

Abstract
High levels of striatal-enriched protein tyrosine phosphatase (STEP) activity are observed in a number of neuropsychiatric disorders such as Alzheimer’s disease. Over-expression of STEP results in the dephosphorylation and inactivation of many key neuronal signaling molecules, including ionotropic glutamate receptors. Moreover, genetically reducing STEP levels in AD mouse models significantly reversed cognitive deficits and decreased glutamate receptor internalization. These results support STEP as a potential target for drug discovery for the treatment of Alzheimer’s disease. Herein, a substrate-based approach for the discovery and optimization of fragments called substrate activity screening (SAS) has been applied to the development of low molecular weight (<450 Da) and non-peptidic, single-digit micromolar mechanism-based STEP inhibitors with greater than 20-fold selectivity across multiple tyrosine and dual specificity phosphatases. Significant levels of STEP inhibition in rat cortical neurons are also observed.
INTRODUCTION
Synaptic connections provide the physical basis for communication within the brain, and synaptic plasticity, the ability for synapses to strengthen or weaken between neurons as a result of molecular signals, is critical to maintaining proper cognitive function. Therefore, disruptions in synaptic function can lead to impairments in cognition. Synaptic dysregulation has been implicated in a range of neuropsychiatric disorders,1 including Alzheimer’s disease (AD),2 schizophrenia,3 depression,4 fragile X syndrome,5 and drug addiction.6
One protein that has been implicated in the dysregulation of synaptic plasticity is STriatal-Enriched protein tyrosine Phosphatase (STEP), which is encoded by the PTPN5 gene and is found in striatum, hippocampus, cortex and related regions. High levels of STEP activity result in the dephosphorylation and inactivation of several neuronal signaling molecules, including extracellular signal-regulated kinases 1 and 2 (ERK1/2),7 proline-rich tyrosine kinase 2 (Pyk2),8 mitogen-activated protein kinase p38,9 and the GluN2B subunit of the N-methyl-d-aspartate receptor (NMDARs). 10, 11 Dephosphorylation of the kinases inactivates them while dephosphorylation of GluN2B results in internalization of NMDA receptors. To test the hypothesis that over expression of STEP might contribute to cognitive deficits in AD mouse models, STEP levels were reduced genetically in AD mice. Progeny null for STEP exhibited significant cognitive improvements as well as increased receptor levels on synaptic membranes. 11 These results support STEP as a potential target for drug discovery for the treatment of AD.
We have previously reported on the use of a fragment-based inhibitor discovery and optimization approach termed Substrate Activity Screening (SAS) for the identification of low molecular weight inhibitors of the tyrosine phosphatases of the bacterium Mycobacterium tuberculosis, PtpA and PtpB.12 To date, there is a single reported ancillary example of a STEP inhibitor having modest activity and without selectivity or cell data.13 Herein, we have applied the SAS method to identify low molecular weight (<450 Da), non-peptidic STEP inhibitors with single-digit micromolar inhibition, 20-fold selectivity over multiple human PTPs, and significant activity in rat cortical neurons.
RESULTS AND DISCUSSION
In the SAS fragment approach as applied to PTPs,12 a library of low molecular weight O-aryl and -heteroaryl phosphates are screened as potential PTP substrates using a simple and continuous coupled assay with purine nucleotide phosphorylase used to detect the release of inorganic phosphate. Active substrates are then converted to inhibitors through the replacement of the phosphate group with a non-hydrolyzable phosphate mimetic. Key advantages of this approach for the identification of mechanism-based inhibitors are: (1) the ease of preparing a library of diverse low molecular weight phosphate substrates, (2) the inherent “turn on” nature of a substrate screen, which minimizes the high percentage of false positives typical to PTP inhibitor screens,14,15 and (3) the flexibility to explore different phosphate mimetics after the identification of substrate fragments.
Identification of Substrates
A library of O-aryl phosphate substrate fragments was previously generated and screened to identify Mycobacterium tuberculosis PtpB and PtpA inhibitors.12 Screening this library of phosphates against STEP yielded several promising fragment substrates (Figure 1). Of note, fragment substrates 6 to 10 had much improved KM values relative to the phosphotyrosine derivative 4, which much more closely resembles naturally occurring PTP substrates.
Figure 1.

Selected initial substrate hits obtained against STEP.
Conversion of Substrates to Inhibitors
The two substrate scaffolds 6 and 8 were identified as initial starting points for further optimization because the biphenyl scaffold has been regarded as a “privileged scaffold” with drug-like properties and because analog preparation is straightforward using cross-coupling methodology.16 Inhibitors 11 and 12 (Figure 2) were first prepared by replacing the phosphate group of each substrate with the non-hydrolyzable phosphate mimetic difluoromethylphosphonic acid (DFMP).17 The Ki values of the two inhibitors were measured using a standard, continuous in vitro inhibition assay, with para-nitrophenyl phosphate (pNPP) serving as the chromogenic substrate.18 Triton-X 100, 0.01% v/v, was added to the assay buffer to prevent non-productive inhibition by micelle formation.19 Importantly, the relative Ki values of inhibitors 11 20 and 1212b were determined to be 337 and 120 µM, respectively, which correlate reasonably well with the KM values of the corresponding substrates 6 and 8.21
Figure 2.

DFMP inhibitors 11 and 12 based on privileged substrate scaffolds 6 and 8.
Optimization of Inhibitor Potency
Introduction of diverse substitution onto the biphenyl cores of inhibitors 11 and 12 was next performed. For fragment 11, a series of substitutions was first introduced on the distal aromatic ring (Table 1). Although substitution at the para position of the distal ring was beneficial for inhibition (11a), any substitution larger than a methyl group resulted in decreased potency (11b). Alkyl substitution at the meta position also led to an increase in potency of the inhibitors, with the α-branched and more bulky isopropyl group outperforming the methyl group (11d versus 11c). The presence of an oxygen atom at the ortho position was also beneficial to the potency of the inhibitors, with the free hydroxyl resulting in greater inhibition than the methoxy derivative (11e and 11f). Combining a meta-alkyl group and an ortho hydroxyl group in a 2,3-orientation (11g) led to an increase in potency while the 2,5-substitution was not advantageous (11h).
Table 1.
