healthy neurons. This raises the question whether expanded polyQ amyloids may also play a role in the propagation of the pathology from initially localized foci [57, 58]. Importantly, the aggregates also sequester other proteins, and it is possible that misfolded expanded HTT protein also recruits the normal, non-expanded HTT protein, analogous to the situation in prion disease. Support for this notion comes from work by Ren and coworkers [59], who showed that fibrillar polyQ aggregates can enter cells from the extracellular space. Furthermore, employing cellular reporters (HTT–gfp fusion proteins), these internalized polyQ fibrils recruit soluble, wild-type-length polyQ proteins and induce them to aggregate [59]. Since trans-splicing converts the mutant allele to wild-type, it presumably not only prevents expression of the mutant allele but also increases the level of the wild-type form. The excision of an exon 1 with expanded polyQ tract results in a truncated RNA species lacking a polyadenylation signal. Consequently, this fragment will be susceptible to degradation in the nucleus and is unlikely to be exported and processed by the ribosome. Therefore, the load of mutant protein will be reduced, and the associated cellular pathology, such as protein aggregation and recruitment of wild-type protein, should be reduced. Conceptually, the repair process is applicable to varying repeat lengths, which could potentially allow the development of a single therapeutic molecule for all HD patients. Furthermore, trans-splicing confers essentially functional allele specificity. While the trans-exon 1 may integrate into the pre-mRNA derived from either allele, only the repair of the expanded molecule will have a (positive) functional effect, and the wild-type mRNA will remain normal. A potential limitation of this technique is the relatively low efficiency of trans-splicing and consequently the necessity of optimizing the PTM either empirically or by screening procedures [60].
To begin to address this issue, we performed proof-of-principle experiments to explore whether trans-splicing can be used for the replacement of the 5′ exon of HTT. We generated a prototype PTM that contained exon 1 of HTT with 21 CAG repeats and several engineered features to enhance its activity, including intronic splice enhancers and a U1 snRNP binding sequence at the 3′ end of exon 1. A tether of 100-bp complementary sequence was used to direct the PTM construct to the start of intron 1. The tether forms a double strand at the very 5′ end of intron 1 and masks the U1 snRNP binding site on the minigene pre-mRNA, thus favoring the strong intron 1 5′ splice site complex on the PTM RNA. A polyadenylation signal was omitted to avoid nuclear export and translation of the PTM RNA. When co-transfected with a splice-competent minigene, the PTM RNA was able to productively interact with minigene pre-mRNA, resulting in the generation of a chimeric mRNA, which we detected by RT-PCR using specific primers. This trans-splicing reaction occurred in all three cell lines tested, HEK293, U2OS, and DBTRG, as well as HD patient-derived fibroblasts and cultured neurons from YAC128 transgenic mice expressing human HTT with 128 CAG repeats, suggesting that trans-splicing is applicable to a range of cell types. Endogenous HTT is expressed ubiquitously. Although brain lesions are prominent in HD, many peripheral tissues are also affected [61], and targeting additional cell types should be a consideration in the development of therapeutic regimens.
The co-transfection system allowed us to demonstrate that 5′ exon replacement of HTT is feasible in principle. The HTT minigene target contained the complete exons 1 through 3 while the intervening introns were shortened to allow handling in a plasmid vector. The endogenous intron 1 of human HTT is over 11 kb long. RNA polymerase II interactions with subunits of the splicing machinery are thought to sequester exons near the polymerase, and very large introns may be looped away from this complex [62, 63]. It is conceivable that the functionality or the spatial organization of such a long intron is different from that of the shortened, 0.86-kb minigene intron 1, which may affect interactions of the PTM RNA with the HTT pre-mRNA. Therefore, we administered the PTM without the minigene to investigate trans-splicing of endogenous HTT pre-mRNA. Using RT-PCR, we were able to detect specifically the trans-spliced mRNA species whose abundance titrated with the amount of PTM plasmid transfected. This demonstrates that, using the prototype PTM, the endogenous HTT pre-mRNA can be repaired successfully. Furthermore, expression of the PTM via a lentiviral system resulted in successful trans-splicing of both the minigene and the endogenous HTT pre-mRNA, suggesting that this strategy is amenable to HTT trans-splicing in future in vivo studies.
