

Abstract
De novo assembly of RNA-Seq data allows us to study transcriptomes without the need for a genome sequence, such as in non-model organisms of ecological and evolutionary importance, cancer samples, or the microbiome. In this protocol, we describe the use of the Trinity platform for de novo transcriptome assembly from RNA-Seq data in non-model organisms. We also present Trinity’s supported companion utilities for downstream applications, including RSEM for transcript abundance estimation, R/Bioconductor packages for identifying differentially expressed transcripts across samples, and approaches to identify protein coding genes. In an included tutorial we provide a workflow for genome-independent transcriptome analysis leveraging the Trinity platform. The software, documentation and demonstrations are freely available from http://trinityrnaseq.sf.net.
Introduction
High throughput sequencing of genomes (DNA-Seq) and transcriptomes (RNA-Seq) has opened the way to study the genetic and functional information stored within any organism at an unprecedented scale and speed. For example, RNA-Seq allows in principle for the simultaneous study of transcript structure (such as alternative splicing), allelic information (e.g., SNPs), and expression with high resolution and large dynamic range1. These advances greatly facilitate functional genomics research in species for which genetic or financial resources are limited, including many ‘non-model’ organisms, which are nevertheless of substantial ecological or evolutionary importance.
While many genomic applications have traditionally relied on the availability of a high-quality genome sequence, such sequences have only been determined for a very small portion of known organisms. Furthermore, sequencing and assembling a genome is still a costly endeavor in many cases, due to genome size and repeat content. Conversely, since the transcriptome is only a fraction of the total genomic sequence, RNA-Seq data can provide a rapid and cheaper ‘fast track’, within reach of any lab, to delineating a reference transcriptome for downstream applications such as alignment, phylogenetics or marker construction. Indeed, even within a whole genome sequencing project, RNA-Seq has become an essential source of evidence for transcribed gene identification and exon structure annotation.
Realizing the full potential of RNA-Seq requires computational methods that can assemble a transcriptome even when a genome sequence is not available. There are primarily two ways to convert raw RNA-Seq data to transcript sequences: with the guidance of assembled genomic sequences or via de novo assembly2, 3. The genome-guided approach to transcriptome studies has quickly become a standard approach to RNA-Seq analysis for model organisms, and several software packages exist for this purpose4, 5. It cannot, however, be applied to organisms without a well-assembled genome, and even if one is present, the results may vary across genome assembly versions. In such cases, a de novo transcriptome assembler is required. However, the process of assembling a transcriptome violates many of the assumptions of assemblers written for genomic DNA. For example, uniform coverage and the ‘one locus – one contig’ paradigm are not valid for RNA: an accurate transcriptome assembler will produce one contig per distinct transcript (isoform) rather than per locus, and different transcripts will have different coverage, reflecting their different expression levels.
Several tools are now available for de novo assembly of RNA-Seq. Trans-ABySS 6, Velvet-Oases7, and SOAPdenovo-trans (http://soap.genomics.org.cn/SOAPdenovo-Trans.html) are all extensions of earlier developed genome assemblers. We previously described an alternative and novel method for transcriptome assembly called Trinity8. Trinity partitions RNA-Seq data into many independent de Bruijn graphs, ideally one graph per expressed gene, and uses parallel computing to reconstruct transcripts from these graphs, including alternatively spliced isoforms. Trinity can leverage strand-specific Illumina Paired-End (PE) libraries, but can also accommodate non-strand-specific and single-end (SE) read data. Trinity reconstructs transcripts accurately with a simple and intuitive interface that requires little to no parameter tuning. Several independent studies have demonstrated that Trinity is highly effective compared to alternative methods (e.g.9-11, The DREAM Project’s Alternative Splicing Challenge (http://www.the-dream-project.org/result/alternative-splicing)). Indicating Trinity’s utility, since its publication in May 2011, it has acquired an avid user base with ~200 citations from May 2011 to March 2013 (http://scholar.google.com/scholar?oi=bibs&hl=en&cites=14735674943942667509). Trinity users study a broad range of model and non-model organisms from all Kingdoms, and come from small labs and large genome projects alike (e.g., the pea aphid genome annotation v2; Fabrice Legeai, INRA and Terence Murphy, RefSeq NCBI, personal communications).
Trinity also has an active developer community, which has greatly enhanced its performance and utility (see http://trinityrnaseq.sourceforge.net). For example, while the runtime performance of the first release was not computationally efficient11, the Trinity developer community has since improved its efficiency, halving memory requirements and increasing processing speed through increased parallelization and improved algorithms (12; M. Ott, personal communication). Furthermore, Trinity was converted into a modular platform that seamlessly uses third-party tools, such as Jellyfish13 for building the initial k-mer catalog. Other third party tools integrated into Trinity have enhanced the utility of its reconstructed transcriptomes. For example, as described below, Trinity now supports tools (e.g., RSEM14, edgeR15 and DESeq 16) that take its output transcripts and test for differential expression, while accounting for both technical and biological sources of variation17-19 and correcting for multiple hypothesis testing. Given Trinity’s popularity and substantial enhancements since publication, it is important to provide detailed protocols that leverage its various features. The protocols we present below will maximize Trinity’s utility to users for studies in non-model organisms, and inform the broad developer community on areas for future enhancements.
Overview of the Trinity RNA-Seq assembler
Trinity’s assembly pipeline consists of three consecutive modules: Inchworm, Chrysalis, and Butterfly (Figure 1). We strongly encourage users to first read Trinity’s first publication8 for an extensive description of the method, which we present here more briefly.
Figure 1. Overview of Trinity assembly and analysis pipeline.

Shown are the key sequential steps in Trinity (left) and the associated compute resources (right). Trinity takes as input short reads (top left) and first uses the Inchworm module to construct contigs. This requires a single high-memory server (~1G RAM per 1M paired reads, but varies based on read complexity; top right). Chrysalis (middle left) clusters related Inchworm contigs, often generating tens to hundreds of thousands of Inchworm contig clusters, each of which is processed to a de Bruijn graph component independently and in parallel on a computing grid (bottom right). Butterfly (bottom left) then extracts all probable sequences from each graph component, which can be parallelized as well.
First, all overlapping k-mers are extracted from the RNA-Seq reads. Inchworm then examines each unique k-mer in decreasing order of abundance, and generates transcript contigs using a greedy extension based on (k-1)-mer overlaps. Inchworm often generates full-length transcripts for a dominant isoform, but reports just the unique portions of alternatively spliced transcripts. This works well for datasets largely deficient in repetitive sequences, such as transcriptomes.
Next, Chrysalis first clusters related Inchworm contigs into components, using the raw reads to group transcripts based on shared read support and paired reads links, when available. This clusters together regions that have likely originated from alternatively spliced transcripts or closely related gene families. Chrysalis then encodes the structural complexity of clustered Inchworm contigs by building a de Bruijn graph for each cluster and partitions the reads amongst the clusters. The partitioning of the Inchworm contigs and RNA-Seq reads into disjoint clusters (“components”) allows massively parallel processing of subsequent computations.
Finally, Butterfly processes the individual graphs in parallel, ultimately reporting full-length transcripts for alternatively spliced isoforms and teasing apart transcripts that correspond to paralogous genes. Butterfly traces the RNA-Seq reads through the graph, and determines connectivity based on the read sequence and on further support from any available paired end data. When connections cannot be verified by traced reads, Butterfly will split the graph into several disconnected sub-graphs and process each separately. Finally, Butterfly traverses the supported graph paths and reconstructs transcript sequences in a manner that reflects the original cDNA molecules.
We describe key issues related to Trinity’s operation in Boxes 1-4 including: requirements of the input sequence data and the optional use of in silico normalization to reduce the quantity of the input reads to be assembled and to improve assembly efficiency (Box 1); computing requirements and the availability of computing resources to users for running Trinity (Box 2); the basics of running Trinity (Box 3); and advanced operations, such as leveraging strand-specific RNA-Seq (Box 4). Additional issues relevant to evaluating de novo transcriptome assemblies, including examining the completeness of an assembly and estimating the potential impact of deeper sequencing are addressed in Supplementary Text Section S1 and in Supplementary Fig. 1 and 2.
Box 1. Input sequence data requirements for assembly.
Users are required to supply short-read data in the form of either FASTQ or FASTA formats. These reads may be either paired-end (PE) or single-end (SE); paired-end sequence data are preferable, particularly when coupled with a strand-specific sequencing protocol (below). If multiple sequencing runs were conducted for a single experiment, these reads may be concatenated into a single read file for single-end sequencing, or into two files (e.g., merging all ‘left’ and all ‘right’ reads into single ‘left.fq’ and ‘right.fq’ files, respectively) in the case of paired-end sequencing. Similarly, if multiple biological or technical replicates have been sequenced, these data can be concatenated into individual files. Trinity may be used with data of any read length commonly produced by next-generation sequencers, but most of our experience stems from the use of either 76 b or 101 b Illumina reads.
For paired reads, Trinity must identify those reads that correspond to opposite ends of a sequenced molecule. Trinity expects reads in either FASTQ or FASTA format to have read names (accessions) that include a /1 or /2 suffix to indicate the left or right end of the sequenced fragment. For example:
(first entry of the ‘left.fq’ file)
@61DFRAAXX100204:1:100:10494:3070/1 ACTGCATCCTGGAAAGAATCAATGGTGGCCGGAAAGTGTTTTTCAAATACAAGAGTGACAAT GTGCCCTGTTGTTT + ACCCCCCCCCCCCCCCCCCCCCCCCCCCCCBC?CCCCCCCCC@@CACCCCCACCCCCCCCCC CCCCCCCCCCCCCC
(first entry of the ‘right.fq’ file)
@61DFRAAXX100204:1:100:10494:3070/2 CTCAAATGGTTAATTCTCAGGCTGCAAATATTCGTTCAGGATGGAAGAACATTTTCTCAGTA TTCCATCTAGCTGC + C<CCCCCCCACCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCBCCCCCCCCCCCCC CCCACCCCCACCC=
If the Casava 1.8 format for FASTQ is used (below), Trinity will reconstruct the read name from:
@HWI-ST896:156:D0JFYACXX:5:1101:1652:2132 1:N:0:GATCAG
to
HWI-ST896:156:D0JFYACXX:5:1101:1652:2132/1
Preprocessing sequence data
While pre-assembly sequence quality control steps are not required, performing a few easy steps can improve performance. First, if the reads include barcodes used for multiplexing on the sequencing instrument, all barcodes must be removed before running Trinity, for example, by running Trimmomatic41 or cutadapt42.