Optimization of distal aryl ring substation for inhibitor 11a
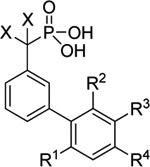 | ||||||
|---|---|---|---|---|---|---|
| X,X | R1 | R2 | R3 | R4 | Ki (µM) | |
| 11 | F,F | H | H | H | H | 337 ± 60 |
| 11a | F,F | H | H | H | Me | 205 ± 1 |
| 11b | F,F | H | H | H | i-Pr | 222 ± 1 |
| 11c | F,F | H | H | Me | H | 137± 8 |
| 11d | F,F | H | H | i-Pr | H | 81 ± 2 |
| 11e | F,F | H | OH | H | H | 168 ± 17 |
| 11f | F,F | H | OMe | H | H | 271 ± 17 |
| 11g | F,F | H | OH | i-Pr | H | 69 ± 9 |
| 11h | F,F | OH | H | i-Pr | H | 95 ± 1 |
| 11i | F,F | H | OH | Et | H | 124 ± 3 |
| 11j | F,F | H | OH | t-Bu | H | 63 ± 1 |
| 11k | F,F | H | OH | cyclobutyl | H | 45 ± 2 |
| 11l | F,F | H | OH | cyclopentyl | H | 25 ± 4 |
| 11m | F,F | H | OH | cyclohexyl | H | 20 ± 3 |
| 11n | F,F | H | OH | cycloheptyl | H | 62 ± 1 |
| 11o | OH,H | H | OH | cyclohexyl | H | 36 ± 1 |
Ki values were determined using at least 2 independent measurements.
We next investigated the effect of altering the meta-alkyl group (11i–11n) on inhibitory potency. The ethyl group (11i) resulted in two-fold reduction in inhibitory potency, while the tert-butyl group provided a modest enhancement (11j). A more significant increase in potency was achieved by introducing cycloalkyl groups, with cyclopentyl (11l) and cyclohexyl (11m) providing the optimal ring size. Decreasing the ring size to cyclobutyl (11k) or increasing it to cycloheptyl (11n) was met with diminished potency compared to the optimal cyclohexyl substituted derivative (11m, Ki = 20 µM). Finally, introduction of the hydroxymethylphosphonic acid phosphate mimetic (11o) in place of difluoromethylphosphonic acid resulted in an approximate two-fold reduction in potency.
The effect of substitution on the distal aromatic ring of inhibitor 12 was also evaluated (Table 2). The electron deficient cyano group proved to be detrimental to inhibition at the ortho (12a), meta (12b) and para (12c) sites. Alkoxy groups also reduced inhibition when placed at the ortho (12d) and meta (12e) positions. Although tolerated, a modest decrease in potency was observed with simple alkyl substitution at the meta (12f) and para (12g) positions. Introduction of H-bond donors were detrimental when placed at the ortho (12h) and para (12k) positions, but were tolerated at the meta position (12i, 12j and 12l), with the hydroxyethyl group (12j) providing modestly increased inhibition. However, the greatest increase in potency was observed for benzyl substitution at the meta position (12m), which resulted in a two-fold enhancement.
Table 2.
Optimization of distal aryl ring substation for inhibitor 12a
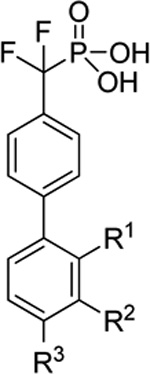 | ||||
|---|---|---|---|---|
| R1 | R2 | R3 | Ki (µM) | |
| 12 | H | H | H | 120 ± 7 |
| 12a | CN | H | H | 375 ± 30 |
| 12b | H | CN | H | 400 ± 48 |
| 12c | H | H | CN | 617 ± 45 |
| 12d | H | O-i-Pr | H | 200 ± 1 |
| 12e | H | H | O-i-Pr | 231 ± 6 |
| 12f | H | i-Pr | H | 132 ± 3 |
| 12g | H | H | i-Pr | 185 ± 33 |
| 12h | NHMs | H | H | 673 ± 5 |
| 12i | H | NHMs | H | 113 ± 1 |
| 12j | H | CH(OH)CH3 | H | 88 ± 3 |
| 12k | H | H | CH(OH)CH3 | 450 ± 183 |
| 12l | H | NHPh | H | 112 ± 9 |
| 12m | H | Bn | H | 73 ± 3 |
Ki values were determined using at least 2 independent measurements.
Further modification of the benzyl substituted inhibitor 12m was next performed (Figure 3). The introduction of halogens on the benzyl group was found to be beneficial for the potency of the inhibitors (12n and 12o), with the 3,4-dichlorobenzyl group being optimal (12p). The hydroxyl group, which was previously observed to be well tolerated at the benzylic position (see 12j, Table 2), increased the solubility of the inhibitor with only a moderate reduction in potency (12q). Replacing the difluoromethylphosphonic acid group with a α-hydroxyphosphonic acid group maintained the desired potency (12r).
Figure 3.

Additional analogs based upon inhibitor 12m.
We next explored the effect of combining the hydroxyl groups in inhibitors 12q and 12r by the preparation and assay of the four possible stereoisomers, 12s–12v. While the potency is not affected by the stereochemistry of the distal biarylmethanol (12s versus 12t), the (S)-configuration at the α-hydroxyphosphonic acid is crucial for inhibitory activity (see 12u and 12v). Further evidence of the importance of this hydroxyl group was the dramatic loss in potency with its absence as demonstrated by compound 12w.
Inhibitor Selectivity
There is high structural homology among PTP active sites, which often leads to difficulty in achieving high selectivity. We therefore tested the best inhibitors from the two biaryl series, inhibitors 11m, 12s and 12t, against a panel of tyrosine and dual-specificity phosphatases (Table 3). Notably, our most potent inhibitors from the 1,4-biphenyl series 12s and 12t displayed greater than 20-fold selectivity against all phosphatases tested.
Table 3.
Selectivity of most potent inhibitors against a panel of PTPsa
| STEP | TC-Ptp | CD45 | LAR | MKP5 | ||
|---|---|---|---|---|---|---|
| 11m | Ki, µM | 20 ± 3 | 71 ± 2 | >500 | >900 | 150 ± 20 |
| selectivity | -- | 3.6 | >25 | >45 | 7.5 | |
| 12s | Ki, µM | 8.9 ± 1.2 | 164 ± 9 | 340 ± 40 | 360 ± 30 | 340 ± 45 |
| selectivity | -- | 18 | 38 | 40 | 41 | |
| 12t | Ki, µM | 7.8 ± 0.7 | 170 ± 20 | 270 ± 20 | 390 ± 110 | >500 |
| selectivity | -- | 22 | 35 | 50 | >65 |
Ki values were determined using at least 2 independent measurements.
STEP Inhibition in Neuronal Cultures
The most promising inhibitors, 12s and 12t, were also evaluated for their ability to inhibit STEP in rat cortical neurons by monitoring the phosphorylation levels of the known STEP substrates, GluN2B, Pyk2 and ERK1/2.7,8,10,11 Clear increases in the phosphorylation levels of each of the substrates were observed for inhibitor 12s with more modest effects observed for 12t (Figure 4).
Scheme 4.

Synthesis of Diarylmethane-based Boron Reagents 22 and 24a
aReagents: (a) PhMgBr, Et2O or 1-bromo-3-iodobenzene, n-butyllithium, THF; aldehyde; (b) Et3SiH, F3CCOOH, CH2Cl2; (c) Pd(dppf)Cl2, KOAc, B2pin2, DMSO; (d) n-butyllithium, THF; 3,4-dichlorobenzaldehyde.
Figure 4.

