

Abstract
Purpose
Array comparative genomic hybridization (array-CGH) is a technique used to analyze quantitative increase or decrease of chromosomes by competitive DNA hybridization of patients and controls. This study aimed to evaluate the benefits and yield of array-CGH in comparison with conventional karyotyping in pediatric neurology patients.
Materials and Methods
We included 87 patients from the pediatric neurology clinic with at least one of the following features: developmental delay, mental retardation, dysmorphic face, or epilepsy. DNA extracted from patients and controls was hybridized on the Roche NimbleGen 135K oligonucleotide array and compared with G-band karyotyping. The results were analyzed with findings reported in recent publications and internet databases.
Results
Chromosome imbalances, including 9 cases detected also by G-band karyotyping, were found in 28 patients (32.2%), and at least 19 of them seemed to be causally related to the abnormal phenotypes. Regarding each clinical symptom, 26.2% of 42 developmental delay patients, 44.4% of 18 mental retardation patients, 42.9% of 28 dysmorphic face patients, and 34.6% of 26 epilepsy patients showed abnormal array results.
Conclusion
Although there were relatively small number of tests in patients with pediatric neurologic disease, this study demonstrated that array-CGH is a very useful tool for clinical diagnosis of unknown genome abnormalities performed in pediatric neurology clinics.
Keywords: Comparative genomic hybridization, nervous system disease, child
INTRODUCTION
Pediatric neurologists employ many diagnostic methods to identify and classify the causes of the diseases often encountered in pediatric neurology clinics. These diseases include developmental delay or mental retardation, dysmorphic face, intractable epilepsy, and multiple congenital anomalies. Developmental delay indicates deficits of motor function, cognitive abilities, and/or language development in children less than 5 years of age. Mental retardation is defined by intellectual disability noted in developmentally delayed infants and young children well after they reach school age. In most of these cases of developmental delay and mental retardation, the causes are not fully known, although they can be partially due to intrauterine infection, premature birth, or genetic disorders, etc. G-band karyotyping is generally used to detect chromosomal imbalance since it is the most common known cause of mental retardation. However, conventional G-band karyotyping analysis cannot reliably detect rearrangements of genomic segments smaller than 3-5 million base pairs (Mb).1 Recently, several new methods based on fluorescence in situ hybridization (FISH), polymerase chain reaction (PCR) and array techniques have been developed to increase early detection rates of microdeletion and microduplication. A major limitation of FISH is that it is difficult to determine which probe to use for the broad spectrum of syndromic face or developmental delay. Due to the limited amount of available material, only a small number of targets can be reasonably investigated. Array comparative genomic hybridization (array-CGH) has recently been used to define genomic imbalance, especially in pediatric neurologic diseases. In the present study, to evaluate the benefits and yield of array CGH, we analyzed 87 pediatric patients with neurologic disease using NimbelGen 135K CGX-3 microarray platforms.
MATERIALS AND METHODS
Patients
During the period from April 2010 to February 2012, 87 patient samples were obtained from the pediatric neurology clinic of Guro and Ansan Korea University Medical Center (KUMC). All patients had at least one of the following clinical manifestations: developmental delay, mental retardation, intractable seizures, or dysmorphic features. Karyotyping by G-banding and array-CGH were performed in all patients. All patients agreed with the study, and this study was approved by the ethical Review Board Hospital District of KUMC.
Methods
The medical records were retrospectively reviewed and collected, then returned to their respective clinics, in order to obtain clinical information about dysmorphic features, developmental delay, mental retardation and epilepsy. The dysmorphic features of the patients were presented by reference from the publications on Am J Med Genet about "Elements of morphology" published in 2009.2,3
Cytogenetic analyses were performed using G-band karyotyping and array CGH simultaneously.
In array CGH, the patients' DNA and normal control DNA (Human Genomic DNA: Male/Female, Promega, Madison, WI, USA) were labelled with Cy3 and Cy5 each through a random priming method, using Klenow fragment (NimbleGen Dual-Color DNA Labeling Kit, Roche NimbleGen, Inc., Madison, WI, USA). An array hybridization and washing were then performed on NimbelGen 135K CGX-3 Array (Roche NimbleGen, Inc., Madison, WI, USA) according to the manufacturer's instructions.