The efficiency of the trans-splicing reaction was approximately 1–5 % using the prototype PTM in this study. It is not clear at present how much reduction of mutant HTT is necessary to achieve a long-term clinical benefit, and whether the level of trans-splicing observed here would be sufficient to prolong the time to disease onset or ameliorate the disease phenotype in an animal model of HD. Nevertheless, this study is a proof-of-principle that measurable levels of trans-splicing can be achieved in disease-relevant cell types, and future work will be directed towards optimization of this process.
Functional correction using trans-splicing has been reported in several models of human disease, including cystic fibrosis, hemophilia A, X-linked immunodeficiency, and various cancers [64–69]. For example, Liu et al. repaired the gene defect Δ508 in CFTR, which encodes the cystic fibrosis transmembrane conductance regulator, in explants of human cystic fibrosis airway epithelia in a xenograft model [64, 65]. They achieved a partial restoration of conductance in airway epithelial cells, thereby demonstrating functional improvements in a disease-relevant cell type. We developed the first trans-splicing strategy to increase the expression of full-length mRNA and functional SMN protein from the SMN2 gene in the context of spinal muscular atrophy [34]. The first in vivo RNA repair by trans-splicing was performed in factor VIII hemophilia A-knockout mice, demonstrating the feasibility of transferring this methodology into an animal model [66]. A recent study by Wally et al. [70] demonstrated the potential power of trans-splicing for the allele-specific correction of a dominant-negative mutation in the plectin gene. A mutation in exon 9 of plectin causes increased protein aggregation and degradation, leading to the blistering skin disease epidermolysis bullosa. Combining an exon replacement strategy with viral delivery, the level of full-length plectin protein was increased >50 % in patient fibroblasts. These examples show that meaningful levels of repair can be achieved, suggesting that trans-splicing might be a promising new tool for the treatment of autosomal-dominant genetic disorders.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Supplementary Fig. 1 Sequence of the trans-splicing product. The F2-R4 product shown in Fig. 3 was cloned and sequenced to confirm correct splicing. Exon junctions are indicated by the thick vertical bars. (PPTX 95 kb)
Acknowledgments
This work was supported by a Faculty Research grant from the University of Missouri College of Veterinary Medicine, the Huntington Disease Foundation of Canada, and the National Institutes of Health (1R21NS070072).
References
- 1.Vonsattel JP, Myers RH, Stevens TJ, Ferrante RJ, Bird ED, Richardson EP., Jr Neuropathological classification of Huntington’s disease. J Neuropathol Exp Neurol. 1985;44:559–577. doi: 10.1097/00005072-198511000-00003. [DOI] [PubMed] [Google Scholar]
- 2.Nasir J, Floresco SB, O’Kusky JR, Diewert VM, Richman JM, Zeisler J, Borowski A, Marth JD, Phillips AG, Hayden MR. Targeted disruption of the Huntington’s disease gene results in embryonic lethality and behavioral and morphological changes in heterozygotes. Cell. 1995;81:811–823. doi: 10.1016/0092-8674(95)90542-1. [DOI] [PubMed] [Google Scholar]
- 3.Zeitlin S, Liu JP, Chapman DL, Papaioannou VE, Efstratiadis A. Increased apoptosis and early embryonic lethality in mice nullizygous for the Huntington’s disease gene homologue. Nat Genet. 1995;11:155–163. doi: 10.1038/ng1095-155. [DOI] [PubMed] [Google Scholar]