Second, removing reads (or regions of reads) with likely sequencing errors may reduce the complexity of the resulting de Bruijn graph, and hence improve RAM usage and program runtime. Specifically, the program Trimmomatic41 can successfully remove terminal nucleotides that are less than a user-supplied minimum quality threshold (e.g., Q15).
Third, if more than 200 million PE sequences are to be assembled, the user may consider performing an in silico normalization of the sequencing reads. Deep RNA-Seq produces vast numbers of reads for transcripts that are already well represented at lower sequencing depths in addition to providing increased sensitivity for the detection of rarer transcripts36, 43. Normalization improves run-time by reducing the total number of reads, while largely maintaining transcriptome complexity and capability for full-length transcript reconstruction44. Trinity includes an in silico read normalization utility inspired by the algorithm described for Diginorm44, except that here each RNA-Seq fragment (single read or pair of reads) is probabilistically selected based on its median k-mer coverage value (C) and the targeted maximum coverage value (M), specifically with probability min(1, M/C) (Supplementary Text Section S4). Analyzing RNA-Seq data sets from fission yeast8, 45 and mouse8, we have found that normalization to as low as 30X k-mer (k=25) coverage (23% to 31% of the reads) results in full-length reconstruction to an extent approaching that based on the entire read set, and far exceeds the full-length reconstruction performance when simply subsampling the same number of reads from the total data set (Fig. 4). Although in silico normalization can better enable
Trinity assembly of large RNA-Seq data sets, and is clearly better than the alternative of subsampling reads, the sensitivity for full-length transcript reconstruction may be affected, such as leading to the 6% decrease in full-length transcript reconstruction observed for the 30X max. coverage normalization obtained based on our mouse RNA-Seq data (Fig. 4). However, the relative impact on the percent of alternatively spliced reference transcript isoforms detected remains largely unchanged (data not shown).
Box 4. Advanced Trinity Operations.
Leveraging strand-specific RNA-Seq data
Trinity’s preferred input is strand-specific RNA-Seq data, which allows us to distinguish between sense and antisense transcripts and minimizes erroneous fusions between neighboring transcriptional units that are encoded on opposite strands. This is particularly useful when applied to dense microbial genomes, where overlapping transcriptional units are common. Several methods are available for generating strand-specific RNA-Seq46, 47.
When given strand-specific data, Trinity first converts all the input reads to the transcribed strand orientation, reverse-complementing if required. Users need to indicate strand-specificity to Trinity.pl via the ‘--SS_lib_type’ parameter, with ‘F’ or ‘R’ values for single-end reads to indicate reads originating from the transcribed or opposite-strand, respectively. Similarly, ‘FR’ or ‘RF’ values are used for paired-end reads to reflect both ends (Fig. 6). For example, the dUTP-based strand-specific sequencing generates ‘RF’ paired-end sequences, where the right read (name ending with /2) corresponds to the transcribed strand and the left read (name ending with /1) exists in the opposite-strand. A corresponding Trinity assembly command would be constructed as:
For single-end reads
% $TRINITY_HOME/Trinity.pl --seqType fq --single single.fq \
--JM 20G --SS_lib_type F
For paired-end reads
% $TRINITY_HOME/Trinity.pl --seqType fq --left left.fq \
--right right.fq --JM 20G --SS_lib_type RF
During the FASTA conversion, Trinity reverse-complements the read sequences that are specified to exist in the ‘R’ orientation.
Using strand-specific data can cause a small increase in running time, due to the increased k-mer complexity of the data. Specifically, in strand-specific data, the forward and reverse-complemented k-mers are stored individually, whereas in non-strand-specific data, there is no distinction between a k-mer in either orientation and it is stored in a single canonical representation. However, this small increase yields several benefits, including a small overall improvement in transcript reconstruction compared to non-strand-specific data (Fig. 7), a substantial reduction in the number of falsely fused transcripts in species with compact genomes such as fission yeast or Drosophila melanogaster (Fig. 7), and the ability to distinguish sense and antisense transcripts, thus revealing otherwise concealed mechanisms for transcriptional regulation8, 45.
Mitigating falsely fused transcripts
To further resolve erroneous fusion of overlapping transcripts from close neighboring genes, Trinity can leverage mate-pair information (with the ‘--jaccard_clip’ parameter) to identify and dissect regions within assembled contigs, consistent with overlapping yet distinct transcripts (Fig. 8). In our experiments, roughly half of fused transcripts in fission yeast and D. melanogaster are resolved using this method, albeit at a cost of doubling to tripling of the total runtime (Fig. 7). Since this operation has little effect on the quality of transcriptomes reconstructed from gene-sparse genomes, such as many vertebrates (e.g., mouse) (Fig. 7), users are encouraged to use ‘--jaccard_clip’ in the case of transcriptome assemblies for organisms expected to have compact genomes, such as microbial eukaryotes, where erroneous fusions are more likely to be generated.
Additional parameters and approaches to consider
Several other options can be tuned to further improve accuracy and reduce runtime. First, while Trinity handles substitution-type sequencing errors well, it cannot detect adaptors or contaminants that survive the poly-dT enrichment (poly-A capture) protocols. In Trinity’s assemblies, it is not uncommon to find sequences from other species (such as viruses and bacteria) or native non-polyadenylated transcripts that are highly abundant (such as rRNA). While some such sequences can yield important insights (e.g., viral genomes in tumors48), in other cases users will opt to pre-filter them. In particular, read pre-processing such as in silico normalization of read quantities, quality trimming, or read filtering can reduce graph complexity and resulting runtimes (Box 1).
Second, when sequencing very deeply (e.g., in order to identify rare transcripts), the larger number of observed sequencing errors contributes to increased graph complexity and longer runtimes. In most cases, setting the minimum k-mer coverage requirement to 2 instead of the default of 1 via the ‘--min_kmer_cov 2’ parameter setting will effectively handle such cases, eliminating the singly occurring k-mers that are heavily enriched in sequencing errors, and vastly decreasing the complexity of the graph. In cases of very deep sequencing (beyond several hundred million PE reads), users can (i) normalize their data (Box 1) and/or (ii) consider performing a Trinity assembly with ‘--min_kmer_cov 2’; and then (iii) align the original read dataset back to the final Trinity transcripts for abundance estimation.
Third, Trinity’s final phase, Butterfly, operates in parallel on graphs from individual clusters and, by default, uses identical parameters for each cluster. As most clusters can be processed by Butterfly quite rapidly (seconds to a few minutes), users who have domain-specific knowledge can explore a number of parameter settings relating to graph traversal to better understand how well assembled is a particular cluster of transcripts. Such parameter adjustments may include redefining the read overlap requirements for path extension, or tuning the minimum edge weight thresholds at branchpoints in the graph (details are provided in the Supplementary Text Section S5 and Supplementary Figures 5-9).
Box 2. Computing requirements.
The Trinity software is designed for Unix-type operating systems (primarily Linux), provides a command-line interface, and is best run on a high memory multi-core computer or in a high-performance computing environment. In general, we recommend having approximately 1 GB of RAM per 1M PE reads. A typical configuration is a multi-core server with 256 GB to 1 TB of RAM, and such systems have become more affordable in the recent years (~ $15,000 to $40,000 – significantly less expensive than many high-performance instruments used in molecular biology, and definitely within reach of a departmental core facility). Smaller datasets can be executed in computing environments with reduced memory resources (e.g., see Tutorial which can be completed on a laptop with 8 GB of RAM). For those that lack the required computing resources, such resources are freely accessible to eligible researchers by the Data Intensive Academic Grid (DIAG, http://diagcomputing.org), and services are available to researchers in the US (and their international collaborators) through the eXtreme Science and Engineering Discovery Environment (XSEDE), on systems such as “Blacklight” at the Pittsburgh Supercomputing Center (http://www.psc.edu/index.php/trinity).
As Trinity is executed from command-line, users should have a basic familiarity with operating in a Unix environment. Each of Trinity’s three core modules has a different characteristic runtime, memory usage, and parallelization (Fig. 1). Although the entire Trinity compute pipeline can be executed on a single high-memory machine, the later stages, including Chrysalis’ ‘QuantifyGraph’ section and the Butterfly computations, benefit from access to a compute farm, where they can be massively parallelized. To this end, Trinity’s final massively parallel section integrates the ability to submit to Load Sharing Facility (LSF), a grid scheduling system. This is accomplished through the use of the command line parameter ‘--grid_computing_module’ identifying a user-defined module for submitting commands to the grid for execution. A submission script (‘trinity_pbs.sh’) is also provided for using Trinity with the Portable Batch System (PBS) Torque and PBSpro cluster job schedulers.
Box 3. Basic Trinity operation.
The assembly pipeline for a typical Trinity assembly is executed from the command line via a single PERL script called ‘Trinity.pl’, with options describing the sequencing protocol and the names of the files containing RNA-Seq reads. For the Unix-style commands shown throughout, we use the initial character ‘%’ as a command prompt, and the $TRINITY_HOME variable corresponds to the location of the Trinity software installation directory. Commands that span multiple lines are separated by a trailing ‘\’ character.
If the RNA-Seq protocol used a non strand-specific method, then Trinity.pl is executed as follows:
For single-end reads:
% $TRINITY_HOME/Trinity.pl --seqType fq \
--single single.fq --JM 20G
For paired-end reads:
% $TRINITY_HOME/Trinity.pl --seqType fq \
--left left.fq \
--right right.fq --JM 20G
The --seqType parameter indicates the file format for the sequenced reads, which can be in FASTQ (‘fq’) or FASTA (‘fa’) format (See Box 1 and the Tutorial for more detailed descriptions of input requirements). Trinity will also detect which flavor of FASTQ is used (Sanger, Illumina 1.3, CASAVA 1.8+) and convert them to FASTA files; Trinity does not presently use the base quality scores provided in the FASTQ file format. If the data are paired, the new FASTA file will have /1 and /2 header information to indicate ‘pairedness’. When other formats of FASTQ or if paired-end FASTA files are provided to Trinity, users must ensure that this pairedness is represented using the /1 and /2 read name suffix notation.