The array was scanned at scanner resolution of 2.5 µm using NimbleGen MS 200 Microarray Scanner and analyzed using NimbleScan v2.6 software (Roche NimbleGen Inc., Madison, WI, USA) and Genoglyphix analysis software (http://www.uk.genoglyphix.com) (Signature Genomics, Spokane, WA, USA) with the use of aberration filter of three contiguous probes. The criteria for Quality Control metrics were determined using standard deviation (SD) <0.14 and median absolute deviation 1 difference <0.23 as major parameters for high-quality array reference values. Log2 values of <0.3 were excluded from the analysis, and the patient to control ratio of 2 : 3 or greater were considered to be copy number variantions (CNVs).
Public databases, such as American College of Medical Genetics Practice Guidelines, University of California Santa Cruz Genome Browser (http://genome.ucsc.edu), Decipher (http://decipher.sanger.ac.uk), the Database of Genomic Variants (DGV, http://projects.tcag.ca/variation/), the International Standards for Cytogenomic Arrays (http://iscaconsortium.org), the National Center for Biotechnology Information, and the Genoglyphix Chromosome Aberration Database, a genoglyphix-user database provided by genolgyphix, were used to classify and interpret the identified CNVs into either benign CNVs (CNVs of uncertain clinical significance) or pathogenic CNVs.
When possible, CNVs detected by microarray analysis were confirmed by FISH using bacterial artificial chromosome probes located in the regions of gain or loss and other regions.
RESULTS
Characteristics of patients
Array-CGH tests were performed in a total of 87 patients, comprising 47 males and 40 females. The age at the time of inspection varied from 1 month to 22 years; the mean age was 61.8±127.1 (2 SD) months.
Comparison of array-CGH results in each clinical manifestation
Each patient showed at least one clinical manifestation. The average age of each patient with a clinical symptom was 44.0±95.3 months in patients with developmental delay, 135.1±121.1 months in patients with mental retardation, 64.3±127.0 in patients with dysmorphic face, and 52.9±90.4 months in patients with epilepsy. Abnormal array-CGH cases were found in 11 (26.2%) of 42 patients with developmental delay, 10 (44.4%) of 18 mental retardation patients, 12 (42.9%) of 28 dysmorphic face patients, and 9 (34.6%) of 26 epilepsy patients. In addition, genetic imbalance was detected in 1 of 3 lissencephaly patients and 1 of 7 cardiac anomaly patients. However, array-CGH abnormality was not found in a small number of patients with multiple congenital anomalies, delayed language development, autism spectrum disorder, microcephaly, paroxysmal kinesigenic dyskinesia and cryptochidism. The positive rates were similar with values of 30.8% and 33.3% for the one manifestated patient, 12/39 and two or more conditional patients, 16/48 each.
Aberration of array-CGH results
Array-CGH analysis revealed genetic aberrations related to known syndromes in 19 (21.8%) of a total of 87 patients (Table 1). In addition, there were potential clinically significant aberrations of array-CGH in 9 (10.3%) patients (Table 2). Furthermore, all patients had more than one benign aberration which totaled 385 CNVs and the average size was 140.6±280.9 kb. From those CNVs, there were 77 aberrations not listed in DGV and may thus be CNVs specific for the Korean population. Twenty-three patients had one aberration each and 5 patients had two aberrations each (patients No. 17, 20, 23, 25, and 26). There were 18 deletion cases, 4 duplication cases, and 6 cases of both deletion and duplication. All duplication only cases were of unclear clinical significance. In the present study, 6 patients had an anomaly in pericentromeric regions (patients No. 2, 6, 11, 12, 26, and 27), while 2 patients had an anomaly in peritelomere regions (patients No. 7 and 10). The total size of these alterations varied between 0.15 Mb and 8.92 Mb in patients with normal karyotyping. In 9 patients (patients No. 8, 12, 13, 14, 16, 17, 18, 20, 27) with abnormal G-band karyotyping, 3 of them (patients No. 17, 20, 27) had additional precise information from array-CGH.
Table 1.