- 4.Duyao MP, Auerbach AB, Ryan A, Persichetti F, Barnes GT, McNeil SM, Ge P, Vonsattel JP, Gusella JF, Joyner AL, et al. Inactivation of the mouse Huntington’s disease gene homolog Hdh. Science. 1995;269:407–410. doi: 10.1126/science.7618107. [DOI] [PubMed] [Google Scholar]
- 5.MacDonald ME. Huntingtin: alive and well and working in middle management. Sci STKE. 2003;207:pe48. doi: 10.1126/stke.2003.207.pe48. [DOI] [PubMed] [Google Scholar]
- 6.Li SH, Li XJ. Huntingtin-protein interactions and the pathogenesis of Huntington’s disease. Trends Genet. 2004;20:146–154. doi: 10.1016/j.tig.2004.01.008. [DOI] [PubMed] [Google Scholar]
- 7.Ferrante RJ, Gutekunst CA, Persichetti F, McNeil SM, Kowall NW, Gusella JF, MacDonald ME, Beal MF, Hersch SM. Heterogeneous topographic and cellular distribution of huntingtin expression in the normal human neostriatum. J Neurosci. 1997;17:3052–3063. doi: 10.1523/JNEUROSCI.17-09-03052.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Fusco FR, Chen Q, Lamoreaux WJ, Figueredo-Cardenas G, Jiao Y, Coffman JA, Surmeier DJ, Honig MG, Carlock LR, Reiner A. Cellular localization of huntingtin in striatal and cortical neurons in rats: lack of correlation with neuronal vulnerability in Huntington’s disease. J Neurosci. 1999;19:1189–1202. doi: 10.1523/JNEUROSCI.19-04-01189.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.DiFiglia M, Sapp E, Chase K, Schwarz C, Meloni A, Young C, Martin E, Vonsattel JP, Carraway R, Reeves SA, et al. Huntingtin is a cytoplasmic protein associated with vesicles in human and rat brain neurons. Neuron. 1995;14:1075–1081. doi: 10.1016/0896-6273(95)90346-1. [DOI] [PubMed] [Google Scholar]
- 10.Velier J, Kim M, Schwarz C, Kim TW, Sapp E, Chase K, Aronin N, DiFiglia M. Wild-type and mutant huntingtins function in vesicle trafficking in the secretory and endocytic pathways. Exp Neurol. 1998;152:34–40. doi: 10.1006/exnr.1998.6832. [DOI] [PubMed] [Google Scholar]
- 11.Kegel KB, Meloni AR, Yi Y, Kim YJ, Doyle E, Cuiffo BG, Sapp E, Wang Y, Qin ZH, Chen JD, Nevins JR, Aronin N, DiFiglia M. Huntingtin is present in the nucleus, interacts with the transcriptional corepressor C-terminal binding protein, and represses transcription. J Biol Chem. 2002;277:7466–7476. doi: 10.1074/jbc.M103946200. [DOI] [PubMed] [Google Scholar]
- 12.Wexler NS, Lorimer J, Porter J, Gomez F, Moskowitz C, Shackell E, Marder K, Penchaszadeh G, Roberts SA, Gayan J, Brocklebank D, Cherny SS, Cardon LR, Gray J, Dlouhy SR, Wiktorski S, Hodes ME, Conneally PM, Penney JB, Gusella J, Cha JH, Irizarry M, Rosas D, Hersch S, Hollingsworth Z, MacDonald M, Young AB, Andresen JM, Housman DE, De Young MM, Bonilla E, Stillings T, Negrette A, Snodgrass SR, Martinez-Jaurrieta MD, Ramos-Arroyo MA, Bickham J, Ramos JS, Marshall F, Shoulson I, Rey GJ, Feigin A, Arnheim N, Acevedo-Cruz A, Acosta L, Alvir J, Fischbeck K, Thompson LM, Young A, Dure L, O’Brien CJ, Paulsen J, Brickman A, Krch D, Peery S, Hogarth P, Higgins DS, Jr, Landwehrmeyer B. Venezuelan kindreds reveal that genetic and environmental factors modulate Huntington’s disease age of onset. Proc Natl Acad Sci USA. 2004;101:3498–3503. doi: 10.1073/pnas.0308679101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Walker FO. Huntington’s disease. Lancet. 2007;369:218–228. doi: 10.1016/S0140-6736(07)60111-1. [DOI] [PubMed] [Google Scholar]
- 14.Cha JH. Transcriptional dysregulation in Huntington’s disease. Trends Neurosci. 2000;23:387–392. doi: 10.1016/S0166-2236(00)01609-X. [DOI] [PubMed] [Google Scholar]
- 15.Bates G, Benn C (2002) The polyglutamine diseases. In: Bates G, Harper P and Jones L (eds) Huntington’s disease, Oxford University Press, London, p 429–474
- 16.Hickey MA, Chesselet MF. Apoptosis in Huntington’s disease. Prog Neuropsychopharmacol Biol Psychiatry. 2003;27:255–265. doi: 10.1016/S0278-5846(03)00021-6. [DOI] [PubMed] [Google Scholar]