Users can include additional parameter settings (see below) to tune any of the three assembly steps according to the characteristics of the dataset, but Trinity usually performs well with the default parameters. Further, some settings, such as --JM 20G above (20G of memory to be allocated to Jellyfish for building the initial k-mer catalog), relate to the hardware being used and Box 2 describes how users can select optimal settings for different hardware configurations.
Trinity output
The final output from running Trinity is a single FASTA-formatted file containing the reconstructed transcript sequences, found as ‘trinity_out_dir/Trinity.fasta’. To understand the output format, recall that Chrysalis divides sequences into graph components based on subsequences shared between them, and Butterfly further refines the sequence’s classification by partitioning components into sets of transcript contigs based on read support within the graph (Fig. 1).
An example of a pair of entries found in the Trinity FASTA-formatted output is (Fig. 5):
>comp0_c0_seq1 len=5528 path=[3647:0-3646 129:3647-3775 1752:3776-5527] AATTGAATCCCTTTTTGTATTGAAAAAGTTGAAATGAAAGACATATACAGAT TGAATGTG…TCCTCTGATACACAGCCTCGCAGGGTTCATTTCAAGCCGTGGG GCTGCGCCACGGGTGCTAAGTCAACTGCATTCGATGCGGCTTTTAAACCCCC AGGGGACACCTCGGCCAGCTGTTTGCCTGCAGTA…TTGTGTTTCTTCAACAG TTTATCAGCTTGCTGAATTGCCATTTTATTATTTCCATTATCAAGATAATCG TAAATGGGCCGGAGGCGCCGGTCGTTAGGGTCCTGCACATGGCCCCGCGTCG CCATGATGACAAGCGCAGAACCTCAGT >comp0_c0_seq2 len=5399 path=[3647:0-3646 1752:3647-5398] AATTGAATCCCTTTTTGTATTGAAAAAGTTGAAATGAAAGACATATACAGAT TGAATG…TGGTGATTGCAAAATATAATGCAATTTCGAACAATTAAAATTATG AAAATATAC…TTGTGTTTCTTCAACAGTTTATCAGCTTGCTGAATTGCCATT TTATTATTTCCATTATCAAGATAATCGTAAATGGGCCGGAGGCGCCGGTCGT TAGGGTCCTGCACATGGCCCCGCGTCGCCATGATGACAAGCGCAGAACCTCA GT
The FASTA header describes how the transcript was structurally represented as reconstructed by Butterfly. For example, in the header of the first reported sequence, the accession value ‘comp0_c0_seq1’ corresponds to i) Chrysalis component ‘comp0’, ii) Butterfly disconnected subgraph ‘c0’, iii) Butterfly-reconstructed sequence ‘seq1’, and iv) having a length of 5,528 bases. The path of the sequence traversed by Butterfly through nodes in the sequence graph (Fig. 5a, blue, red and green nodes) is provided in the header as “path=[3647:0-3646 129:3647-3775 1752:3776-5527]”, listing the identifiers of the ordered nodes (3647, 129, 1752) and the ranges within the reconstructed transcript sequence that correspond to each respective node.
The second sequence above is a second transcript derived from the same component, identified by the accession ‘comp0_c0_seq2’ that corresponds to a sequence ‘seq2’ output from the same Chrysalis component and Butterfly subgraph ‘comp0_c0’. In this case, the path traverses only two of the nodes 3647 and 1752; Fig. 5a, blue and green nodes), through the edge connecting them directly. Thus, ‘seq1’ differs from ‘seq2’ only by the addition of the sequence in the internal node ‘129’ (highlighted subsequence; Fig. 5a, red node). Such variations can result from alternative splicing. Here, for example, comparison to the reference mouse genome shows that the internally unique sequence in ‘seq1’ corresponds to a cassette exon that is skipped in ‘seq2’ (Fig. 5c, where Trinity ‘seq1’ and ‘seq2’ are shown as ‘Isoform B’ and ‘Isoform A’, respectively). Validating such transcripts can be done by comparison to an annotated reference genome (even of a related species) or experimentally. In some cases, alternative paths reflect the shared and distinct portions in paralogous genes or alternative alleles. Mate pairing information and sufficiently long reads allow Trinity to resolve phased variations and correctly reconstruct the individual isoforms or paralogous transcripts from the more complex graphs8.
Transcriptome analysis package for non-model organisms
Generating a de novo RNA-Seq assembly is only the first step towards transcriptome analysis. Common goals for studying transcriptomes in both model and non-model organisms include identifying transcripts, characterizing transcript structural complexity and coding content, and understanding which genes and isoforms are expressed in different samples (tissues, environmental conditions, etc.). Trinity supports this by leveraging additional popular software already likely to be installed in a bioinformatics environment, by incorporating additional Open Source software as plug-in components that are directly included in the Trinity software suite, and by providing easy-to-use scripts that aim to provide a familiar and friendly command-line interface to otherwise complex analysis modules. Results are often provided as tab-delimited files that users can import into their favorite spreadsheet programs, and visualizations are generated in PDF format. Below, we describe the details of the individual protocol steps and identify the currently supported software utilities.
Comparing transcriptomes across samples
In many cases, a user will wish to compare the type or level of transcripts between samples, for example for differentially expressed genes. There are two possible routes in this case. One option is to assemble reads corresponding to each of the sample types separately, and then compare the results from each of the assemblies. This, however, is complicated by the need to match the ‘same’ transcripts derived from the independent assemblies. A more straightforward and recommended alternative is to first combine all reads across all samples and biological replicates into a single RNA-Seq data set, assemble the reads to generate a single ‘reference’ Trinity assembly (Fig. 2), and then quantify the level of each of these transcripts in each sample, by aligning each sample’s (not normalized) reads to the reference transcriptome assembly and counting the number of reads that align to each transcript (over-simplified here, detailed below). Finally, statistical tests are applied to compare the counts of reads observed for each transcript across the different samples, and those transcripts observed to have significantly different representation by reads across samples are reported. Further analysis of the differentially expressed transcripts can reveal patterns of gene expression and yield insights into relationships among the investigated samples.
Figure 2. De novo transcriptome assembly and analysis workflow.

Reads from multiple samples (e.g., different tissues, top) are combined into a single data set. Reads may be optionally normalized to reduce read counts while retaining read diversity and sample complexity. The combined read set is assembled by Trinity to generate a ‘reference’ de novo transcriptome assembly (right). Protein coding regions can be extracted from the reference assembly using TransDecoder and further characterized according to likely functions based on sequence homology or domain content. Separately, sample-specific expression analysis is performed by aligning the original sample reads to the reference transcriptome assembly on a per sample basis, followed by abundance estimation using RSEM. Differentially expressed transcripts are identified by applying Bioconductor software, such as edgeR, to a matrix containing the RSEM abundance estimates (number of RNA-Seq fragments mapped to each transcript from each sample). Differentially expressed transcripts can then be further grouped according to their expression patterns.
Transcript abundance estimation
Transcript quantification is a prerequisite to many downstream investigations. Several metrics have been proposed for measuring transcript abundance levels based on RNA-Seq data, normalizing for depth of sequencing and the length of transcripts. These metrics include Reads Per Kilobase of target transcript length per Million reads mapped (RPKM20) for single-end sequences, and an analogous computation based on counting whole Fragments (FPKM21) for paired-end RNA-Seq data.
To calculate the number of RNA-Seq reads or fragments that were derived from transcripts, the reads must first be aligned to the transcripts. When working with a reference genome and an annotated transcriptome, reads are usually aligned to one or both4. In a de novo assembly setting, the reads are re-aligned to the assembled transcripts. However, alternatively spliced isoforms and recently duplicated genes may share subsequences longer than the length of a read (or read pair), and these reads will map equally well to multiple targets. Several methods4, 14, 22 were recently developed to estimate how to correctly ‘allocate’ such reads to transcripts in a way that best approximates the transcripts’ true expression levels. Among these is the RSEM (RNA-Seq by Expectation-Maximization) software14, which uses an iterative process to fractionally assign reads to each transcript based on the probabilities of the reads being derived from each transcript (Fig. 3), taking into account positional biases created by RNA-Seq library-generating protocols.
Figure 3. Abundance estimation via Expectation Maximization by RSEM.
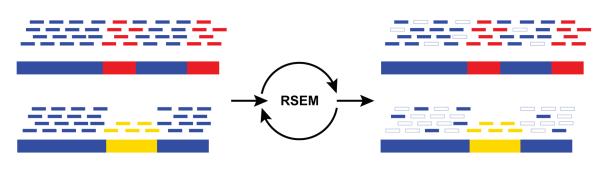
Shown is an illustrative example of abundance estimation for two transcripts with shared (blue) and unique (red, yellow) sequences. To estimate transcript abundances, RNA-Seq reads (short bars) are first aligned to the transcript sequences (long bars, bottom). Unique regions of isoforms will capture uniquely-mapping RNA-Seq reads (red and yellow short bars), and shared sequences between isoforms will capture multiply-mapping reads (blue short bars). An Expectation Maximization algorithm, implemented in the RSEM software, estimates the most likely relative abundances of the transcripts and then fractionally assigns reads to the isoforms based on these abundances. The assignments of reads to isoforms resulting from iterations of expectation maximization are illustrated as filled short bars (right), and those assignments eliminated are shown as hollow. Note that assignments of multiply-mapped reads are in fact performed fractionally according to a maximum likelihood estimate. Thus, in this example, a higher fraction of each read is assigned to the more highly expressed top isoform than to the bottom isoform.
RSEM comes bundled with the Trinity software distribution. The RSEM protocol currently requires gap-free alignments of RNA-Seq reads to Trinity-reconstructed transcripts, such as alignments generated by the Bowtie software23. Given the Trinity-assembled transcripts and the RNA-Seq reads generated from a sample, RSEM will directly execute Bowtie to align the reads to the Trinity transcripts and then compute transcript abundance, estimating the number of RNASeq fragments corresponding to each Trinity transcript, including normalized expression values as FPKM. In addition to estimating the expression levels of individual transcripts, RSEM computes ‘gene-level’ estimates using the Trinity component as a proxy for the gene. In order to compare expression levels of different transcripts or genes across samples, a Trinity-included script invokes edgeR to perform an additional TMM (Trimmed Mean of M-values) scaling normalization that aims to account for differences in total cellular RNA production across all samples24, 25.