Aberrations Result Related to Known Syndromes in 19 Patients
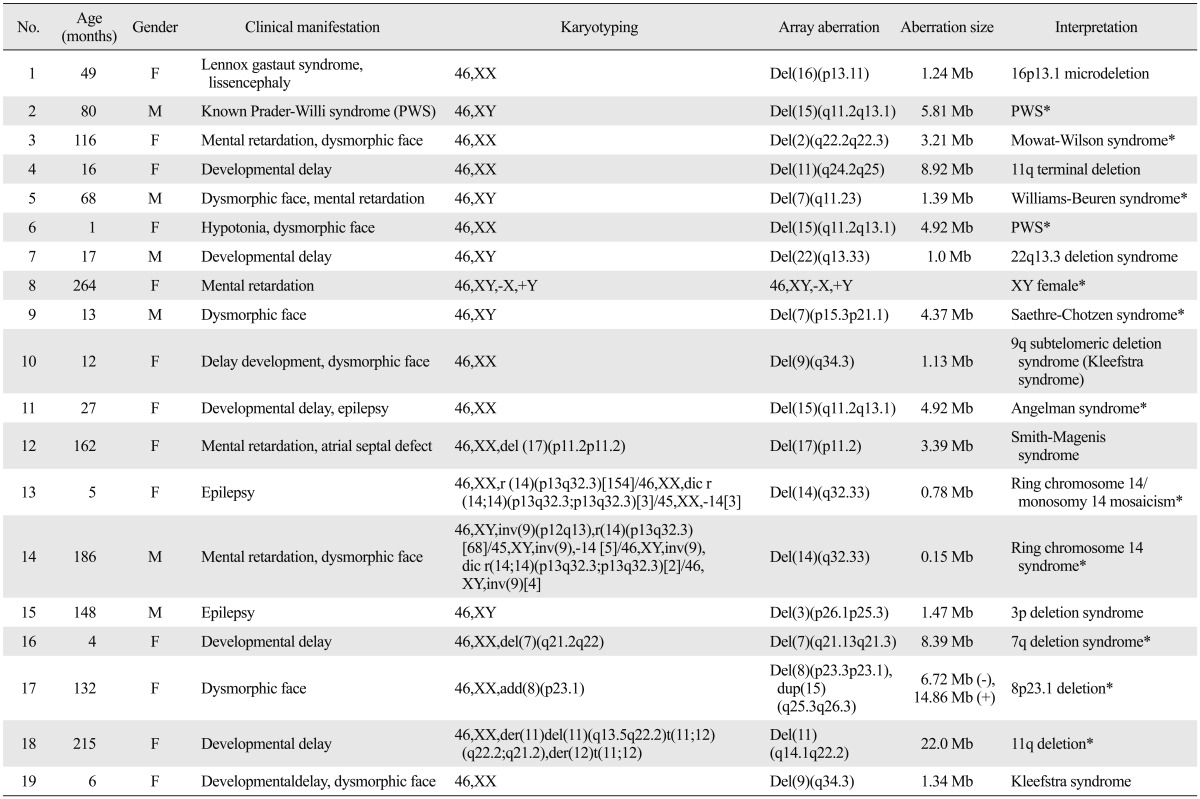
*Confirmed with other methods.
Table 2.