Rat cortical neurons were treated with vehicle or 12s (a) or 12t (b) (concentrations of 0.1, 1 or 10 µM) for 1 h and analyzed by Western blotting. (*p<0.05; **p<0.01; ***p<0.001 one-way ANOVA, Dunnett’s post hoc). Data represent the phospho-signal normalized to the total protein signal and GAPDH ± s.e.m. (n = 3–5 each group).
Blood-Brain Barrier Permeability
Given the promising cell data, the best compound, 12s, was evaluated for its ability to passively permeate the blood-brain barrier (BBB) using the well validated parallel artificial membrane permeability assay (PAMPA) technique (Pion, Inc., Billerica, MA).22 Although the compound can cross cell membranes, as evidenced in the above cell data, compound 12s did not possess the ability to passively cross the BBB in this model system (data in SI). As such, BBB permeability remains a real challenge for future work. The highly polar nature of the α-hydroxyphosphonic acid motif is likely to be one of the key factors limiting BBB permeability. Replacement of this motif with less polar, non-hydrolyzable phosphate mimetics might overcome this problem.15 Alternatively, effective prodrug strategies have also been developed to mask polar functionality in order to enable BBB permeability. 23
CHEMISTRY
The biaryl difluoromethylphosphonic acid (DFMP) inhibitors could be conveniently prepared by relying on the Suzuki-Miyaura cross-coupling reaction (Scheme 1). The commercial availability of many diverse arylboronic acids enabled rapid access to the initial set of biaryl inhibitors. However, further optimization of the inhibitor structures required that we prepare the requisite more complex arylboronic acid inputs.
Scheme 1.

Inhibitor Synthesis through Suzuki-Miyaura Cross-Coupling.
aReagents: (a) ArBLn, PdCl2, CyJohnPhos, K2CO3, dioxane/H2O; (b) ArBLn, Pd(PPh3)4, Na2CO3, DME/EtOH/H2O.
For the preparation of 3-biphenyl inhibitors 11g, 11i–j, 11l–11m and 11o, the synthesis of 2-hydroxyphenylboronic acids was required (Scheme 2). Selective ortho-bromination of the 2-alkylphenols 15a–e gave the aryl bromides 16a–e,24 upon which ortholithiation, treatment with B(OMe)3 and aqueous workup provided the desired arylboronic acids 17a–e.25
Scheme 2.

Synthesis of Arylboronic Acids 17a
aReagents: (a) N-bromosuccinimide, N,N-diisopropylamine, CH2Cl2; (b) n-butyllithium, Et2O; B(OMe)3; aqueous HCl.
Starting phenols 15f and 15g were not commercially available and were synthesized from 2-bromophenol in a three-step sequence (Scheme 3). Dilithiation was followed by addition of the desired ketones resulting in benzylic alcohols, which upon treatment with Et3SiH under acidic conditions afforded the 2-alkylphenols 15. The phenols were then converted to the aryltrifluoroborates 18 utilizing an iridium-catalyzed one-pot silyl-directed ortho borylation approach.26
Scheme 3.

Synthesis of Aryltrifluoroborates 18a
aReagents: (a) n-butyllithium, Et2O; (b) ketone; aqueous NH4Cl; (c) Et3SiH, F3CCOOH, CH2Cl2; (d) Et2SiH2, [Ir(cod)Cl]2, benzene; B2pin2, HBpin, [Ir(cod)Cl]2, dtbpy, THF; aqueous KHF2.
The synthesis of the arylboron reagents needed to prepare inhibitors 12m–p and 12r started with the addition of PhMgBr or in situ generated 3-bromophenyllithium to aldehydes 19 to give diarylmethanols 20 (Scheme 4). Acid mediated reductive removal of the hydroxyl group to give 21 was followed by Miyaura borylation reactions to afford boronic esters 22.27 Alternatively, boronic acid 24 was conveniently prepared from the previously reported intermediate 23.28
The α-hydroxymethylphosphonic acid inhibitors 11o and 12r were also prepared by Suzuki cross-coupling reaction (Scheme 5). Ketones 26 and 28 were first obtained by cross coupling ketophosphonic acids 2529 and 27 with arylboronic acids 17e and 22d, respectively. Subsequent reduction then led to the α-hydroxymethylphosphonic acid inhibitors 11o and 12r.
Scheme 5.

Synthesis of α-Hydroxymethylphosphonic Acid Inhibitors 11o and 12ra
aReagents: (a) Pd(PPh3)4, Na2CO3, DME/EtOH/H2O; (b) NaBH4, MeOH.
The four stereoisomeric α-hydroxymethylphosphonic acids 12s to 12v were prepared from the enantiomerically pure α-hydroxymethylphosphonic acids 30 and the enantiomerically enriched boronic esters 32 by Suzuki-Miyaura cross-coupling (Scheme 6). The synthesis of the enantiomerically pure phosphonic acids 30 started with the addition of tris[(1R,2S,5R)-menth-2-yl]phosphite to 4-bromobenzaldehyde to give a mixture of diastereomers, 29a and 29b, which were separated by recrystallization according to literature procedures for analogous compounds.30 Removal of the menthyl groups by treatment with TMSCl and NaI afforded phosphonic acids 30a and 30b. The absolute stereochemistry of the α-hydroxyphosphonic acids was confirmed by chemical correlation through hydrodebromination of 30b to the corresponding α-hydroxy-phenylmethylphosphonic acid, for which the absolute configuration had previously been determined.31 The catalytic asymmetric addition of a 3,4-dichlorophenylzinc reagent to 3-bromobenzaldehyde using each enantiomer of 3-exo-(morpholino)isoborneol (MIB)32 gave the enantiomerically enriched diarylmethanols 31a and 31b in 90% ee. Subsequent Miyaura borylation led to the boronic esters 32a and 32b.33 Final Suzuki-Miyaura cross-coupling afforded the four diastereomeric inhibitors, 12s–v.
Scheme 6.

Synthesis of the Four Stereoisomeric α-Hydroxymethylphosphonic Acid Inhibitors 12s to 12va
aReagents (a) P(OMnt)3, TMSCl, 0 °C; separation of diastereomers by recrystallization; (b) TMSCl, NaI, CH3CN; (c) i. n-butyllithium, tBuOMe; ii. ZnCl2; iii. n-butyllithium; iv. tetraethylethylenediamine, toluene; v. (+)-MIB or (−)-MIB (10% mol); vi. 3-bromobenzaldehyde; (d) Pd(dppf)Cl2, KOAc, B2Pin2, DMSO; (e) Pd(PPh3)4, Na2CO3, DME/EtOH/H2O.