- 17.DiProspero NA, Chen EY, Charles V, Plomann M, Kordower JH, Tagle DA. Early changes in Huntington’s disease patient brains involve alterations in cytoskeletal and synaptic elements. J Neurocytol. 2004;33:517–533. doi: 10.1007/s11068-004-0514-8. [DOI] [PubMed] [Google Scholar]
- 18.Leoni V, Mariotti C, Tabrizi SJ, Valenza M, Wild EJ, Henley SM, Hobbs NZ, Mandelli ML, Grisoli M, Bjorkhem I, Cattaneo E, Di Donato S. Plasma 24S-hydroxycholesterol and caudate MRI in pre-manifest and early Huntington’s disease. Brain. 2008;131:2851–2859. doi: 10.1093/brain/awn212. [DOI] [PubMed] [Google Scholar]
- 19.Duennwald ML, Lindquist S. Impaired ERAD and ER stress are early and specific events in polyglutamine toxicity. Genes Dev. 2008;22:3308–3319. doi: 10.1101/gad.1673408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Dragatsis I, Levine MS, Zeitlin S. Inactivation of Hdh in the brain and testis results in progressive neurodegeneration and sterility in mice. Nat Genet. 2000;26:300–306. doi: 10.1038/81593. [DOI] [PubMed] [Google Scholar]
- 21.Zuccato C, Ciammola A, Rigamonti D, Leavitt BR, Goffredo D, Conti L, MacDonald ME, Friedlander RM, Silani V, Hayden MR, Timmusk T, Sipione S, Cattaneo E. Loss of huntingtin-mediated BDNF gene transcription in Huntington’s disease. Science. 2001;293:493–498. doi: 10.1126/science.1059581. [DOI] [PubMed] [Google Scholar]
- 22.Puttaraju M, Jamison SF, Mansfield SG, Garcia-Blanco MA, Mitchell LG. Spliceosome-mediated RNA trans-splicing as a tool for gene therapy. Nat Biotechnol. 1999;17:246–252. doi: 10.1038/6986. [DOI] [PubMed] [Google Scholar]
- 23.Mansfield SG, Kole J, Puttaraju M, Yang CC, Garcia-Blanco MA, Cohn JA, Mitchell LG. Repair of CFTR mRNA by spliceosome-mediated RNA trans-splicing. Gene Ther. 2000;7:1885–1895. doi: 10.1038/sj.gt.3301307. [DOI] [PubMed] [Google Scholar]
- 24.Kikumori T, Cote GJ, Gagel RF. Promiscuity of pre-mRNA spliceosome-mediated trans splicing: a problem for gene therapy? Hum Gene Ther. 2001;12:1429–1441. doi: 10.1089/104303401750298580. [DOI] [PubMed] [Google Scholar]
- 25.Puttaraju M, DiPasquale J, Baker CC, Mitchell LG, Garcia-Blanco MA. Messenger RNA repair and restoration of protein function by spliceosome-mediated RNA trans-splicing. Mol Ther. 2001;4:105–114. doi: 10.1006/mthe.2001.0426. [DOI] [PubMed] [Google Scholar]
- 26.Labrador M, Corces VG. Extensive exon reshuffling over evolutionary time coupled to trans-splicing in Drosophila . Genome Res. 2003;13:2220–2228. doi: 10.1101/gr.1440703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Flouriot G, Brand H, Seraphin B, Gannon F. Natural trans-spliced mRNAs are generated from the human estrogen receptor-alpha (hER alpha) gene. J Biol Chem. 2002;277:26244–26251. doi: 10.1074/jbc.M203513200. [DOI] [PubMed] [Google Scholar]
- 28.Dorn R, Krauss V. The modifier of mdg4 locus in Drosophila: functional complexity is resolved by trans splicing. Genetica. 2003;117:165–177. doi: 10.1023/A:1022983810016. [DOI] [PubMed] [Google Scholar]
- 29.Finta C, Zaphiropoulos PG. Intergenic mRNA molecules resulting from trans-splicing. J Biol Chem. 2002;277:5882–5890. doi: 10.1074/jbc.M109175200. [DOI] [PubMed] [Google Scholar]
- 30.Caudevilla C, Serra D, Miliar A, Codony C, Asins G, Bach M, Hegardt FG. Natural trans-splicing in carnitine octanoyltransferase pre-mRNAs in rat liver. Proc Natl Acad Sci USA. 1998;95:12185–12190. doi: 10.1073/pnas.95.21.12185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bruzik JP, Maniatis T. Spliced leader RNAs from lower eukaryotes are trans-spliced in mammalian cells. Nature. 1992;360:692–695. doi: 10.1038/360692a0. [DOI] [PubMed] [Google Scholar]