Aberration Result Potentially Causative in 9 Patients
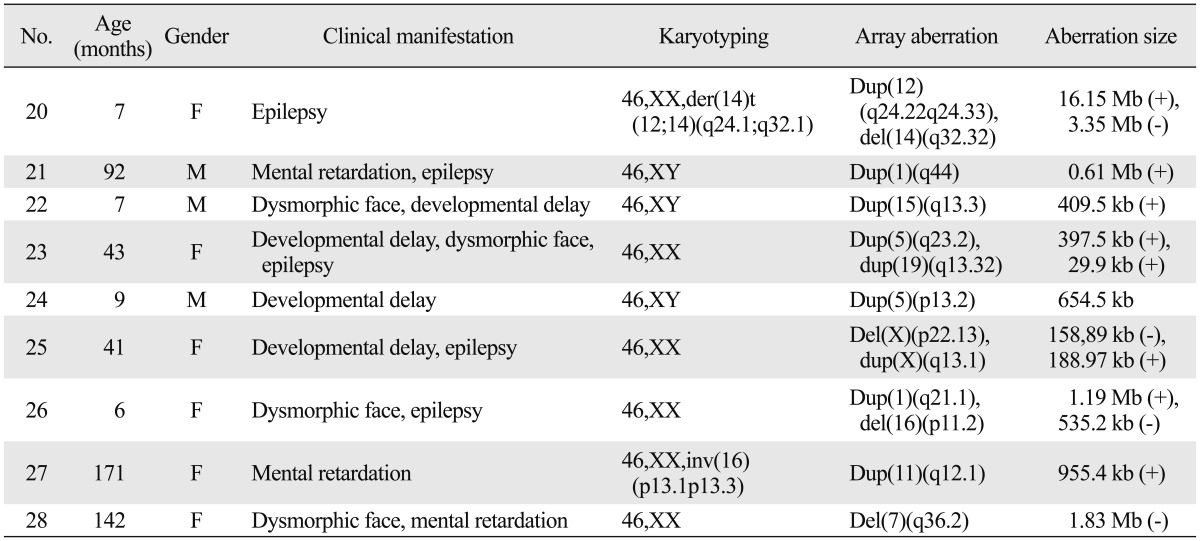
Clinically significant cases
Nineteen patients had aberrations associated with known syndromic regions (Table 1). These conditions included well-established syndromes such as Prader-Willi syndrome (PWS), Angelman syndrome, 16p13.1 microdeletion, 22q13.3 deletion syndrome and Williams-Beuren syndrome. Except for one known PWS case (No. 2), 2 additional cases were detected using the array-CGH test and were later revealed by methylation test to be one case of PWS and one case of Angelmann syndrome. Patient No. 1 with lissencephaly, with development of intractable seizures at an early age, and known Lennox-Gastaut syndrome was revealed to have 12.4 Mb deletion in the 16p13.1 region. Other study found that the 16p13.1 deletion is related with intellectual disability and epilepsy.4 Patient No. 3 with developmental delay, hypertelorism, broad nasal bridge, narrow chin and open mouth had a 3.21 Mb deletion in the 2q22.2-22.3 region. Patient No. 5 with broad nasal bridge, long philtrum, prominent ears and moderate mental retardation had a 1.39 Mb deletion in the 7q11.23 region that included the Elastine gene (OMIM 130160). Patient No. 7 with developmental delay was revealed to have a 1.0 Mb deletion in the 22q13.3 region that included the ZEB2 gene (OMIM 605802) which is known to be related to global developmental delay.5,6 Patient No. 9 with asymmetric face, protruded unilateral forehead, asymmetric craniosynostosis, underdevelopment of sinuses, and intellectual disability had a 4.37 Mb deletion that included the TWIST gene (OMIM 601622), which is consistent with diagnosis of Saethre-Chotzen syndrome. These three patients (No. 3, 5, 9) were confirmed with each syndrome by FISH. Patient No. 10 and 19 with developmental delay, dysmorphic face as hypertelorism, a short nose and down turned corners of the mouth had a 1.13 Mb and 1.34 Mb deletion each that included the EHMT1 gene (OMIM 607001) and the phenotype was consistent with Kleefstra syndrome.7 Patient No. 12 with mental retardation, behavior problems, and scoliosis had a 3.39 Mb deletion that included the RAI1 gene (OMIM 607642) and the phenotype was consistent with Smith-Magenis syndrome. Patient No. 15 had a deletion in 3p26.1p25.3 which is related with epilepsy.8 The 7 patients (patients No. 8, 12, 13, 14, 16, 17, 18) with chromosomal anomalies also had aberrations in CGH array. An additional material of chromosome 8 in patient No. 17 was newly defined in array-CGH which was not possible by conventional karyotype. On the other hand, the precise mosaicisms of patients No. 13 and 14 were not reveled in array-CGH which was documented by conventional karyotype.