CONCLUSION
Although high STEP activity has been observed in many neuropsychiatric disorders, such as Alzheimer’s disease, there have been no selective and potent inhibitors reported for potential treatment and study of these diseases. This report describes the first dedicated effort for the identification of selective small-molecule mechanism-based inhibitors of STEP. A library of low molecular weight O-aryl and -heteroaryl phosphate fragments were screened, which identified both the 4- and the 3-biaryl scaffolds as promising templates for inhibitor development. After conversion to inhibitors, SAR of the scaffolds was explored to identify potent inhibitors from each series, with the most potent inhibitors (11m, 12s and 12t) all showing promising selectivity over other human phosphatases tested. Importantly, the most selective inhibitors 12s and 12t were able to inhibit STEP in rat cortical neurons as indicated by the significant increase in phosphorylation levels of STEP substrates. Future efforts will focus on increasing the inhibitory activity and enhancing the blood-brain barrier permeability of the STEP inhibitors.
EXPERIMENTAL SECTION
Previously reported substrates and inhibitors
Substrates 1–1012a and inhibitors 1120 and 1212b have been previously reported.
General Methods
Unless otherwise noted, all reagents were obtained from commercial suppliers and used without further purification. Tetrahydrofuran (THF), dioxane, CH2Cl2, and diethyl ether were passed through a column of activated alumina (type A2, 12 × 32, Purify Co.) under nitrogen pressure immediately prior to use. All 1H, 19F and 31P NMR spectra were obtained at room temperature on a Bruker AVB-400 or AVB-500 spectrometer. NMR chemical shifts are reported in ppm relative to TMS (0.00), CHCl3 (7.26), or CH3OH (3.31) for 1H, trifluoroacetic acid (−76.55) for 19F, and H3PO4 (0.00) for 31P. Mass spectrometry (HRMS, ESI) are reported in m/z. Chromatography was performed with SiliCycle SiliaFlash P60 230–400 mesh silica gel, or by utilizing a Biotage SP1 Flash Purification System (Biotage No. SP1-B1A) or a Teledyne Isco CombiFlash Rf System. Reversed-phase purifications were conducted with a Teledyne Isco CombiFlash Rf System equipped with HP C18 Gold cartridges. Product yields are not optimized. Enzymatic assays were carried out on a BioTek Synergy 2 Multi-Mode Microplate Reader. All of the tested substrates and inhibitors displayed ≥95% purity as determined by HPLC or UPLC. BBB PAMPA studies were conducted by Pion, Inc (Billerica, MA).
General procedures for determination of substrate KM
To facilitate screening in a high-throughput fashion, 96-well plates were used with reaction volumes of 100 µL. 30 µL of water was added to each well, followed by 5 µL of 20× buffer (1.0 M imidazole, 1.0 M NaCl, 0.2% Triton-X 100, pH 7.0), 10 µL of 10× DTT (50 mM; 5 mM in assay), 40 µL of 2-amino-6-mercapto-7-methylpurine riboside (MESG) solution (1 mM; 400 µM in assay), and 5 µL of purine nucleotide phosphorylase (PNP) solution (20 U/mL; 1 U/mL in assay). 5 µL of STEP phosphatase (2 µM; 100 nM in assay) was added and the 96-well assay plate was incubated at 27 °C for 3 min. The coupled assay was started by addition of 5 µL of the appropriate substrate dilution in DMSO (typically: 3.00, 1.20, 0.480, 0.192, 0.077, 0.031, 0.012, 0 mM in assay). The plate was then immediately placed into a spectrophotometric plate reader and 20 minutes of kinetic data was obtained (360 nm, 27 °C). The initial rate data collected was used for Michaelis-Menten kinetic analysis where the KM was obtained using the substrate-velocity data with the equation V = (Vmax *[S])/(KM +[S]).
General procedures for determination of inhibitor Ki
Reaction volumes of 100 µL were used in 96-well plates. 65 µL of water was added to each well, followed by 5 µL of 20× buffer (1.0 M imidazole, 1.0 M NaCl, 0.2% Triton-X 100, pH 7.0), 10 µL of 10× DTT (50 mM; 5 mM in assay), 5 µL of STEP phosphatase (2 µM; 100 nM in assay) and 5 µL of the appropriate inhibitor dilution in DMSO (with two- or three-fold serial dilutions). The assay plate was incubated for 5 min at 27 °C at which point the reaction was started by addition of 10 µL of a 10× pNPP substrate (5 mM; 500 µM in assay). The plate was then immediately placed into a spectrophotometric plate reader and 20 minutes of kinetic data was obtained (405 nm, 27 °C). The initial rate data collected was used for determination of Ki values. For Ki determination, the kinetic values were obtained directly from nonlinear regression of substrate-velocity curves in the presence of various concentrations of inhibitor. Assays were run in at least duplicate using the same inhibitor stock solutions.
For the selectivity assays, the KM of pNPP toward each of the enzymes was determined in the above assay buffer and used for data analysis. For the assays with the dual-specificity MKP5, due to poor turnover of pNPP, the chromogenic substrate 6,8-difluoro-4-methylumbelliferyl phosphate (DiFMUP) was used instead. In addition, the assay buffer used was a 10× buffer (0.2 M Tris-HCl, 0.2% Triton-X 100, pH 8.0) and 5 mM DTT was used as in the other assays.
Cell culture and Western blotting
The Yale University Institutional Animal Care and Use Committee approved all procedures. Primary cortical neurons were isolated from Sprague Dawley rat embryos (E18) (Charles River Laboratories, Wilmington, MA) as previously described.34 Briefly, cells were dissociated with trypsin, resuspended in Hanks' Balanced Salt Solution and then plated on poly-D-lysine-coated plates (1 × 106 cells/well) in Neurobasal media supplemented with 2% B27 (Invitrogen, San Diego, CA). Neurons were allowed to grow for 18–21 days at 37°C in a CO2 incubator. Compounds to be tested were diluted in DMSO and added to the media at final concentrations of 0, 0.1, 1 and 10 µM and incubated for 1 h at 37°C in a CO2 incubator. The percentage of DMSO remained constant (0.1%) in all wells. After incubation, neurons were lysed in RadioImmuno Precipitation Assay (RIPA) buffer supplied with protease inhibitor cocktail (Roche Applied Science, Indianapolis, IN) and phosphatase inhibitors (NaF and Na3VO4). Protein concentration of the samples were estimated using the BCA assay kit (Pierce, Thermo Scientific).
Samples were prepared and resolved by SDS-PAGE, transferred to nitrocellulose membrane and incubated with phospho-specific antibodies (anti-phospho-pY204/187 ERK1/2, anti-pY402 Pyk2, anti-pY1472 GluN2B) or total protein antibodies, Pyk2, anti-GluN2B, anti-ERK1/2) overnight at 4°C. All antibodies used in this study are listed in the supporting information. Membranes were washed and incubated in peroxidase-conjugated secondary antibodies (GE Healthcare, Waukesha, WI). The immunoreactivity was visualized using a Chemiluminescent substrate kit (Pierce Biotechnology, Rockford, IL) and detected using a G:BOX with the image program GeneSnap (Syngene, Cambridge, UK). All densitometric quantifications were performed using the Image J (NIH) Software.