Both full-length and partially reconstructed Trinity transcripts can be useful for estimating gene expression, as compared to expression levels estimated using a high quality reference genome-based transcript annotation. However, the more completely reconstructed transcripts tend to be more highly correlated with the expression levels estimated for reference transcripts (Supplementary Text Section S2, Supplementary Fig. 3).
Analysis of differentially expressed transcripts
To estimate differential gene expression between two types of samples, we would ideally obtain at least 3 biological replicates of each sample. The replicates allow us to test whether the observed differences in expression are significantly different from expected biological variation under the null hypothesis that transcripts are not differentially expressed. In the absence of biological replicates, it is still possible to identify differentially expressed transcripts by using statistical models of expected variation, such as under the Poisson or negative binomial distribution. The Poisson distribution well models variation expected between technical replicates26, whereas the negative binomial distribution better accounts for the increased variation observed between biological replicates, and is the favored model for identifying differentially expressed transcripts by leading software tools15, 16.
Trinity transcriptome assemblies can serve as a useful substrate for evaluating changes in gene expression between samples, with results largely consistent with studies based on reference transcriptomes (Supplementary Text Section S2, Supplementary Fig. 4). To this end, we rely on tools from the Bioconductor project for identifying differentially expressed transcripts, including edgeR15 and DESeq16. Bioconductor requires the R software for statistical computing, which includes a command-line environment and programming language syntax that can pose a significant barrier to new users or those lacking more extensive bioinformatics training. To facilitate use of Bioconductor tools for transcriptome studies, the Trinity software suite includes easy-to-use scripts that leverage the R software to identify differentially expressed transcripts, generate tab-delimited output files listing differentially expressed transcripts including fold-change and statistical significance values, and generate visualizations such as MA-plots, volcano-plots, correlation plots, and clustered heatmaps in PDF format (see Tutorial).
Protein prediction and functional annotation of Trinity transcripts
Most transcripts assembled from eukaryotic RNA-Seq data derived from polyadenylated RNA are expected to code for proteins. A sequence homology search, such as by BLASTX, against sequences from a well-annotated, phylogenetically-related species is the most practical way to identify likely coding transcripts and to predict their functions. Unfortunately, such well-annotated ‘relative’ species are often not available for newly targeted transcriptomes. In such cases, using the latest non-redundant protein database (eg. NCBI’s ‘nr’ (ftp://ftp.ncbi.nlm.nih.gov/blast/db/FASTA/nr.gz) or Uniprot (ftp://ftp.uniprot.org/pub/databases/uniprot/current_release/knowledgebase/complete/uniprot_tre mbl.fasta.gz)) is an appropriate alternative.
Newly targeted transcriptomes may also encode proteins that are insufficiently represented by detectable homologies to known proteins. To capture those coding regions requires methods that predict coding regions based on metrics tied to sequence composition. One such utility is TransDecoder (Supplemental Text Section S3), which we developed and include with Trinity to assist in the identification of potential coding regions within reconstructed transcripts. When run on the Trinity-reconstructed transcripts, TransDecoder identifies candidate protein-coding regions based on nucleotide composition, open reading frame (ORF) length, and (optional) Pfam domain content.
Dynamic graphical user interfaces, such as IGV27 or GenomeView28, are especially useful in studying transcript reconstructions. Although originally designed for genomes, these can be readily used for viewing read alignments to transcripts, putative coding regions, regions of protein sequence homology, and short read alignments with pair links.
Limitations of the Trinity approach to transcriptome analysis
Although Trinity is highly effective for reconstructing transcripts and alternatively spliced isoforms, in the absence of a reference genome, it can be difficult if not impossible to fully understand the structural basis for the observed transcript variations, such as whether they are due to one or more skipped exons, alternative donor or acceptor spliced sites, or retained introns. We are currently exploring experimental and computational strategies to better enable such insights from RNA-Seq data in the absence of reference genomes.
The RNA-Seq reads and pairing information allow Trinity to resolve isoforms and paralogs8, but its success depends on finding sequence variations that can be properly phased by individual reads or through pair-links. Sequence variations that cannot be properly phased can result in erroneous chimeras between isoforms or paralogs that are impossible to discern from short read data alone. Improvements in long read technologies 29 should help address these challenges. Notably, although Trinity currently only officially supports Illumina RNA-Seq, efforts are underway to explore the use of transcript sequencing reads generated from alternative technologies, including those from Pacific Biosciences 30 and Ion Torrent31.
Finally, as in high throughput genome sequences, evidence for polymorphisms can be mined from the Illumina RNA-Seq data mapped to Trinity assemblies (as in Ref. 32) and visualized within the display. However, one must be particularly cautious in evaluating polymorphisms in the context of RNA-Seq and de novo transcriptome assembly data, since incorrect transcript assembly or isoform misalignment can be easily misinterpreted as evidence for polymorphism. In particular, when transcripts are very highly expressed, sequencing errors can yield substantially expressed ‘variants’. Determining the best practices for calling SNPs in de novo transcriptome assemblies and examining allele-specific expression is an open area of research. Indeed as bioinformatic software become easier to use, it is essential for the research community to develop and use best practices in order to ensure that controversial results are not the result of multiple sources of error33.
Alternative analysis packages
Excellent tools are available and in widespread use for transcriptome studies in organisms for which a high-quality reference genome sequence is available, including those provided in the Tuxedo software suite (Bowtie, Tophat, Cufflinks, Cuffdiff, and CummeRbund 4, 21, 23, 34) or Scripture 5. Available de novo RNA-Seq assembly software include among others Oases7, SOAPdenovo-trans, and TransABySS6. The recently published eXpress software22 implements a highly efficient algorithm for estimating transcript expression levels, and leverages the Bowtie2 software34 for short read alignments, providing an alternative to using RSEM for estimating transcript measurements. New methods for differential expression analysis based on RNA-Seq data are also emerging35-39. As we continue to maintain and enhance the Trinity software and support related downstream analyses, we will explore the impact of new tools as they become available, and integrate those found to be most useful into future analysis pipelines, and we encourage users to explore alternative methods independently. In addition, we encourage users to explore the currently supported tools, including edgeR and RSEM, independently from using the Trinity-provided helper utilities, since they include additional capabilities that may not be fully exposed through the Trinity wrappers.
Future Trinity developments are planned to not only support genome-free de novo transcriptome assembly, but also to be able to leverage reference genome sequences and transcript annotations where available. In addition to providing effective methods to assist in genome annotation, such developments should expand upon our abilities to explore the transcriptional complexity of model organisms, particularly in those cases where genomes are modified or rearranged, such as in cancer 40.
Introduction to the Trinity RNA-Seq tutorial
The following protocol will show you how to:
Run Trinity to assemble a transcriptome reference from RNA-Seq from multiple samples.
Estimate expression levels for each transcript in each sample.
Identify transcripts that are differentially expressed between the different samples.
This tutorial provides a walk-through to some standard operations used to generate and analyze Trinity assemblies, including de novo RNA-Seq assembly, abundance estimation, and differential expression. For simplicity, our tutorial will not use libraries from biological replicates, but note that at least three biological replicates per sample or condition are required in order to test for significance given observed biological and technical variation.
All the methods and tools for interrogating assemblies are described on the Trinity software website (http://trinityrnaseq.sf.net), and here we provide a selection of these operations that strikes a balance between showcasing the breadth of capabilities and length of this tutorial. This tutorial was produced with version Trinityrnaseq_r2013-02-25, and readers should keep in mind that as Trinity is a continually evolving research software, some parameters and filenames might change in future software releases. The most recent version of this tutorial is maintained at (http://trinityrnaseq.sf.net/trinity_rnaseq_tutorial.html). A typical use case for Trinity is not very different from the tutorial and we highly recommend users complete the tutorial successfully before applying the protocol to their own data.
Before executing the steps described in the tutorial below, we encourage you to first read through the entire tutorial document.
Materials
EQUIPMENT
Data (requirements vary according to your experimental goals)
- Hardware (64-bit computer running Linux; ~1G of RAM per ~1M PE reads)
- Trinity version trinityrnaseq_r2013-02-25: http://trinityrnaseq.sourceforge.net
- Bowtie version 0.12.9: http://bowtie-bio.sourceforge.net (RSEM is currently not compatible with Bowtie 2, so be sure to obtain the latest release for Bowtie 1, which is currently v. 0.12.9 as released on December 16, 2012.)
- Samtools version 0.1.18: http://sourceforge.net/projects/samtools/files/samtools/
- R version 2.15: http://www.r-project.org
- Blast+ version 2.2.27: ftp://ftp.ncbi.nlm.nih.gov/blast/executables/blast+/LATEST/
Ensure that each of the above installed software tools (excepting Trinity) are available within your unix PATH setting. For example, if you have tools installed in a ‘/usr/local/tools’ directory, you can update your PATH setting to include this directory in the search path by:
% export PATH=/usr/local/tools:$PATH
EQUIPMENT SETUP
Optionally and only for simplicity, define the environmental variable TRINITY_HOME, replacing ‘/software/trinityrnaseq’ below with the path to your Trinity software installation. Otherwise you can write the full path where $TRINITY_HOME appears in this tutorial.
% export TRINITY_HOME=/software/trinityrnaseq
After you install R, you will need to install the following R packages:
Bioconductor: http://www.bioconductor.org
edgeR: http://www.bioconductor.org/packages/release/bioc/html/edgeR.html
gplots
The simplest way to do this is from within R:
% R > source(“http://bioconductor.org/biocLite.R”) > biocLite() > biocLite(“edgeR”) > biocLite(“ctc”) > biocLite(“Biobase”) > biocLite(“ape”) > install.packages(“gplots”)
TUTORIAL DATA
For this tutorial, you will need to acquire strand-specific RNA-Seq data from Schizosaccharomyces pombe grown in four conditions, described in Ref. 45: log growth (log), plateau phase (plat), diauxic shift (ds), and heat shock (hs), each with 1M Illumina paired-end strand-specific RNA-Seq data, to a total of 4M paired-end reads.