Unclear clinically significant cases
Patient No. 20 had relatively large abberations which might be enough to cause disease, but did not exhibit a certain syndrome. Patient No. 21 had a 0.61 Mb microduplication in the 1q44 region, which is related to previously reported distal 1q trisomy syndrome.9 Patient No. 22 had a 409.5 kb duplication in 15q13.3(29,816,893-30,226,405), a region that includes part of the CHRNA7 gene (OMIM 118511). Duplication of this gene was previously reported to accompany defects in cognitive function.10 Patient No. 23 had a 397.5 kb duplication in 5q23.2(125,723,529-126,121,061), a region that includes the ALDH7A1 gene (OMIM 107323), which is related to epilepsy and some cases of developmental delay.11 Patient No. 24 had a 654.5 kb duplication in 5p13.2(37,082,186-37,736,722), which is involved in a part of 15p13 duplication syndrome or Cornelia de Lange syndrome.12 Patient No. 25 had a 158.89 kb deletion in Xp22.13 (18,500,733-18,659,624) and a 188.97 kb duplication in the Xq13.1(71,661,331-71,850,300) region. The deletion in Xp22.13 included CDKL5 gene, which is well known as a causative gene with variant Rett syndrome with early onset seizure.13 Patient No. 26 clinically presented with arched eyebrows, deeply set eyes, thin lips, micrognathia and epilepsy, and had a 1.19 Mb duplication in 1q21.1(144,998, 070-146,193,043), which is related to mental retardation, dysmorphic face, and epilepsy.14,15 She had another 535.2 kb deletion in the 16p11.2(29,564,890-30,100,123) region, which was reported to be related to developmental delay, seizure, and dysmorphic face such as broad forehead, micrognathia, hypertelorism, and a flat midface.16 Patient No. 27 had a 955.4 kb duplication in the 11q12.1(55,657,058-56,612,480) region, which was associated with mental retardation in a previous report.17 No. 28 had lesions of her phenotype correlated to locus aberration in 7q36.3 referenced with Decipher. Unclear significant clinical cases were not available in the parents analysis in this study.
DISCUSSION
Patients with developmental delay or mental retardation account for 2-3% of the entire population. Chromosomal anomalies found by the G-band karyotype test are responsible for 10-15% of these patients,18,19 and the causes can be additionally identified by the FISH test in 2-5% of previously diagnostically unexplained patients.19-22 However, causes are still unknown in more than half of the patients (60-80%). Over 40% of all genes are expressed in the human brain, and CNVs might contribute significantly to neurologic problems such as cognitive, behavioral, and psychological variations.23 A recent review by Hochstenbach, et al.24 showed additional 13.6% detection by array-CGH, which was between 9-50%. In the present study, the detection rate of array-CGH was 24.4% for 78 cases that were normal in G-band karyotyping, and highly significant 12 cases comprised 15.4% of 78 cases. This detection rate was slightly higher than that of previous studies, which is considered to be attributable to selection bias for the following two reasons: 1) a patient with known PWS and severe patients were included, and 2) array-CGH was used as a last resort after all other diagnostic tests, due to its high cost. Epilepsy has a genetically complex disease entity affecting up to 3% of the population. Previous studies had proven chromosomal abnormalities in epilepsy, but the use and diagnostic yield of array-CGH are not well known.
The ability to identify and interpret factors that are highly likely to be associated with disorders by comparing the accumulated information variation with a relevant database are important in clinical practice. Benign CNVs that are normal in phenotype but have numerical diversity have occasionally been reported. These benign CNVs account for about 12% of the entire genome,25 and they are diverse in size, ranging from 1 kb to several Mb, thus, making it difficult to differentiate benign CNVs from pathogenic CNVs. In the present study, the size of 500 kb was used as a relative criterion to distinguish CNVs, as in previous studies.26-30 Generally, if CNVs were relatively small and found in normal parents, they are interpreted as benign CNVs. Primary CNVs are interpreted to be very likely to undergo morbid variations. However, even if the same CNV is found in the normal parent, gene penetrance or variable expressivity cannot be ruled out. When deletion and duplication are found simultaneously in a patient, the possibility of balanced translocation or inversion of chromosome in the parents should be considered. In this study, the patients No. 3 and 18 were suggestive of balanced translocation in their parents with normal phenotype, and if a patient had CNVs that contained a large number of genes and were previously reported to be pathogenic, the condition was interpreted to be clinically meaningful even if the CNVs were smaller than 500 kb. Since numerous CNVs can be detected in one person and the phenotypes can be diverse even for an anomaly of the same loci,31 the quality of the test and experience are very important. If it is difficult to judge the result of CNVs, other methods such as FISH of specific locus, real-time PCR, multiplex ligation dependent probe amplification and function tests are needed for verification. And parents analysis are also important to correlate genomic imbalances with the phenotypes. In the present study, however, aforementioned tests were performed only in some cases because of the limitations of cost and the consent of parents or guardians.