All data are presented as means ± SEM. Differences among multiple groups were evaluated using one-way ANOVA with Dunnett’s post hoc test using Graph Pad Prism 6 software. For all analyses, a p value of <0.05 indicated a statistically significant difference.
General procedure for selective o-bromination of phenols (A)
Selective ortho-bromination of alkylphenols was accomplished via a modified literature procedure.24 A solution of N-bromosuccinimide (1.0 equiv) in CH2Cl2 (0.2 M), was added dropwise via cannula to a stirred solution of the phenol in CH2Cl2 (0.5 M) containing catalytic N,N-diisopropylamine (0.1 equiv); the reaction flask having been placed in an ambient temperature water bath. The reaction mixture was stirred for 90 min at which point it was acidified with 1 N HCl to pH < 2.0. The reaction mixture was diluted with 1 volume of water, the layers were separated, and the organic layer was dried over MgSO4. The volatile material was removed with a rotary evaporator under vacuum to afford the crude products, generally as yellow to colorless liquids, which were typically used without further purification.
General procedure for boronic acid formation from o-bromophenols (B)
Borylation of the phenols was accomplished following a literature procedure.25 Butyllitium (2.5 M in hexanes, 2.15 equiv) was added dropwise via syringe to a stirred solution of the starting ortho-bromophenol in diethyl ether (0.2 M) in a dry ice-acetone cold temperature bath. The cold bath was removed and the reaction solution was allowed to warm to ambient temperature in air for 2.5 h. The reaction flask was resuspended in the dry ice-acetone cold temperature bath. After 10 min stirring in the cold bath, a solution of trimethyl borate (1.67 equiv, 1.5 M in diethyl ether) was added. After stirring in the cold bath for 30 min, the reaction flask was stirred at ambient temperature for 18 h. The reaction mixture was quenched with 0.5 volumes of 1 N HCl, the layers were separated and the organic layer was dried over MgSO4. The volatile material was removed with a rotary evaporator under vacuum to afford the crude products as orange to brown viscous oils or sticky solids. Although crude products show multiple sets of peaks in the aromatic region (potentially the hydrolytic boroxine species), the crude products show only one major peak on LCMS which corresponds to the mass of the boronic acids, and the products were used without further purification.
General procedure for Suzuki-Miyaura Cross-Coupling (C)
A 1 dram oven dried vial was charged with a stir bar, the appropriate aryl bromide (1.0 equiv), organoboron species (1.5 equiv), potassium carbonate (5 equiv), palladium (II) chloride or palladium (II) acetate (5 mol%) and (2-biphenyl)dicyclohexylphosphine (10 mol%). After addition of all reagents, solvent (4:1 dioxane:water) was added to the vial (0.25 M in aryl bromide). The vial was sealed with a screw top containing a Teflon septum and placed in a pre-heated heating block to 80 °C to stir 18 h with vigorous stirring. The vial was then removed from the heating block and allowed to cool to room temperature, followed by addition of 0.3 volumes of 10 N HCl open to air. The reaction mixture was diluted with 1 volume of water and 1 volume of methanol, filtered through a Kimwipe and purified by reversed-phase gradient column chromatography (5% to 100% acetonitrile in water with 0.1% trifluoroacetic acid). Volatile components were removed, and the resulting water solutions were lyophilized to afford the products.
General procedure for Suzuki-Miyaura Cross-Coupling (D)
A 1 dram oven dried vial was charged with a stir bar, the appropriate aryl bromide (1.0 equiv), organoboron species (1.5 equiv), sodium carbonate (6 equiv), and tetrakis(triphenylphosphine)palladium(0) (10 mol%). After addition of all reagents, solvent (4:1:1 dimethoxyethane:ethanol:water) was added to the vial (0.10–0.25 M in aryl bromide). The vial was sealed with a screw top containing a Teflon septum and placed in a pre-heated heating block to 80 °C to stir for 4 to 18 h with vigorous stirring. The vial was then removed from the heating block and allowed to cool to room temperature, followed by addition of 0.3 volumes of 10 N HCl open to air. The reaction mixture was diluted with 1 volume of water and 1 volume of methanol, filtered through a Kimwipe and purified by reversed-phase gradient column chromatography (5% to 100% acetonitrile in water with 0.1% trifluoroacetic acid). Volatile components were removed, and the resulting water solutions were lyophilized to afford the products.
((3-Bromophenyl)difluoromethyl)phosphonic Acid (13)
Iodotrimethylsilane (5.22 mL, 36.6 mmol, 2.2 equiv) was added to a stirred solution of diethyl ((3-bromophenyl)difluoromethyl)phosphonate21b (5.71 g, 16.6 mmol), in CH2Cl2 (50 mL, 0.3 M). The reaction solution was stirred at ambient temperature for 14 h. The volatile components were then removed by a rotary evaporator under vacuum. Sodium hydroxide (664 mg, 16.6 mmol) in methanol (20 mL) was added to the resulting residue, and this solution was stirred at ambient temperature for 1 h, allowing the mono-sodium salt of the desired compound to precipitate from the solution. The solvent was removed by filtration. LCMS showed the presence of an undesired byproduct in the collected crude product, thus it was purified via reversed-phase gradient column chromatography (5% to 100% acetonitrile in water with 0.1% trifluoroacetic acid buffer). Volatile components were removed, and the resulting water solution was lyophilized to afford the product as an off-white powder (2.75 g, 9.44 mmol, 57%). 1H NMR (400 MHz, CD3OD) δ 7.41 (t, J = 7.9 Hz, 1H), 7.58 (d, J = 7.8 Hz, 1H), 7.67 (d, J = 8.0 Hz, 1H), 7.74 (s, 1H). 19F NMR (376 MHz, CD3OD) δ −110.13 (d, JFP = 110.2 Hz). 31P NMR (162 MHz, CD3OD) δ 6.76 (br). HRMS (ESI) m/z : calcd 284.9133 (M − H+); found: 284.9129.
(Difluoro(4'-methyl-[1,1'-biphenyl]-3-yl)methyl)phosphonic Acid (11a)
For the Suzuki coupling, general procedure C was followed with 51 mg (0.375 mmol) of 4-methyl-phenylboronic acid, and 71 mg (0.25 mmol) of aryl bromide 13. The procedure yielded 42 mg (56%) of the desired compound as a white powder. 1H NMR (400 MHz, CD3OD) δ 2.38 (s, 3H), 7.28 (d, J = 8.0 Hz, 2H), 7.49–7.60 (m, 4H), 7.72 (d, J = 7.2 Hz, 1H), 7.82 (s, 1H). 19F NMR (376 MHz, CD3OD) δ −109.69 (d, J = 112.9 Hz). 31P NMR (162 MHz, CD3OD) δ 7.05 (t, J = 113.0 Hz). HRMS (ESI) m/z : calcd 297.0498 (M − H+); found: 297.0500.