- 32.Li H, Wang J, Mor G, Sklar J. A neoplastic gene fusion mimics trans-splicing of RNAs in normal human cells. Science. 2008;321:1357–1361. doi: 10.1126/science.1156725. [DOI] [PubMed] [Google Scholar]
- 33.Rickman DS, Pflueger D, Moss B, VanDoren VE, Chen CX, de la Taille A, Kuefer R, Tewari AK, Setlur SR, Demichelis F, Rubin MA. SLC45A3-ELK4 is a novel and frequent erythroblast transformation-specific fusion transcript in prostate cancer. Cancer Res. 2009;69:2734–2738. doi: 10.1158/0008-5472.CAN-08-4926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Coady TH, Baughan TD, Shababi M, Passini MA, Lorson CL. Development of a single vector system that enhances trans-splicing of SMN2 transcripts. PLoS One. 2008;3:e3468. doi: 10.1371/journal.pone.0003468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Gropp M, Itsykson P, Singer O, Ben-Hur T, Reinhartz E, Galun E, Reubinoff BE. Stable genetic modification of human embryonic stem cells by lentiviral vectors. Mol Ther. 2003;7:281–287. doi: 10.1016/S1525-0016(02)00047-3. [DOI] [PubMed] [Google Scholar]
- 36.DiFiglia M, Sena-Esteves M, Chase K, Sapp E, Pfister E, Sass M, Yoder J, Reeves P, Pandey RK, Rajeev KG, Manoharan M, Sah DW, Zamore PD, Aronin N. Therapeutic silencing of mutant huntingtin with siRNA attenuates striatal and cortical neuropathology and behavioral deficits. Proc Natl Acad Sci USA. 2007;104:17204–17209. doi: 10.1073/pnas.0708285104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.McBride JL, Boudreau RL, Harper SQ, Staber PD, Monteys AM, Martins I, Gilmore BL, Burstein H, Peluso RW, Polisky B, Carter BJ, Davidson BL. Artificial miRNAs mitigate shRNA-mediated toxicity in the brain: implications for the therapeutic development of RNAi. Proc Natl Acad Sci USA. 2008;105:5868–5873. doi: 10.1073/pnas.0801775105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Drouet V, Perrin V, Hassig R, Dufour N, Auregan G, Alves S, Bonvento G, Brouillet E, Luthi-Carter R, Hantraye P, Deglon N. Sustained effects of nonallele-specific Huntingtin silencing. Ann Neurol. 2009;65:276–285. doi: 10.1002/ana.21569. [DOI] [PubMed] [Google Scholar]
- 39.Mochizuki H, Yasuda T, Mouradian MM. Advances in gene therapy for movement disorders. Neurotherapeutics. 2008;5:260–269. doi: 10.1016/j.nurt.2008.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Danos O. AAV vectors for RNA-based modulation of gene expression. Gene Ther. 2008;15:864–869. doi: 10.1038/gt.2008.69. [DOI] [PubMed] [Google Scholar]
- 41.Harper SQ. Progress and challenges in RNA interference therapy for Huntington disease. Arch Neurol. 2009;66:933–938. doi: 10.1001/archneurol.2009.180. [DOI] [PubMed] [Google Scholar]
- 42.Slow EJ, van Raamsdonk J, Rogers D, Coleman SH, Graham RK, Deng Y, Oh R, Bissada N, Hossain SM, Yang YZ, Li XJ, Simpson EM, Gutekunst CA, Leavitt BR, Hayden MR. Selective striatal neuronal loss in a YAC128 mouse model of Huntington disease. Hum Mol Genet. 2003;12:1555–1567. doi: 10.1093/hmg/ddg169. [DOI] [PubMed] [Google Scholar]
- 43.Wood M, Yin H, McClorey G. Modulating the expression of disease genes with RNA-based therapy. PLoS Genet. 2007;3:e109. doi: 10.1371/journal.pgen.0030109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Boado RJ, Kazantsev A, Apostol BL, Thompson LM, Pardridge WM. Antisense-mediated down-regulation of the human huntingtin gene. J Pharmacol Exp Ther. 2000;295:239–243. [PubMed] [Google Scholar]
- 45.Nellemann C, Abell K, Norremolle A, Lokkegaard T, Naver B, Ropke C, Rygaard J, Sorensen SA, Hasholt L. Inhibition of Huntington synthesis by antisense oligodeoxynucleotides. Mol Cell Neurosci. 2000;16:313–323. doi: 10.1006/mcne.2000.0872. [DOI] [PubMed] [Google Scholar]