A normal array does not mean that most of the genetic disorders are normal. This is because there are balanced chromosome rearrangements, mosaicism of less than 20-30%, severe ploidy, single and multiple base polymorphisms, numerical increase or decrease, and tribasic repeat sequence that cannot be detected by array-CGH. In this study, the mosaicisms of patient No. 13 and 14 were detected in karyotype, but not in array-CGH. Also, other abnormalities as well as uniparental disomy, loss of heterozygosity, or a 2 : 1 allele ratio which can be shown in single nucleotide polymorphism (SNP) array could not be detected in the present study. Furthermore, there are many other genetic factors of general diseases that include SNP, multi-nucleotide polymorphism, insertion and deletion of genes, morphological variation (inversion and translocation), rare variation, and gene interaction etc. It is reported that most of ring chromosome 14 patients have seizures, which are thought to be contributed by the terminal deletion of chromosome 14 (q32.31-qter).32 However, seizures were not found in patients with the same breakpoint linear deletion of 14q compared with ring chromosomes.33 This may be due to the control of 14q genes by the adjacent 14q telomere region. This is a good case which shows that gene interaction may induce the phenotype, not the deletion.
In this study, we were able to take more precise genetic information by array-CGH for microdeletion and microduplication in patients with developmental delay and/or idiopathic mental retardation and/or dysmorphic faces and/or epilepsy even in the normal G-band karyotype. The study revealed aberrations in 19 of 78 normal karyotype patients, yielding a 24.4% detection rate. These findings would be helpful in early diagnosis, family genetic counseling, and further development of new drugs and gene therapy. Clinical application of array-CGH still has some limitations in its obscure interpretation of CNVs, cost, and no specific treatment for most genetic diseases. However, we expect it to provide further information on species specificity, identification of the cause of idiopathic pediatric neurologic disease, and further development of gene therapy.
ACKNOWLEDGEMENTS
This work was supported by a grant from the Department of Pediatrics alumni fund, Korea University College of Medicine.
Footnotes
The authors have no financial conflicts of interest.
References
- 1.Slater HR, Bailey DK, Ren H, Cao M, Bell K, Nasioulas S, et al. High-resolution identification of chromosomal abnormalities using oligonucleotide arrays containing 116,204 SNPs. Am J Hum Genet. 2005;77:709–726. doi: 10.1086/497343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Allanson JE, Cunniff C, Hoyme HE, McGaughran J, Muenke M, Neri G. Elements of morphology: standard terminology for the head and face. Am J Med Genet A. 2009;149A:6–28. doi: 10.1002/ajmg.a.32612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hall BD, Graham JM, Jr, Cassidy SB, Opitz JM. Elements of morphology: standard terminology for the periorbital region. Am J Med Genet A. 2009;149A:29–39. doi: 10.1002/ajmg.a.32597. [DOI] [PubMed] [Google Scholar]
- 4.de Kovel CG, Trucks H, Helbig I, Mefford HC, Baker C, Leu C, et al. Recurrent microdeletions at 15q11.2 and 16p13.11 predispose to idiopathic generalized epilepsies. Brain. 2010;133(Pt 1):23–32. doi: 10.1093/brain/awp262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Koolen DA, Reardon W, Rosser EM, Lacombe D, Hurst JA, Law CJ, et al. Molecular characterisation of patients with subtelomeric 22q abnormalities using chromosome specific array-based comparative genomic hybridisation. Eur J Hum Genet. 2005;13:1019–1024. doi: 10.1038/sj.ejhg.5201456. [DOI] [PubMed] [Google Scholar]