(Difluoro(4'-isopropyl-[1,1'-biphenyl]-3-yl)methyl)phosphonic Acid (11b)
For the Suzuki coupling, general procedure C was followed with 62 mg (0.375 mmol) of 4-isopropylphenylboronic acid, and 71 mg (0.25 mmol) of aryl bromide 13. The procedure yielded 45 mg (55%) of the desired compound as a yellow solid. 1H NMR (400 MHz, CD3OD) δ 1.28 (d, J = 6.9 Hz, 6H), 2.95 (hept, J = 6.9 Hz, 1H), 7.33 (d, J = 8.3 Hz, 2H), 7.48–7.60 (m, 4H), 7.73 (d, J = 7.3 Hz, 1H), 7.83 (s, 1H).19F NMR (376 MHz, CD3OD) δ −109.67 (d, J = 112.9 Hz). 31P NMR (162 MHz, CD3OD) δ 7.11 (br t).HRMS (ESI) m/z : calcd 325.0811 (M − H+); found: 325.0816.
(Difluoro(3'-methyl-[1,1'-biphenyl]-3-yl)methyl)phosphonic Acid (11c)
For the Suzuki coupling, general procedure C was followed with 51 mg (0.375 mmol) of 3-methyl-phenylboronic acid, and 71 mg (0.25 mmol) of aryl bromide 13. The procedure yielded 51 mg (68%) of the desired compound as a colorless oil. 1H NMR (400 MHz, CD3OD) δ 2.40 (s, 3H), 7.18 (d, J = 7.5 Hz, 1H), 7.32 (t, J = 7.6 Hz, 1H), 7.41 (d, J = 7.7 Hz, 1H), 7.45 (s, 1H), 7.53 (t, J = 7.7 Hz, 1H), 7.58 (d, J = 8.0 Hz, 1H), 7.71 (d, J = 7.5 Hz, 1H), 7.83 (s, 1H). 19F NMR (376 MHz, CD3OD) δ −109.64 (d, J = 113.4 Hz). 31P NMR (162 MHz, CD3OD) δ 7.09 (t, J = 113.3 Hz). HRMS (ESI) m/z : calcd 297.0498 (M − H+); found: 297.0498.
(Difluoro(3'-isopropyl-[1,1'-biphenyl]-3-yl)methyl)phosphonic Acid (11d)
For the Suzuki coupling, general procedure C was followed with 62 mg (0.375 mmol) of 3-isopropylphenylboronic acid, and 71 mg (0.25 mmol) of aryl bromide 13. The procedure yielded 13 mg (16%) of the desired compound as a yellow viscous oil. 1H NMR (400 MHz, CD3OD) δ 1.29 (d, J = 7.0 Hz, 6H), 2.97 (hept, J = 6.9 Hz, 1H), 7.25 (d, J = 7.5 Hz, 1H), 7.37 (t, J = 7.6 Hz, 1H), 7.43 (d, J = 7.6 Hz, 1H), 7.48 (s, 1H), 7.54 (t, J = 7.7 Hz, 1H), 7.59 (d, J = 7.8 Hz, 1H), 7.73 (d, J = 7.5 Hz, 1H), 7.83 (s, 1H). 19F NMR (376 MHz, CD3OD) δ −109.67 (d, JFP = 113.4 Hz). 31P NMR (162 MHz, CD3OD) δ 7.16 (br t). HRMS (ESI) m/z : calcd 325.0811 (M − H+); found: 325.0805.
(Difluoro(2'-hydroxy-[1,1'-biphenyl]-3-yl)methyl)phosphonic Acid (11e)
For the Suzuki coupling, general procedure C was followed with 52 mg (0.375 mmol) of 2-hydroxy-phenylboronic acid, and 71 mg (0.25 mmol) of aryl bromide 13. The procedure yielded 35 mg (47%) of the desired compound as an off white hygroscopic powder. 1H NMR (400 MHz, CD3OD) δ 6.87–6.94 (m, 2H), 7.17 (td, J = 7.7, 1.6 Hz, 1H), 7.27 (dd, J = 7.5, 1.7 Hz, 1H), 7.48 (t, J = 7.7 Hz, 1H), 7.54 (d, J = 7.6 Hz, 1H), 7.71 (d, J = 7.5 Hz, 1H), 7.79 (s, 1H). 19F NMR (376 MHz, CD3OD) δ −109.53 (d, JFP = 113.8 Hz). 31P NMR (162 MHz, CD3OD) δ 7.24 (t, JPF = 113.9 Hz). HRMS (ESI) m/z : calcd 299.0290 (M − H+); found: 299.0285.
(Difluoro(2'-methoxy-[1,1'-biphenyl]-3-yl)methyl)phosphonic Acid (11f)
For the Suzuki coupling, general procedure C was followed with 57 mg (0.375 mmol) of 2-methoxy-phenylboronic acid, and 71 mg (0.25 mmol) of aryl bromide 13. The procedure yielded 35 mg (45%) of the desired compound as a yellow solid. 1H NMR (400 MHz, CD3OD) δ 3.79 (s, 3H), 7.02 (t, J = 7.4 Hz, 1H), 7.08 (d, J = 8.2 Hz, 1H), 7.30 (d, J = 9.1 Hz, 1H), 7.35 (d, J = 7.9 Hz, 1H), 7.47 (t, J = 7.7 Hz, 1H), 7.55 (d, J = 8.0 Hz, 1H), 7.62 (d, J = 7.7 Hz, 1H), 7.73 (s, 1H). 19F NMR (376 MHz, CD3OD) δ −109.54 (d, JFP = 114.3 Hz). 31P NMR (162 MHz, CD3OD) δ 7.18 (t, JPF = 114.0 Hz). HRMS (ESI) m/z : calcd 313.0447 (M + H+); found: 313.0445.
(Difluoro(2'-hydroxy-3'-isopropyl-[1,1'-biphenyl]-3-yl)methyl)phosphonic Acid (11g)
General procedure A was followed for the ortho-bromination of 2-isopropylphenol starting with 1.35 mL of the phenol (10.0 mmol). The procedure yielded 1.49 g (69%) of the brominated phenol, 16b, as a liquid which was used without purification. This bromophenol (495 mg) was converted to the boronic acid through general procedure B, yielding 315 mg (76%) of a very viscous brown oil with molecular mass corresponding to the boronic acid, 17b. The crude boronic acid was used without further purification. For the final Suzuki coupling, general procedure C was followed with 136 mg (0.75 mmol) of 17b and 142 mg (0.50 mmol) of aryl bromide 13. The procedure yielded 43 mg (25%) of the desired compound as a white, hygroscopic powder. 1H NMR (400 MHz, CD3OD) δ 1.26 (d, J = 6.9 Hz, 6H), 3.38 (hept, J = 6.9 Hz, 1H), 6.92 (t, J = 7.6 Hz, 1H), 7.04 (dd, J = 7.6, 1.7 Hz, 1H), 7.19 (dd, J = 7.8, 1.7 Hz, 1H), 7.52 (t, J = 7.6 Hz, 1H), 7.57 (d, J = 7.1 Hz, 1H), 7.62 (d, J = 7.5 Hz, 1H), 7.72 (s, 1H). 19F NMR (376 MHz, CD3OD) δ −109.81 (d, JFP = 114.0 Hz). 31P NMR (162 MHz, CD3OD) δ 7.27 (t, JPF = 113.5 Hz). HRMS (ESI) m/z : calcd 341.0760 (M − H+); found: 341.0755.