- 46.Hu J, Matsui M, Gagnon KT, Schwartz JC, Gabillet S, Arar K, Wu J, Bezprozvanny I, Corey DR. Allele-specific silencing of mutant huntingtin and ataxin-3 genes by targeting expanded CAG repeats in mRNAs. Nat Biotechnol. 2009;27:478–484. doi: 10.1038/nbt.1539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Boudreau RL, McBride JL, Martins I, Shen S, Xing Y, Carter BJ, Davidson BL. Nonallele-specific silencing of mutant and wild-type huntingtin demonstrates therapeutic efficacy in Huntington’s disease mice. Mol Ther. 2009;17:1053–1063. doi: 10.1038/mt.2009.17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.White JK, Auerbach W, Duyao MP, Vonsattel JP, Gusella JF, Joyner AL, MacDonald ME. Huntingtin is required for neurogenesis and is not impaired by the Huntington’s disease CAG expansion. Nat Genet. 1997;17:404–410. doi: 10.1038/ng1297-404. [DOI] [PubMed] [Google Scholar]
- 49.Dietrich P, Shanmugasundaram R, Shuyu E, Dragatsis I. Congenital hydrocephalus associated with abnormal subcommissural organ in mice lacking huntingtin in Wnt1 cell lineages. Hum Mol Genet. 2009;18:142–150. doi: 10.1093/hmg/ddn324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.van Bilsen PH, Jaspers L, Lombardi MS, Odekerken JC, Burright EN, Kaemmerer WF. Identification and allele-specific silencing of the mutant huntingtin allele in Huntington’s disease patient-derived fibroblasts. Hum Gene Ther. 2008;19:710–719. doi: 10.1089/hum.2007.116. [DOI] [PubMed] [Google Scholar]
- 51.Schwarz DS, Ding H, Kennington L, Moore JT, Schelter J, Burchard J, Linsley PS, Aronin N, Xu Z, Zamore PD. Designing siRNA that distinguish between genes that differ by a single nucleotide. PLoS Genet. 2006;2:e140. doi: 10.1371/journal.pgen.0020140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Zhang Y, Engelman J, Friedlander RM. Allele-specific silencing of mutant Huntington’s disease gene. J Neurochem. 2009;108:82–90. doi: 10.1111/j.1471-4159.2008.05734.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Pfister EL, Kennington L, Straubhaar J, Wagh S, Liu W, DiFiglia M, Landwehrmeyer B, Vonsattel JP, Zamore PD, Aronin N. Five siRNAs targeting three SNPs may provide therapy for three-quarters of Huntington’s disease patients. Curr Biol. 2009;19:774–778. doi: 10.1016/j.cub.2009.03.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Warby SC, Doty CN, Graham RK, Shively J, Singaraja RR, Hayden MR. Phosphorylation of huntingtin reduces the accumulation of its nuclear fragments. Mol Cell Neurosci. 2009;40:121–127. doi: 10.1016/j.mcn.2008.09.007. [DOI] [PubMed] [Google Scholar]
- 55.Carroll JB, Warby SC, Southwell AL, Doty CN, Greenlee S, Skotte N, Hung G, Bennett CF, Freier SM, Hayden MR. Potent and selective antisense oligonucleotides targeting single-nucleotide polymorphisms in the Huntington disease gene/allele-specific silencing of mutant huntingtin. Mol Ther. 2011;19:2178–2185. doi: 10.1038/mt.2011.201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.McBride JL, Pitzer MR, Boudreau RL, Dufour B, Hobbs T, Ojeda SR, Davidson BL. Preclinical safety of RNAi-mediated HTT suppression in the rhesus macaque as a potential therapy for Huntington’s disease. Mol Ther. 2011;19:2152–2162. doi: 10.1038/mt.2011.219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Lee SJ, Lim HS, Masliah E, Lee HJ. Protein aggregate spreading in neurodegenerative diseases: problems and perspectives. Neurosci Res. 2011;70:339–348. doi: 10.1016/j.neures.2011.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Jucker M, Walker LC. Pathogenic protein seeding in Alzheimer disease and other neurodegenerative disorders. Ann Neurol. 