- 6.Phelan MC. Deletion 22q13.3 syndrome. Orphanet J Rare Dis. 2008;3:14. doi: 10.1186/1750-1172-3-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kleefstra T, van Zelst-Stams WA, Nillesen WM, Cormier-Daire V, Houge G, Foulds N, et al. Further clinical and molecular delineation of the 9q subtelomeric deletion syndrome supports a major contribution of EHMT1 haploinsufficiency to the core phenotype. J Med Genet. 2009;46:598–606. doi: 10.1136/jmg.2008.062950. [DOI] [PubMed] [Google Scholar]
- 8.Malmgren H, Sahlén S, Wide K, Lundvall M, Blennow E. Distal 3p deletion syndrome: detailed molecular cytogenetic and clinical characterization of three small distal deletions and review. Am J Med Genet A. 2007;143A:2143–2149. doi: 10.1002/ajmg.a.31902. [DOI] [PubMed] [Google Scholar]
- 9.Lenzini E, Ballarati L, Drigo P, Carrozzi M, Gambel-Benussi D, Giardino D, et al. 1q44-qter trisomy: clinical report and review of the literature. Genet Test Mol Biomarkers. 2009;13:79–86. doi: 10.1089/gtmb.2008.0075. [DOI] [PubMed] [Google Scholar]
- 10.Wisniewski L, Hassold T, Heffelfinger J, Higgins JV. Cytogenetic and clinical studies in five cases of inv dup(15) Hum Genet. 1979;50:259–270. doi: 10.1007/BF00399391. [DOI] [PubMed] [Google Scholar]
- 11.Scharer G, Brocker C, Vasiliou V, Creadon-Swindell G, Gallagher RC, Spector E, et al. The genotypic and phenotypic spectrum of pyridoxine-dependent epilepsy due to mutations in ALDH7A1. J Inherit Metab Dis. 2010;33:571–581. doi: 10.1007/s10545-010-9187-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Yan J, Zhang F, Brundage E, Scheuerle A, Lanpher B, Erickson RP, et al. Genomic duplication resulting in increased copy number of genes encoding the sister chromatid cohesion complex conveys clinical consequences distinct from Cornelia de Lange. J Med Genet. 2009;46:626–634. doi: 10.1136/jmg.2008.062471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Mari F, Azimonti S, Bertani I, Bolognese F, Colombo E, Caselli R, et al. CDKL5 belongs to the same molecular pathway of MeCP2 and it is responsible for the early-onset seizure variant of Rett syndrome. Hum Mol Genet. 2005;14:1935–1946. doi: 10.1093/hmg/ddi198. [DOI] [PubMed] [Google Scholar]
- 14.Mefford HC, Sharp AJ, Baker C, Itsara A, Jiang Z, Buysse K, et al. Recurrent rearrangements of chromosome 1q21.1 and variable pediatric phenotypes. N Engl J Med. 2008;359:1685–1699. doi: 10.1056/NEJMoa0805384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sahoo T, Theisen A, Rosenfeld JA, Lamb AN, Ravnan JB, Schultz RA, et al. Copy number variants of schizophrenia susceptibility loci are associated with a spectrum of speech and developmental delays and behavior problems. Genet Med. 2011;13:868–880. doi: 10.1097/GIM.0b013e3182217a06. [DOI] [PubMed] [Google Scholar]
- 16.Shinawi M, Liu P, Kang SH, Shen J, Belmont JW, Scott DA, et al. Recurrent reciprocal 16p11.2 rearrangements associated with global developmental delay, behavioural problems, dysmorphism, epilepsy, and abnormal head size. J Med Genet. 2010;47:332–341. doi: 10.1136/jmg.2009.073015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Abou Jamra R, Wohlfart S, Zweier M, Uebe S, Priebe L, Ekici A, et al. Homozygosity mapping in 64 Syrian consanguineous families with non-specific intellectual disability reveals 11 novel loci and high heterogeneity. Eur J Hum Genet. 2011;19:1161–1166. doi: 10.1038/ejhg.2011.98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ropers HH. Genetics of intellectual disability. Curr Opin Genet Dev. 2008;18:241–250. doi: 10.1016/j.gde.2008.07.008. [DOI] [PubMed] [Google Scholar]
- 19.Rauch A, Hoyer J, Guth S, Zweier C, Kraus C, Becker C, et al. Diagnostic yield of various genetic approaches in patients with unexplained developmental delay or mental retardation. Am J Med Genet A. 2006;140:2063–2074. doi: 10.1002/ajmg.a.31416. [DOI] [PubMed] [Google Scholar]