Reads for assembly
The data can be downloaded by visiting this URL in a web browser, or directly from the command line using ‘wget’, by:
% wget \ http://sourceforge.net/projects/trinityrnaseq/files/misc/Trinity NatureProtocolTutorial.tgz/download
The file downloaded should be called ‘TrinityNatureProtocolTutorial.tgz’ and is 540 MB in size. Unpack this file by:
tar –xvf TrinityNatureProtocolTutorial.tgz
which should generate the following files in a TrinityNatureProtocolTutorial/ directory with the following contents:
S_pombe_refTrans.fasta # reference transcriptome for S. pombe 1M_READS_sample/Sp.hs.1M.left.fq # PE reads for heatshock 1M_READS_sample/Sp.hs.1M.right.fq 1M_READS_sample/Sp.log.1M.left.fq # PE reads for log phase 1M_READS_sample/Sp.log.1M.right.fq 1M_READS_sample/Sp.ds.1M.right.fq # PE reads for diauxic shock 1M_READS_sample/Sp.ds.1M.left.fq 1M_READS_sample/Sp.plat.1M.left.fq # PE reads for plateau phase 1M_READS_sample/Sp.plat.1M.right.fq samples_n_reads_described.txt # tab-delimited description file.
This protocol expects the raw data to be of high quality, free from adaptor, barcodes and other contaminating subsequences.
Procedure
De novo RNA-Seq assembly using Trinity (Timing: ~60 to 90 minutes)
1. Create a working folder and place the ‘TrinityNatureProtocolTutorial/’ directory contents there (as per the Materials section).
2. In order to facilitate downstream analyses, concatenate the RNA-Seq data across all samples into a single set of inputs to generate a single reference Trinity assembly. Combine all ‘left’ reads into a single file, and combine all ‘right’ reads into a single file by:
% cat 1M_READS_sample/*.left.fq > reads.ALL.left.fq % cat 1M_READS_sample/*.right.fq > reads.ALL.right.fq
3. Now, assemble the reads into transcripts using Trinity by:
% $TRINITY_HOME/Trinity.pl --seqType fq --JM 10G \ --left reads.ALL.left.fq --right reads.ALL.right.fq -- SS_lib_type RF --CPU 6
The --JM option allows the user to control the amount of RAM used during Jellyfish kmer counting, in this case, 10 Gb of RAM. The --CPU option controls the number of parallel processes. Feel free to change these depending on your system. The Trinity reconstructed transcripts will exist as FASTA-formatted sequences in the output file ‘trinity_out_dir/Trinity.fasta’.
4. The following ‘TrinityStats.pl’ will report the number of transcripts, components, and the transcript contig N50 value based on the ‘Trinity.fasta’ file. The contig N50 value, defined as the maximum length whereby at least 50% of the total assembled sequence resides in contigs of at least that length, is a commonly used metric for evaluating the contiguity of a genome assembly. Note that, unlike genome assemblies, maximizing N50 is not appropriate for transcriptomes; it is more appropriate to use an index based on a reference dataset (from the same or a closely related species) and to estimate the number of reference genes recovered and how many can be deemed to be full-length49, 50 (see below). The N50 value is, however, useful for confirming that the assembly succeeded (you will expect a value that is near the average transcript length of S. pombe, avg. = 1397 bases). Use the script ‘$TRINITY_HOME/utilities/TrinityStats.pl’ to examine this statistic for the Trinity assemblies:
% $TRINITY_HOME/util/TrinityStats.pl trinity_out_dir/Trinity.fasta
Quality Assessment (Recommended, but Optional) (Timing: ~90 minutes)
5. Examine the breadth of genetic composition and transcript contiguity by leveraging a reference data set. The annotated reference transcriptome of Schizosaccharomyces pombe is included as file ‘S_pombe_refTrans.fasta’. Use megablast and our included analysis script to analyze its representation by the Trinity assembly as described below:
- Prepare the reference transcriptome fasta file as a BLAST database:
% makeblastdb -in S_pombe_refTrans.fasta -dbtype nucl
- Run megablast to align the known transcripts to the Trinity assembly:
% blastn -query trinity_out_dir/Trinity.fasta \ -db S_pombe_refTrans.fasta \ -out Trinity_vs_S_pombe_refTrans.blastn \ -evalue 1e-20 -dust no -task megablast -num_threads 2 \ -max_target_seqs 1 -outfmt 6 - Once BLAST is finished, run the script below examine the length coverage of top database hits.
% $TRINITY_HOME/util/analyze_blastPlus_topHit_coverage.pl \ Trinity_vs_S_pombe_genes.blastn \ trinity_out_dir/Trinity.fasta \ S_pombe_refTrans.fasta
6. Examine the number of input RNA-Seq reads that are well represented by the transcriptome assembly. Trinity provides a script ‘alignReads.pl’ that executes Bowtie to align the left and right fragment reads separately to the Trinity contigs and then groups the reads together into pairs while retaining those single read alignments that are not found to be properly paired with their mate.
Run ‘alignReads.pl’ by:
% $TRINITY_HOME/util/alignReads.pl --seqType fq \ --left reads.ALL.left.fq --right reads.ALL.right.fq \ --SS_lib_type RF --retain_intermediate_files \ --aligner bowtie \ --target trinity_out_dir/Trinity.fasta -- -p 4
7. When ‘alignReads.pl’ is run using strand-specific data, as indicated above with the ‘--SS_lib_type RF’ parameter setting, it will separate the alignments into those that align to the sense strand (‘+’) from the antisense strand (‘−’). All output files including coordinate-sorted and read-name-sorted SAM files should exist in a ‘bowtie_out/’ directory. Count the number of reads aligning (at least once) to the sense strand of transcripts by running the utility below on the sense-strand read name-sorted alignment file as shown:
% $TRINITY_HOME/util/SAM_nameSorted_to_uniq_count_stats.pl \ bowtie_out/bowtie_out.nameSorted.sam.+.sam
Abundance Estimation Using RSEM (Timing: ~40 to 60 minutes)
Transcript abundance estimates are obtained by running RSEM separately for each sample, as shown below. The PERL script ‘run_RSEM_align_n_estimate.pl’ simply provides an interface to the RSEM software, translating the familiar Trinity command-line parameters to their RSEM equivalents and then executing the RSEM software.
8. # RSEM for log
% $TRINITY_HOME/util/RSEM_util/run_RSEM_align_n_estimate.pl \
--transcripts trinity_out_dir/Trinity.fasta \
--left Sp.log.1M.left.fq \
--right Sp.log.1M.right.fq \
--SS_lib_type RF \
--prefix LOG
9. # RSEM for ds
% $TRINITY_HOME/util/RSEM_util/run_RSEM_align_n_estimate.pl \
--transcripts trinity_out_dir/Trinity.fasta \
--left Sp.ds.1M.left.fq \
--right Sp.ds.1M.right.fq \
--SS_lib_type RF \
--prefix DS
10. # RSEM for hs
% $TRINITY_HOME/util/RSEM_util/run_RSEM_align_n_estimate.pl \
--transcripts trinity_out_dir/Trinity.fasta \
--left Sp.hs.1M.left.fq \
--right Sp.hs.1M.right.fq \
--SS_lib_type RF \
--prefix HS
11. # RSEM for plat
% $TRINITY_HOME/util/RSEM_util/run_RSEM_align_n_estimate.pl \
--transcripts trinity_out_dir/Trinity.fasta \
--left Sp.plat.1M.left.fq \
--right Sp.plat.1M.right.fq \
--SS_lib_type RF \
--prefix PLAT
Each step generates files ‘${prefix}.isoforms.results’ and ‘${prefix}.genes.results’, containing the abundance estimations for Trinity transcripts (Table 1) and components (Table 2), respectively. The ${prefix} in the filename is set based on the ‘--prefix’ setting in the above commands, which is unique to each sample. Note, the genes and transcripts can be examined separately using their corresponding RSEM abundance estimates in the differential expression analysis guide below. For brevity, we pursue only the transcripts below.
Table 1.
Example contents of RSEM’s ‘isoforms.results’ file
| transcript_id | gene_id | length | effective_length | expected_count | TPM | FPKM | IsoPct |
|---|---|---|---|---|---|---|---|
| comp56_c0_seq1 | comp56_c0 | 3739 | 3443 | 637.65 | 16664.43 | 7008.23 | 11.26 |
| comp56_c0_seq2 | comp56_c0 | 3697 | 3401 | 4966.34 | 131393.38 | 55257.53 | 88.74 |
| comp62_c0_seq1 | comp62_c0 | 7194 | 6898 | 4551.13 | 59364.09 | 24965.59 | 95.54 |
| comp62_c0_seq2 | comp62_c0 | 7076 | 6778 | 208.87 | 2771.95 | 1165.74 | 4.46 |
Transcript_id: Trinity transcript identifier
Gene_id: Trinity component to which the reconstructed transcript was derived.
Length: length of the reconstructed transcript.
Effective length: The mean number of 5′ start positions from which an RNA-Seq fragment could have been derived from this transcript, given the distribution of fragment lengths inferred by RSEM. The value is equal to (transcript_length - mean_fragment_length + 1).
Expected count: number of expected RNA-Seq fragments assigned to the transcript given maximum likelihood transcript abundance estimates.
TPM: transcripts per million
FPKM: number of RNA-Seq fragments per kilobase of transcript effective length per million fragments mapped to all transcripts.
IsoPct: percent of expression for a given transcript compared to all expression from that Trinity component.
Table 2.
Example contents of RSEM’s ‘genes.results’ file
| gene_id | transcript_id(s) | length | effective_length | expected_count | TPM | FPKM |
|---|---|---|---|---|---|---|
| comp56_c0 | comp56_c0_seq1, comp56_c0_seq2 |
3701.73 | 3405.49 | 5604 | 148057.81 | 62265.76 |
| comp62_c0 | comp62_c0_seq1, comp62_c0_seq2 |
7188.74 | 6892.5 | 4760 | 62136.04 | 26131.33 |
The gene’s length and effective length are defined as the IsoPct weighted sum of transcript lengths and effective lengths. The gene’s expected counts, TPM, and FPKM are defined as the sum of its transcripts’ expected counts, TPM and FPKM.
Differential Expression Analysis Using edgeR (Timing: < 5 minutes) Identification of differentially expressed transcripts between pairs of samples
In the Trinity directory you will find the script ‘run_DE_analysis.pl’ (under ‘$TRINITY_HOME/Analysis/DifferentialExpression/’). This is a PERL script that automates many of the tasks of running edgeR or DESeq; in this tutorial, we only leverage edgeR. The input requires only one file: a matrix containing the counts of RNA-Seq fragments per feature in a simple tab-delimited text file. The first column is the name of the transcript. The second, third, etc., are the raw counts for each of the corresponding samples. The program therefore needs a minimum of three columns. The first row contains the column headings including a label for each sample.