2011;70:532–540. doi: 10.1002/ana.22615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Ren PH, Lauckner JE, Kachirskaia I, Heuser JE, Melki R, Kopito RR. Cytoplasmic penetration and persistent infection of mammalian cells by polyglutamine aggregates. Nat Cell Biol. 2009;11:219–225. doi: 10.1038/ncb1830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Mitchell LG, McGarrity GJ. Gene therapy progress and prospects: reprogramming gene expression by trans-splicing. Gene Ther. 2005;12:1477–1485. doi: 10.1038/sj.gt.3302596. [DOI] [PubMed] [Google Scholar]
- 61.Sassone J, Colciago C, Cislaghi G, Silani V, Ciammola A. Huntington’s disease: the current state of research with peripheral tissues. Exp Neurol. 2009;219:385–397. doi: 10.1016/j.expneurol.2009.05.012. [DOI] [PubMed] [Google Scholar]
- 62.Morris DP, Greenleaf AL. The splicing factor, Prp40, binds the phosphorylated carboxyl-terminal domain of RNA polymerase II. J Biol Chem. 2000;275:39935–39943. doi: 10.1074/jbc.M004118200. [DOI] [PubMed] [Google Scholar]
- 63.Goldstrohm AC, Albrecht TR, Sune C, Bedford MT, Garcia-Blanco MA. The transcription elongation factor CA150 interacts with RNA polymerase II and the pre-mRNA splicing factor SF1. Mol Cell Biol. 2001;21:7617–7628. doi: 10.1128/MCB.21.22.7617-7628.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Liu X, Jiang Q, Mansfield SG, Puttaraju M, Zhang Y, Zhou W, Cohn JA, Garcia-Blanco MA, Mitchell LG, Engelhardt JF. Partial correction of endogenous DeltaF508 CFTR in human cystic fibrosis airway epithelia by spliceosome-mediated RNA trans-splicing. Nat Biotechnol. 2002;20:47–52. doi: 10.1038/nbt0102-47. [DOI] [PubMed] [Google Scholar]
- 65.Liu X, Luo M, Zhang LN, Yan Z, Zak R, Ding W, Mansfield SG, Mitchell LG, Engelhardt JF. Spliceosome-mediated RNA trans-splicing with recombinant adeno-associated virus partially restores cystic fibrosis transmembrane conductance regulator function to polarized human cystic fibrosis airway epithelial cells. Hum Gene Ther. 2005;16:1116–1123. doi: 10.1089/hum.2005.16.1116. [DOI] [PubMed] [Google Scholar]
- 66.Chao H, Mansfield SG, Bartel RC, Hiriyanna S, Mitchell LG, Garcia-Blanco MA, Walsh CE. Phenotype correction of hemophilia A mice by spliceosome-mediated RNA trans-splicing. Nat Med. 2003;9:1015–1019. doi: 10.1038/nm900. [DOI] [PubMed] [Google Scholar]
- 67.Nakayama K, Pergolizzi RG, Crystal RG. Gene transfer-mediated pre-mRNA segmental trans-splicing as a strategy to deliver intracellular toxins for cancer therapy. Cancer Res. 2005;65:254–263. [PubMed] [Google Scholar]
- 68.Pergolizzi RG, Ropper AE, Dragos R, Reid AC, Nakayama K, Tan Y, Ehteshami JR, Coleman SH, Silver RB, Hackett NR, Menez A, Crystal RG. In vivo trans-splicing of 5′ and 3′ segments of pre-mRNA directed by corresponding DNA sequences delivered by gene transfer. Mol Ther. 2003;8:999–1008. doi: 10.1016/j.ymthe.2003.08.022. [DOI] [PubMed] [Google Scholar]
- 69.Tahara M, Pergolizzi RG, Kobayashi H, Krause A, Luettich K, Lesser ML, Crystal RG. Trans-splicing repair of CD40 ligand deficiency results in naturally regulated correction of a mouse model of hyper-IgM X-linked immunodeficiency. Nat Med. 2004;10:835–841. doi: 10.1038/nm1086. [DOI] [PubMed] [Google Scholar]
- 70.Wally V, Klausegger A, Koller U, Lochmuller H, Krause S, Wiche G, Mitchell LG, Hintner H, Bauer JW. 5′ trans-splicing repair of the PLEC1 gene. J Invest Dermatol. 2008;128:568–574. doi: 10.1038/sj.jid.5701152. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Fig. 1 Sequence of the trans-splicing product. The F2-R4 product shown in Fig. 3 was cloned and sequenced to confirm correct splicing. Exon junctions are indicated by the thick vertical bars. (PPTX 95 kb)