(Difluoro(2'-hydroxy-5'-isopropyl-[1,1'-biphenyl]-3-yl)methyl)phosphonic Acid (11h)
General procedure A was followed for the ortho-bromination of 4-isopropylphenol starting with 1.36 g of the phenol (10.0 mmol). To remove the dibrominated byproduct, the crude product was purified by column chromatography (9:1 hexanes/ethyl acetate; product Rf: 0.33) to afford the product as a yellow oil (760 mg, 35%). This bromophenol (625 mg) was converted to the boronic acid through general procedure B, yielding 437 mg (84%) of an off white solid with molecular mass corresponding to the boronic acid. The crude boronic acid was used without further purification. For the final Suzuki coupling, general procedure C was followed with 68 mg (0.375 mmol) of this boronic acid and 71 mg (0.25 mmol) of aryl bromide 13. The procedure yielded 34 mg (40%) of the desired compound as a brown, hygroscopic powder. 1H NMR (400 MHz, CD3OD) δ 1.24 (d, J = 6.9 Hz, 6H), 2.87 (hept, J = 6.9 Hz, 1H), 6.83 (d, J = 8.3 Hz, 1H), 7.06 (dd, J = 8.3, 2.3 Hz, 1H), 7.12 (d, J = 2.3 Hz, 1H), 7.48 (t, J = 7.7 Hz, 1H), 7.54 (d, J = 7.8 Hz, 1H), 7.70 (d, J = 7.5 Hz, 1H), 7.78 (s, 1H). 19F NMR (376 MHz, CD3OD) δ −109.48 (d, JFP = 114.1 Hz). 31P NMR (162 MHz, CD3OD) δ 7.39 (t, JPF = 114.7 Hz). HRMS (ESI) m/z : calcd 341.0760 (M − H+); found: 341.0756.
((3'-Ethyl-2'-hydroxy-[1,1'-biphenyl]-3-yl)difluoromethyl)phosphonic Acid (11i)
General procedure A was followed for the ortho-bromination of 2-ethylphenol starting with 1.20 mL of the phenol (10.0 mmol). The procedure yielded 1.86 g (93%) of the brominated phenol, 16a, as a yellow liquid which was used without purification. This bromophenol (1.86 g) was converted to the boronic acid through general procedure B, yielding 870 mg (57%) of an orange sticky solid with molecular mass corresponding to the boronic acid, 17a. The crude boronic acid was used without further purification. For the final Suzuki coupling, general procedure C was followed with 62 mg (0.375 mmol) of 17a and 71 mg (0.25 mmol) of aryl bromide 13. The procedure yielded 14 mg (17%) of the desired compound as a white, hygroscopic powder. 1H NMR (400 MHz, CD3OD) δ 1.23 (t, J = 7.5 Hz, 3H), 2.70 (q, J = 7.5 Hz, 2H), 6.89 (t, J = 7.6 Hz, 1H), 7.06 (dd, J = 7.6, 1.7 Hz, 1H), 7.12 (dd, J = 7.5, 1.7 Hz, 1H), 7.51 (t, J = 7.6 Hz, 1H), 7.57 (d, J = 7.7 Hz, 1H), 7.63 (d, J = 7.4 Hz, 1H), 7.73 (s, 1H). 19F NMR (376 MHz, CD3OD) δ −109.75 (d, JFP = 113.6 Hz). 31P NMR (162 MHz, CD3OD) δ 7.25 (t, JPF = 113.6 Hz). HRMS (ESI) m/z : calcd 327.0603 (M − H+); found: 327.0599.
((3'-(tert-Butyl)-2'-hydroxy-[1,1'-biphenyl]-3-yl)difluoromethyl)phosphonic Acid (11j)
General procedure A was followed for the ortho-bromination of 2-tert-butylphenol starting with 1.52 mL of the phenol (10.0 mmol). The procedure yielded 2.30 g (99%) of the brominated phenol, 16c, as a light yellow solid which was used without purification. This bromophenol (2.30 g) was converted to the boronic acid through general procedure B, yielding 1.09 g (56%) of a very viscous brown oil with molecular mass corresponding to the boronic acid, 17c. The crude boronic acid was used without further purification. For the final Suzuki coupling, general procedure C was followed with 73 mg (0.375 mmol) of 17c and 71 mg (0.25 mmol) of aryl bromide 13. The procedure yielded 11 mg (12%) of the desired compound as a white, hygroscopic powder. 1H NMR (400 MHz, CD3OD) δ 1.44 (s, 9H), 6.87 (t, J = 7.7 Hz, 1H), 7.02 (d, J = 7.5 Hz, 1H), 7.26 (d, J = 7.7 Hz, 1H), 7.50–7.63 (m, 3H), 7.68 (s, 1H). 19F NMR (376 MHz, CD3OD) δ −110.00 (d, JFP = 113.0 Hz). 31P NMR (162 MHz, CD3OD) δ 6.96 (t, JPF = 113.0 Hz). HRMS (ESI) m/z : calcd 355.0916 (M − H+); found: 355.0913.
((3'-Cyclobutyl-2'-hydroxy-[1,1'-biphenyl]-3-yl)difluoromethyl)phosphonic Acid (11k)
For the Suzuki coupling, general procedure D was followed with 90 mg (0.35 mmol) of the potassium trifluoroborate 18a and 100 mg (0.35 mmol) of aryl bromide 13. The procedure yielded 18 mg (15%) of the desired compound as a white hygroscopic powder. 1H NMR (400 MHz, CD3OD) δ 1.75–1.93 (m, 1H), 1.96–2.23 (m, 3H), 2.32–2.43 (m, 2H), 3.82 (p, J = 8.6 Hz, 1H), 6.92 (t, J = 7.5 Hz, 1H), 7.05 (dd, J = 7.6, 1.7 Hz, 1H), 7.20 (dd, J = 7.4, 1.6 Hz, 1H), 7.49 (t, J = 7.7 Hz, 1H), 7.54 –7.61 (m, 2H), 7.71 (s, 1H). 19F NMR (376 MHz, CD3OD) δ −109.20 (d, JFP = 109.4 Hz). 31P NMR (162 MHz, CD3OD) δ 6.97 (br t). HRMS (ESI) m/z : calcd 353.0760 (M − H+); found: 353.0754.