We can create the matrix using the expected fragment count data produced by RSEM. You will see that each of the RSEM ‘*.isoforms.results’ files has a number of columns, but we only need the one called ‘expected_count’.
12. Merge the individual RSEM-estimated fragment counts for each of the samples into a single data table by:
% $TRINITY_HOME/util/RSEM_util/merge_RSEM_frag_counts_single_table.pl\ LOG.isoforms.results DS.isoforms.results HS.isoforms.results \ PLAT.isoforms.results > Sp_isoforms.counts.matrix
13. Now, use edgeR to identify differentially expressed transcripts for each pair of samples assayed, by:
%
$TRINITY_HOME/Analysis/DifferentialExpression/run_DE_analysis.
pl \
--matrix Sp_isoforms.counts.matrix \
--method edgeR \
--output edgeR_dir
All the edgeR results from the pairwise comparisons now exist in the ‘edgeR_dir/’ output directory, and also include the following files of interest:
*.edgeR.DE_results - identified differentially expressed transcripts including fold change and statistical significance (see Table 3 - output format for DE_results)
*.edgeR.DE_results.MA_n_Volcano.pdf - MA and volcano plots from pairwise comparisons (Fig. 9)
Table 3.
Example contents of logarithmic vs. plateau growth edgeR ‘DE_results’ file
| Transcript | logFC | logCPM | PValue | FDR |
|---|---|---|---|---|
| comp5128_c0_seq1 | 10.3 | 11.1 | 2.13e-22 | 1.22e-18 |
| comp5231_c0_seq1 | 10.0 | 10.9 | 1.10e-21 | 3.13e-18 |
| comp5097_c0_seq1 | 8.7 | 11.3 | 5.72e-20 | 1.10e-16 |
| comp1686_c0_seq1 | 9.2 | 10.4 | 1.01e-19 | 1.46e-16 |
| comp1012_c0_seq1 | 8.3 | 11.5 | 2.8e-19 | 3.23e-16 |
logFC: log2(fold change) = log2(plateau_phase/logarithmic_growth)
logCPM: log2(counts per million)
FDR: false discovery rate
Figure 9. Pairwise comparisons of transcript abundance.

Shown are two visualizations for comparing transcript expression profiles between the logarithmic growth and plateau growth samples from S. pombe. (a) MA-plot for differential expression analysis generated by EdgeR, plots for each gene its log2(fold change) between the two samples (A, Y axis) vs. its log2(average expression) in the two samples (M, X axis). (b) Volcano plot comparing false discovery rate (-log10FDR, Y axis) as a function of log2(fold-change) between the samples (logFC, X axis). Transcripts that are identified as significantly differentially expressed at most 0.1% FDR are colored red.
Normalizing expression values across samples
The statistical analyses of differential expression as performed above are based on the analysis of raw counts of fragments corresponding to individual transcripts (or genes) according to different samples. Execute the following steps to perform TMM normalization and generate a matrix of expression values measured in FPKM.
14. First, extract the transcript length values from any one of RSEM’s *.isoform.results files.
% cut -f1,3,4 DS.isoforms.results > Trinity.trans_lengths.txt % $TRINITY_HOME/Analysis/DifferentialExpression/run_TMM_normalization_write_FPKM_matrix.pl \ --matrix Sp_isoforms.counts.matrix \ --lengths Trinity.trans_lengths.txt
The above will generate the following files:
‘Trinity_trans.counts.matrix.TMM_info.txt‘ - containing the effective library size for each sample after TMM normalization
‘Trinity_trans.counts.matrix.TMM_normalized.FPKM’- normalized transcript expression values according to transcript and sample, measured as FPKM. This matrix file will be used for clustering expression profiles for transcripts across samples and generating heatmap visualizations as described below.
Analyzing expression patterns and sample relationships
15. To study expression patterns of transcripts or genes across samples, it is often useful to restrict our analysis to those transcripts that are significantly differentially expressed in at least one pairwise sample comparison. Given a set of differentially expressed transcripts, we can extract their normalized expression values and perform hierarchical clustering to group together transcripts with similar expression patterns across samples, and to group together those samples that have similar expression profiles according to transcripts. For example, enter the ‘edgeR_dir/’ output directory and extract those transcripts that are at least 4-fold differentially expressed with false discovery-corrected statistical significance of at most 0.001 by:
% cd edgeR_dir/
% $TRINITY_HOME/Analysis/DifferentialExpression/analyze_diff_expr.pl \
--matrix../Trinity_components.counts.matrix.TMM_normalized.FPKM \
-C 2 -P 0.001
Please note that the -C parameter takes the log2(fold_change) cutoff, which in this case is log2(4) = 2. A number of files are generated, all with the prefix “diffExpr.P0.001_C2” indicating the parameter choices:
‘diffExpr.P0.001_C2.matrix’ contains the subset of transcripts from the complete matrix ‘matrix.TMM_normalized.FPKM’ that were identified as differentially expressed, as defined by the specified thresholds.
‘diffExpr.P0.001_C2.matrix.heatmap.pdf’ contains a clustered heatmap image showing the relationships among transcripts and samples (Fig. 10a) and a heatmap of the pair-wise Spearman correlations between samples (Fig. 10b).
‘diffExpr.P0.001_C2.matrix.R.all.RData’ is a local storage of all the data generated during this analysis. This is used below with additional analysis tools.
Figure 10. Comparisons of transcriptional profiles across samples.

(a) Hierarchical clustering of transcripts and samples. Shown is a heatmap showing the relative expression levels of each transcript (rows) in each sample (column). Rows and columns are hierarchically clustered. Expression values (FPKM) are log2 transformed and then median-centered by transcript. (b) Heatmap showing the hierarchically clustered Spearman correlation matrix resulting from comparing the transcript expression values (TMM-normalized FPKM) for each pair of samples. (c) Transcript clusters, extracted from the hierarchical clustering using R. X axis: samples (DS: diauxic shift; HS: heat shock; Log: mid-log growth; Plat: plateau growth); Y axis: median-centered log2(FPKM). Grey lines: individual transcripts; Blue line: average expression values per cluster. Number of transcripts in each cluster is shown in a left corner of each plot.
The ‘analyze_diff_expr.pl’ script will directly report the number of differentially expressed transcripts identified at the given thresholds. In addition, you can easily determine the number of differentially expressed transcripts, by counting the number of lines in the file by:
% wc -l diffExpr.P0.001_C2.matrix
and subtracting 1 so that you do not count the column header line as a transcript entry.
Due to the wide dynamic range in expression values of transcripts, it is useful to first log-transform expression values before plotting data points. In addition, to examine common expression patterns that focus on the relative expression of transcripts across multiple samples, it is useful to center each transcript’s expression values by the median (or alternatively, the mean) value. This is done by subtracting each transcript’s median log2(FPKM) value from its log2(FPKM) value in each sample. The hierarchically clustered transcript heatmap generated above uses median-centered log2(FPKM) values. Clusters of transcripts with common expression profiles can be automatically extracted from the earlier generated hierarchical clusters by running the script below, which uses R to cut the tree representing the hierarchically clustered transcripts based on specified criteria, such as to generate a specific number of clusters or by cutting the tree at a certain height. For example, run the following to partition transcripts by cutting the tree at 20% of the tree height:
% $TRINITY_HOME/Analysis/DifferentialExpression/define_clusters_by_cutting_tree.pl \ --Ptree 20 -R diffExpr.P0.001_C2.matrix.R.all.RData
This generates a directory: ‘diffExpr.P0.001_C2.matrix.R.all.RData.clusters_fixed_P_20/’ containing:
‘subcluster_*_log2_medianCentered_fpkm.matrix’ - each auto-defined cluster of transcripts is provided along with expression values that are log2-transformed and median-centered.
‘my_cluster_plots.pdf’ - contains a plot of the log2-transformed, median-centered expression values for each cluster (Fig. 10c).
The above script can be run several times with different values of --Ptree in order to increase or decrease the number of clusters generated.
Automating the required sections of the tutorial
For those interested in executing the sections of the tutorial without manually typing in each command, we include a script that executes the required sections of the tutorial. This includes concatenating all samples’ reads into a single input data set, assembling the reads using Trinity, performing abundance estimation separately for each sample, and running edgeR to identify differentially expressed transcripts. This can be run by the following command, including the -I (optional) parameter for an interactive experience, where the system will pause and wait for user response before proceeding to the next step.
% $TRINITY_HOME/util/run_Trinity_edgeR_pipeline.pl \
--samples_file samples_n_reads_described.txt -I
Troubleshooting
| Step | Section | Problem | Possible reason | Solution |
|---|---|---|---|---|
| 3 | Trinity assembly |
‘bad_alloc’ error |
Insufficient computing resources resulting in a fatal out-of-memory error. |
Ensure you have ~1G of RAM per ~1 M PE reads to be assembled. See Box 2 for computing requirements and services available. |
| 3 | Trinity assembly |
Large numbers of fusion transcripts |
Not using strand- specific RNA-Seq, or applying assembly to a transcriptome derived from a compact genome having (minimally) overlapping transcripts. |
If PE reads are being used, try running Trinity.pl with the ‘--jaccard_clip’ parameter, which uses PE reads to separate minimally overlapping transcripts. |
| 3 | Trinity assembly |
Retained introns are prevalent |
Unprocessed RNA is captured and assembled, or contaminating genomic DNA contributes to the assembly. |
Setting Trinity.pl ‘--min_kmer_cov’ to 2 or higher should reduce the number of retained introns, but will also reduce sensitivity for transcript reconstruction. Alternatively, lowly expressed transcripts (often enriched for retained introns) can be filtered from a given component post- abundance estimation. |
| 5-11 | Quality assessment and abundance estimation |
Cannot find makeblastdb, blastn, or Bowtie |
The additional required software tools were not installed or available via the Unix PATH setting. |
See EQUIPMENT section, be sure software tools are installed as required and that the software utilities are accessible via your PATH setting. Check with a systems administrator as necessary. |
| 13-15 | Differential expression analysis |
Few or no transcripts identified as differentially expressed |
Assuming transcripts are truly differentially expressed, increased sensitivity is required to detect them. |
Adjust the sensitivity thresholds of ‘analyze_diff_expr.pl’, increasing the allowed FDR and lowering the fold- change requirements. Try running Bioconductor tools directly and examine the available options for data exploration. Increase your depth of sequencing to improve upon the detection of lowly expressed transcripts. |
Timing
Steps 1-4, Trinity de novo transcriptome assembly of 4M PE Illumina reads, ~60 to 90 minutes.