- 20.Ravnan JB, Tepperberg JH, Papenhausen P, Lamb AN, Hedrick J, Eash D, et al. Subtelomere FISH analysis of 11 688 cases: an evaluation of the frequency and pattern of subtelomere rearrangements in individuals with developmental disabilities. J Med Genet. 2006;43:478–489. doi: 10.1136/jmg.2005.036350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Knight SJ, Regan R, Nicod A, Horsley SW, Kearney L, Homfray T, et al. Subtle chromosomal rearrangements in children with unexplained mental retardation. Lancet. 1999;354:1676–1681. doi: 10.1016/S0140-6736(99)03070-6. [DOI] [PubMed] [Google Scholar]
- 22.Ballif BC, Sulpizio SG, Lloyd RM, Minier SL, Theisen A, Bejjani BA, et al. The clinical utility of enhanced subtelomeric coverage in array CGH. Am J Med Genet A. 2007;143A:1850–1857. doi: 10.1002/ajmg.a.31842. [DOI] [PubMed] [Google Scholar]
- 23.Stankiewicz P, Beaudet AL. Use of array CGH in the evaluation of dysmorphology, malformations, developmental delay, and idiopathic mental retardation. Curr Opin Genet Dev. 2007;17:182–192. doi: 10.1016/j.gde.2007.04.009. [DOI] [PubMed] [Google Scholar]
- 24.Hochstenbach R, Buizer-Voskamp JE, Vorstman JA, Ophoff RA. Genome arrays for the detection of copy number variations in idiopathic mental retardation, idiopathic generalized epilepsy and neuropsychiatric disorders: lessons for diagnostic workflow and research. Cytogenet Genome Res. 2011;135:174–202. doi: 10.1159/000332928. [DOI] [PubMed] [Google Scholar]
- 25.Redon R, Ishikawa S, Fitch KR, Feuk L, Perry GH, Andrews TD, et al. Global variation in copy number in the human genome. Nature. 2006;444:444–454. doi: 10.1038/nature05329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Xiang B, Zhu H, Shen Y, Miller DT, Lu K, Hu X, et al. Genome-wide oligonucleotide array comparative genomic hybridization for etiological diagnosis of mental retardation: a multicenter experience of 1499 clinical cases. J Mol Diagn. 2010;12:204–212. doi: 10.2353/jmoldx.2010.090115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wagenstaller J, Spranger S, Lorenz-Depiereux B, Kazmierczak B, Nathrath M, Wahl D, et al. Copy-number variations measured by single-nucleotide-polymorphism oligonucleotide arrays in patients with mental retardation. Am J Hum Genet. 2007;81:768–779. doi: 10.1086/521274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hoyer J, Dreweke A, Becker C, Göhring I, Thiel CT, Peippo MM, et al. Molecular karyotyping in patients with mental retardation using 100K single-nucleotide polymorphism arrays. J Med Genet. 2007;44:629–636. doi: 10.1136/jmg.2007.050914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Friedman JM, Baross A, Delaney AD, Ally A, Arbour L, Armstrong L, et al. Oligonucleotide microarray analysis of genomic imbalance in children with mental retardation. Am J Hum Genet. 2006;79:500–513. doi: 10.1086/507471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Fan YS, Jayakar P, Zhu H, Barbouth D, Sacharow S, Morales A, et al. Detection of pathogenic gene copy number variations in patients with mental retardation by genomewide oligonucleotide array comparative genomic hybridization. Hum Mutat. 2007;28:1124–1132. doi: 10.1002/humu.20581. [DOI] [PubMed] [Google Scholar]
- 31.Mosca-Boidron AL, Bouquillon S, Faivre L, Callier P, Andrieux J, Marle N, et al. What can we learn from old microdeletion syndromes using array-CGH screening? Clin Genet. 2012;82:41–47. doi: 10.1111/j.1399-0004.2011.01747.x. [DOI] [PubMed] [Google Scholar]
- 32.Morimoto M, Usuku T, Tanaka M, Otabe O, Nishimura A, Ochi M, et al. Ring chromosome 14 with localization-related epilepsy: three cases. Epilepsia. 2003;44:1245–1249. doi: 10.1046/j.1528-1157.2003.05403.x. [DOI] [PubMed] [Google Scholar]
- 33.van Karnebeek CD, Quik S, Sluijter S, Hulsbeek MM, Hoovers JM, Hennekam RC. Further delineation of the chromosome 14q terminal deletion syndrome. Am J Med Genet. 2002;110:65–72. doi: 10.1002/ajmg.10207. [DOI] [PubMed] [Google Scholar]