Steps 5-7, Quality Assessment (optional), ~60 to 90 minutes
Steps 8-11, Using RSEM for abundance estimation, ~40 to 60 minutes.
Steps 12-15, Differential Expression Analysis Using EdgeR, < 5 minutes.
The amount of time taken for each of the commands executed for the required sections of the tutorial, as reported during the automated execution via ‘run_Trinity_edgeR_pipeline.pl’ described above, and as run on a high performance server at the Broad Institute (hardware specifications included), is provided as Supplementary Text Section S6.
Anticipated results
Since Trinity’s output is not absolutely deterministic, very slight variations in the output may result from Trinity being run at different times or on different hardware.
Trinity assembly statistics are provided in Table 4.
Table 4.
Trinity assembly statistics for the assembly of 4M PE S. pombe reads.
| Assembly statistic | Value |
|---|---|
| Total Trinity transcripts | 9299 |
| Total Trinity components | 8694 |
| Contig N50 | 1585 |
Reference transcript BLASTN mapping results from Step 5: results provided in Table 5. 4,765 of the reference S. pombe transcripts have a BLAST hit with an E-value less than 1e-20 and 3,401 of the 5,163 total reference transcripts are considered approximately ‘full-length’, with the Trinity contigs aligning by greater than 90% of the matching reference transcript’s length.
Table 5.
Distribution of BLASTN hit coverage of reference transcripts
| % Length coverage bin |
Count of reference transcripts in bin |
Cumulative count of reference transcripts at or above bin level. |
|---|---|---|
| 100 | 3401 | 3401 |
| 90 | 194 | 3595 |
| 80 | 165 | 3760 |
| 70 | 197 | 3957 |
| 60 | 224 | 4181 |
| 50 | 203 | 4384 |
| 40 | 158 | 4542 |
| 30 | 140 | 4682 |
| 20 | 83 | 4765 |
| 10 | 0 | 4765 |
| 0 | 0 | 4765 |
The counts of reads mapped to the Trinity assembly via alignReads.pl and using the Bowtie aligner, ascertained from Step 7, are provided in Table 6.
Table 6.
Counts of reads mapped to the Trinity assembly
| Read classification | Count of individual reads | Percent of mapped reads |
|---|---|---|
| Proper pairing | 8102100 | 93.12 |
| Left only | 307933 | 3.54 |
| Right only | 284203 | 3.27 |
| Improper pairing | 6476 | 0.07 |
The number of differentially expressed transcripts identified as having a significant FDR value of at most 0.001 and at least 4-fold difference in expression values, ascertained from Step 15, is 659.
Supplementary Material
Figure 4. Effects of in silico fragment normalization of RNA-Seq data on Trinity full-length transcript reconstruction.

Shown are the number of full-length transcripts reconstructed (Y axis) from a dataset of paired-end strand-specific RNA-Seq in S. pombe (a, 10M paired-end reads) or mouse (b, 100M, paired-end reads), using either the full dataset (Total; 100%) or different samplings (X axis) by either Trinity’s in silico normalization procedure at 5X up to 100X targeted maximum k-mer (k=25) coverage (blue bars) or by random down-sampling of the same number of reads (red bars).
Figure 5. Transcriptome and genome representations of alternatively spliced transcripts.

Shown is an example of the graphical representation generated by Trinity’s Butterfly software (a) along with the corresponding reconstructed transcripts (b) and their exonic structure based on the alignment to the mouse genome (c). Each node in the graph (a) is associated with a sequence, and directed edges connect consecutive sequences from 5′ to 3′ in the same transcript. Bulges (bifurcations) indicate sequence differences between alternative reconstructed transcripts, including alternatively spliced cassette exons; only a single bulge is shown in this transcript graph. Edges are annotated by the number of RNA-Seq fragments supporting the transcript from the 5′ sequence to the 3′ one. In this example, there are two supported paths: one from the blue to the green node (supported by 32 fragments) yielding ‘isoform A’ (b, top), and the other from the blue to the red to the green node, supported by at most 5 fragments, yielding ‘isoform B’ (b, bottom). The red node is a result of an alternatively skipped exon, as apparent in the gene structure (c, red bar, shown in ‘isoform B’). Navigable transcript graphs are optionally generated by Butterfly, provided in ‘dot’ format and can be visualized using graphviz (http://graphviz.org). These details are provided on the Trinity website (http://trinityrnaseq.sourceforge.net/advanced_trinity_guide.html).
Figure 6. Strand-specific library types.
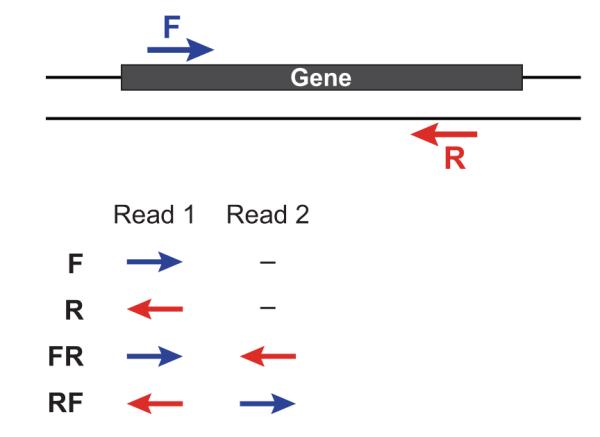
The left (/1) and right (/2) sequencing reads are depicted according to their orientations relative to the sense strand of a transcript sequence. The strand-specific library type (F, R, FR, or RF) depends on the library construction protocol and is user-specified to Trinity via the ‘--SS_lib_type’ parameter.
Figure 7. Full-length transcript reconstruction by Trinity in different organisms, sequencing depths, and parameters.

Shown is the number of fully reconstructed transcripts (bars, left Y axis) for Trinity assemblies of RNA-Seq data derived from fission yeast (Schizosaccharomyces pombe8, 45), Drosophila melanogaster11, and mouse8 with different combinations of parameters: DS – double stranded mode, SS – strand-specific mode, +J – using the ‘--jaccard_clip’ parameter to split falsely fused transcripts. Both SS and DS results are provided for S. pombe and mouse, but only DS results are provided for Drosophila since its RNA-Seq data was not strand-specific. Blue: full-length transcripts; red: full length merged, i.e., transcripts erroneously fused with another (typically neighboring) transcript. The black curve (right Y axis) indicates the run times in each case with a contemporary high-memory (256G to 512G RAM) server using a maximum of 4 threads (‘--CPU 4’, see Tutorial).
Figure 8. Evaluating paired-read support via the Jaccard similarity coefficient.

Read pair support is computed by first counting the number of RNA-Seq fragments (bounds of paired reads) that span each of two outer points of a specified window length (default: 100 bases), and then computing the Jaccard similarity coefficient (intersection/union) comparing the fragments that overlap either point. An example is shown for a neighboring pair of S. pombe transcripts (SPAC23C4.14 and SPAC23C4.15, bottom) that have substantial overlapping read coverage (gray track), resulting in a contiguous (fused) transcript assembled by Inchworm. However, the Jaccard similarity coefficient (blue track) calculated from the paired-reads (grey dumbbells) clearly identifies the position of reduced pair support. Examples of strong (upper left) and weak (upper right) pair support are depicted at top. When using the ‘--jaccard_clip’ parameter, the Inchworm contig is dissected to two separate full-length transcripts, which are then further processed by Chrysalis and Butterfly as part of the Trinity pipeline.
Acknowledgements
We are grateful to David Jaffe and Sarah Young for access to additional computing resources, Zehua Chen for help in R-scripting, to Leslie Gaffney for help with figure illustrations, to C. Titus Brown for essential discussions and inspiration related to digital normalization strategies, to Guillaume Marcais and Carl Kingsford for supporting the use of their Jellyfish software in Trinity, and to Brian Walenz for supporting our earlier use of Meryl. We are grateful to our users and their feedback, in particular Jennifer Wortman and Peter Bain for comments on earlier drafts of the manuscript.
This project has been funded in part with Federal funds from the National Institute of Allergy and Infectious Diseases, National Institutes of Health, Department of Health and Human Services, under Contract No.:HHSN272200900018C. Work was supported by HHMI, an NIH PIONEER award, a Center for Excellence in Genome Science grant 5P50HG006193-02 from the NHGRI and the Klarman Cell Observatory at the Broad Institute (AR). Alexie Papanicolaou was supported by the Commonwealth Scientific and Industrial Research Organization’s (CSIRO) Office of the Chief Executive (OCE). Moran Yassour was supported by the Clore Foundation. Philip Blood was supported by the National Science Foundation grant number OCI-1053575 for the Extreme Science and Engineering Discovery Environment (XSEDE) project. Bo Li and Colin Dewey were partially supported by NIH grant 1R01HG005232-01A1. In addition, Bo Li was partially funded by Dr. James Thomson’s MacArthur Professorship and by Morgridge Institute for Research support for Computation and Informatics in Biology and Medicine. Mathias Lieber was supported by the Bundesministerium für Bildung und Forschung (BMBF) via the project ‘NGSgoesHPC’. Nathalie Pochet was funded by the Fund for Scientific Research - Flanders (FWO Vlaanderen), Belgium. Robert Henschel and Richard D. LeDuc were funded by the National Science Foundation under Grant No. ABI-1062432 and CNS-0521433 to Indiana University, and by Indiana METACyt Initiative, which is supported in part by Lilly Endowment, Inc.
Any opinions, findings, and conclusions or recommendations expressed in this material are those of the authors and do not necessarily reflect the views of any of the funding bodies and institution including the National Science Foundation, the National Center for Genome Analysis Support and Indiana University.
Footnotes
Author contributions statements BJH is the current lead developer of Trinity and is additionally responsible for the development of the companion in silico normalization and TransDecoder utilities described herein. MY contributed to Butterfly software enhancements, generating figures, and to the manuscript text. BL and CND developed RSEM and are responsible for enhancements related to improved Trinity support. BJH and AP wrote the initial draft of the manuscript. AR is the Principal Investigator. All authors contributed to Trinity development and/or writing of the final manuscript, and all approve the final text.
The authors declare that they have no competing financial interests.
References
- 1.Wang Z, Gerstein M, Snyder M. RNA-Seq: a revolutionary tool for transcriptomics. Nature reviews. Genetics. 2009;10:57–63. doi: 10.1038/nrg2484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Haas BJ, Zody MC. Advancing RNA-Seq analysis. Nature biotechnology. 2010;28:421–423. doi: 10.1038/nbt0510-421. [DOI] [PubMed] [Google Scholar]
- 3.Martin JA, Wang Z. Next-generation transcriptome assembly. Nature reviews. Genetics. 2011;12:671–682. doi: 10.1038/nrg3068. [DOI] [PubMed] [Google Scholar]
- 4.Trapnell C, et al. Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks. Nature protocols. 2012;7:562–578. doi: 10.1038/nprot.2012.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Guttman M, et al. Ab initio reconstruction of cell type-specific transcriptomes in mouse reveals the conserved multi-exonic structure of lincRNAs. Nature biotechnology. 2010;28:503–510. doi: 10.1038/nbt.1633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Robertson G, et al. De novo assembly and analysis of RNA-seq data. Nature methods. 2010;7:909–912. doi: 10.1038/nmeth.1517. [DOI] [PubMed] [Google Scholar]
- 7.Schulz MH, Zerbino DR, Vingron M, Birney E. Oases: robust de novo RNA-seq assembly across the dynamic range of expression levels. Bioinformatics. 2012;28:1086–1092. doi: 10.1093/bioinformatics/bts094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Grabherr MG, et al. Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nature biotechnology. 2011;29:644–652. doi: 10.1038/nbt.1883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Duan J, Xia C, Zhao G, Jia J, Kong X. Optimizing de novo common wheat transcriptome assembly using short-read RNA-Seq data. BMC genomics. 2012;13:392. doi: 10.1186/1471-2164-13-392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Xu DL, et al. De novo assembly and characterization of the root transcriptome of Aegilops variabilis during an interaction with the cereal cyst nematode. BMC genomics. 2012;13:133. doi: 10.1186/1471-2164-13-133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zhao QY, et al. Optimizing de novo transcriptome assembly from short-read RNA-Seq data: a comparative study. BMC bioinformatics. 2011;12(Suppl 14):S2. doi: 10.1186/1471-2105-12-S14-S2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Henschel R, et al. Trinity RNA-Seq assembler performance optimization. XSEDE ’12 Proceedings of the 1st Conference of the Extreme Science and Engineering Discovery Environment: Bridging from the eXtreme to the campus and beyond.2012. [Google Scholar]
- 13.Marcais G, Kingsford C. A fast, lock-free approach for efficient parallel counting of occurrences of k-mers. Bioinformatics. 2011;27:764–770. doi: 10.1093/bioinformatics/btr011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Li B, Dewey CN. RSEM: accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC bioinformatics. 2011;12:323. doi: 10.1186/1471-2105-12-323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Robinson MD, McCarthy DJ, Smyth GK. edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics. 2010;26:139–140. doi: 10.1093/bioinformatics/btp616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Anders S, Huber W. Differential expression analysis for sequence count data. Genome biology. 2010;11:R106. doi: 10.1186/gb-2010-11-10-r106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bullard JH, Purdom E, Hansen KD, Dudoit S. Evaluation of statistical methods for normalization and differential expression in mRNA-Seq experiments. BMC bioinformatics. 2010;11:94. doi: 10.1186/1471-2105-11-94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Fang Z, Cui X. Design and validation issues in RNA-seq experiments. Briefings in bioinformatics. 2011;12:280–287. doi: 10.1093/bib/bbr004. [DOI] [PubMed] [Google Scholar]
- 19.Auer PL, Doerge RW. Statistical design and analysis of RNA sequencing data. Genetics. 2010;185:405–416. doi: 10.1534/genetics.110.114983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mortazavi A, Williams BA, McCue K, Schaeffer L, Wold B. Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nature methods. 2008;5:621–628. doi: 10.1038/nmeth.1226. [DOI] [PubMed] [Google Scholar]
- 21.Trapnell C, et al. Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nature biotechnology. 2010;28:511–515. doi: 10.1038/nbt.1621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Roberts A, Pachter L. Streaming fragment assignment for real-time analysis of sequencing experiments. Nature methods. 2013;10:71–73. doi: 10.1038/nmeth.2251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Langmead B, Trapnell C, Pop M, Salzberg SL. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome biology. 2009;10:R25. doi: 10.1186/gb-2009-10-3-r25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Robinson MD, Oshlack A. A scaling normalization method for differential expression analysis of RNA-seq data. Genome biology. 2010;11:R25. doi: 10.1186/gb-2010-11-3-r25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Dillies MA, et al. A comprehensive evaluation of normalization methods for Illumina high-throughput RNA sequencing data analysis. Briefings in bioinformatics. 2012 doi: 10.1093/bib/bbs046. [DOI] [PubMed] [Google Scholar]
- 26.Marioni JC, Mason CE, Mane SM, Stephens M, Gilad Y. RNA-seq: an assessment of technical reproducibility and comparison with gene expression arrays. Genome research. 2008;18:1509–1517. doi: 10.1101/gr.079558.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Robinson JT, et al. Integrative genomics viewer. Nature biotechnology. 2011;29:24–26. doi: 10.1038/nbt.1754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Abeel T, Van Parys T, Saeys Y, Galagan J, Van de Peer Y. GenomeView: a next-generation genome browser. Nucleic acids research. 2012;40:e12. doi: 10.1093/nar/gkr995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Liu L, et al. Comparison of next-generation sequencing systems. Journal of biomedicine & biotechnology. 2012;2012:251364. doi: 10.1155/2012/251364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Eid J, et al. Real-time DNA sequencing from single polymerase molecules. Science. 2009;323:133–138. doi: 10.1126/science.1162986. [DOI] [PubMed] [Google Scholar]
- 31.Rothberg JM, et al. An integrated semiconductor device enabling non-optical genome sequencing. Nature. 2011;475:348–352. doi: 10.1038/nature10242. [DOI] [PubMed] [Google Scholar]
- 32.Van Belleghem SM, Roelofs D, Van Houdt J, Hendrickx F. De novo transcriptome assembly and SNP discovery in the wing polymorphic salt marsh beetle Pogonus chalceus (Coleoptera, Carabidae) PloS one. 2012;7:e42605. doi: 10.1371/journal.pone.0042605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kleinman CL, Majewski J. Comment on “Widespread RNA and DNA sequence differences in the human transcriptome”. Science. 2012;335:1302. doi: 10.1126/science.1209658. author reply 1302. [DOI] [PubMed] [Google Scholar]
- 34.Langmead B, Salzberg SL. Fast gapped-read alignment with Bowtie 2. Nature methods. 2012;9:357–359. doi: 10.1038/nmeth.1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Pounds SB, Gao CL, Zhang H. Empirical bayesian selection of hypothesis testing procedures for analysis of sequence count expression data. Statistical applications in genetics and molecular biology. 2012;11 doi: 10.1515/1544-6115.1773. [DOI] [PubMed] [Google Scholar]
- 36.Tarazona S, Garcia-Alcalde F, Dopazo J, Ferrer A, Conesa A. Differential expression in RNA-seq: a matter of depth. Genome research. 2011;21:2213–2223. doi: 10.1101/gr.124321.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Cumbie JS, et al. GENE-counter: a computational pipeline for the analysis of RNA-Seq data for gene expression differences. PloS one. 2011;6:e25279. doi: 10.1371/journal.pone.0025279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hardcastle TJ, Kelly KA. baySeq: empirical Bayesian methods for identifying differential expression in sequence count data. BMC bioinformatics. 2010;11:422. doi: 10.1186/1471-2105-11-422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Leng N, et al. An empirical Bayes hierarchical model for inference in RNA-seq experiments. Bioinformatics. 2012 doi: 10.1093/bioinformatics/btt087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Tuna M, Amos CI. Genomic sequencing in cancer. Cancer letters. 2012 doi: 10.1016/j.canlet.2012.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lohse M, et al. RobiNA: a user-friendly, integrated software solution for RNA-Seq-based transcriptomics. Nucleic acids research. 2012;40:W622–627. doi: 10.1093/nar/gks540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Martin M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet.journal. 2011;17 [Google Scholar]
- 43.Haas BJ, Chin M, Nusbaum C, Birren BW, Livny J. How deep is deep enough for RNA Seq profiling of bacterial transcriptomes? BMC genomics. 2012;13:734. doi: 10.1186/1471-2164-13-734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Brown CT, Howe A, Zhang Q, Pryrkosz AB, Brom TH. A Reference-Free Algorithm for Computational Normalization of Shotgun Sequencing Data. arXiv:1203.4802 [q-bio.GN] 2012 [Google Scholar]
- 45.Rhind N, et al. Comparative functional genomics of the fission yeasts. Science. 2011;332:930–936. doi: 10.1126/science.1203357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Borodina T, Adjaye J, Sultan M. A strand-specific library preparation protocol for RNA sequencing. Methods in enzymology. 2011;500:79–98. doi: 10.1016/B978-0-12-385118-5.00005-0. [DOI] [PubMed] [Google Scholar]
- 47.Parkhomchuk D, et al. Transcriptome analysis by strand-specific sequencing of complementary DNA. Nucleic acids research. 2009;37:e123. doi: 10.1093/nar/gkp596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Sung WK, et al. Genome-wide survey of recurrent HBV integration in hepatocellular carcinoma. Nature genetics. 2012;44:765–769. doi: 10.1038/ng.2295. [DOI] [PubMed] [Google Scholar]
- 49.Kumar S, Blaxter ML. Comparing de novo assemblers for 454 transcriptome data. BMC genomics. 2010;11:571. doi: 10.1186/1471-2164-11-571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Papanicolaou A, Stierli R, Ffrench-Constant RH, Heckel DG. Next generation transcriptomes for next generation genomes using est2assembly. BMC bioinformatics. 2009;10:447. doi: 10.1186/1471-2105-10-447. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.