((3'-Cyclopentyl-2'-hydroxy-[1,1'-biphenyl]-3-yl)difluoromethyl)phosphonic Acid (11l)
General procedure A was followed for the ortho-bromination of 2-cyclopentylphenol starting with 406 mg of the phenol (2.5 mmol). The procedure yielded 590 mg (98%) of the brominated phenol, 16d, as a liquid which was used without purification. This bromophenol (590 mg) was converted to the boronic acid through general procedure B, yielding 420 mg (83%) of a very viscous brown oil with molecular mass corresponding to the boronic acid, 17d. The crude boronic acid was used without further purification. For the final Suzuki coupling, general procedure C was followed with 77 mg (0.375 mmol) of 17d and 71 mg (0.25 mmol) of aryl bromide 13. The procedure yielded 10 mg (11%) of the desired compound as an off white hygroscopic powder. 1H NMR (400 MHz, CD3OD) δ 1.56–1.78 (m, 4H), 1.78–1.92 (m, 2H), 2.01–2.12 (m, 2H), 3.33–3.46 (m, 1H), 6.91 (t, J = 7.6 Hz, 1H), 7.03 (dd, J = 7.6, 1.7 Hz, 1H), 7.20 (dd, J = 7.6, 1.7 Hz, 1H), 7.52 (t, J = 7.7 Hz, 1H), 7.57 (d, J = 7.7 Hz, 1H), 7.62 (d, J = 7.5 Hz, 1H), 7.72 (s, 1H). 19F NMR (376 MHz, CD3OD) δ −109.57 (d, JFP = 111.9 Hz). 31P NMR (162 MHz, CD3OD) δ 7.19 (br t). HRMS (ESI) m/z : calcd 367.0916 (M − H+); found: 367.0912.
((3'-Cyclohexyl-2'-hydroxy-[1,1'-biphenyl]-3-yl)difluoromethyl)phosphonic Acid (11m)
For the Suzuki coupling, general procedure C was followed with 106 mg (0.375 mmol) of the potassium trifluoroborate, 17e, and 71 mg (0.25 mmol) of aryl bromide 13. The procedure yielded 22 mg (23%) of the desired compound as a white, hygroscopic powder. 1H NMR (400 MHz, CD3OD) δ 1.23–1.56 (m, 5H), 1.73–1.82 (m, 1H), 1.82–1.93 (m, 4H), 2.95–3.08 (m, 1H), 6.91 (t, J = 7.6 Hz, 1H), 7.03 (dd, J = 7.6, 1.7 Hz, 1H), 7.17 (dd, J = 7.6, 1.7 Hz, 1H), 7.51 (t, J = 7.6 Hz, 1H), 7.57 (d, J = 7.6 Hz, 1H), 7.61 (d, J = 7.3 Hz, 1H), 7.72 (s, 1H). 19F NMR (376 MHz, CD3OD) δ −109.61 (d, JFP = 113.3 Hz). 31P NMR (162 MHz, CD3OD) δ 7.29 (t, JPF = 112.4 Hz). HRMS (ESI) m/z : calcd 381.1073 (M − H+); found: 381.1068.
((3'-Cycloheptyl-2'-hydroxy-[1,1'-biphenyl]-3-yl)difluoromethyl)phosphonic Acid (11n)
For the Suzuki coupling, general procedure D was followed with 90 mg (0.30 mmol) of the potassium trifluoroborate 18b and 87 mg (0.30 mmol) of aryl bromide 13. The procedure yielded 20 mg (17%) of the desired compound as a white hygroscopic powder in a 1:0.4 ratio with residual trifluoroacetic acid (as determined by 19F NMR). 1H NMR (400 MHz, CD3OD) δ 1.49–2.00 (m, 12H), 3.16–3.29 (m, 1H), 6.87 (t, J = 7.6 Hz, 1H), 6.99 (dd, J = 7.4, 1.7 Hz, 1H), 7.13 (dd, J = 7.6, 1.7 Hz, 1H), 7.44–7.68 (m, 3H), 7.72 (s, 1H). 19F NMR (376 MHz, CD3OD) δ −109.61 (d, J = 111.5 Hz). 31P NMR (162 MHz, CD3OD) δ 7.01 (br t). HRMS (ESI) m/z : calcd 395.1229 (M − H+); found: 395.1225.
((3'-Cyclohexyl-2'-hydroxy-[1,1'-biphenyl]-3-yl)(hydroxy)methyl)phosphonic Acid (11o)
For the Suzuki coupling, general procedure D was followed with 85 mg (0.30 mmol) of the potassium trifluoroborate 17e and 72 mg (0.25 mmol) of monosodium (3-bromobenzoyl)phosphonate, 25.35 The procedure yielded 21 mg (23%) of the α-ketophosphate, 26, as an off-white hygroscopic powder. To accomplish reduction of the ketone, a 1 dram vial was charged with a stir bar, 10 mg of this α-ketophosphate and 0.3 mL of MeOH. The vial was cooled in an ice water bath and 10 mg of NaBH4 was added as a solid. The mixture was stirred for 10 minutes in the ice bath after which time another portion of 10 mg of NaBH4 was added. After 10 additional minutes stirring, the reaction was quenched with addition of 50 µL of glacial acetic acid. Following removal of the volatile components on a rotary evaporator, the residue was purified by reversed-phase gradient column chromatography (5% to 100% acetonitrile in water with 0.1% trifluoroacetic acid). Volatile components were removed, and the resulting water solution was lyophilized to afford the desired product as a white hygroscopic powder (5 mg, 50%). 1H NMR (400 MHz, CD3OD) δ 1.15–1.44 (m, 5H), 1.63–1.72 (m, 1H), 1.73–1.86 (m, 4H), 2.86–3.01 (m, 1H), 4.84 (d, J = 13.2 Hz, 1H), 6.78 (t, J = 7.5 Hz, 1H), 6.91 (dd, J = 7.6, 1.7 Hz, 1H), 7.04 (dd, J = 7.6, 1.8 Hz, 1H), 7.26–7.35 (m, 2H), 7.40 (d, J = 7.3 Hz, 1H), 7.51 (s, 1H). 31P NMR (162 MHz, CD3OD) δ 22.45 (s). HRMS (ESI) m/z : calcd 361.1210 (M − H+); found: 361.1215
((2'-Cyano-[1,1'-biphenyl]-4-yl)difluoromethyl)phosphonic Acid (12a)
Compound 12a was prepared using general procedure D. Hence, using 80 mg (0.28 mmol) of ((4-bromophenyl)difluoromethyl)phosphonic acid 14, and 61 mg (0.42 mmol) of (2-cyanophenyl)boronic acid, the title compound was obtained in 23% yield (23 mg) as a white solid. 1H NMR (400 MHz, DMSO-d6) δ 7.54–7.59 (m, 1H), 7.60–7.62 (m, 1H), 7.67 (d, J = 8.0 Hz, 2H), 7.74–7.84 (m, 3H), 7.85–7.87 (m, 1H); 31P NMR (162 MHz, DMSO-d6) δ 5.39 (t, J = 110.5 Hz); 19F NMR (376 MHz, DMSO-d6) δ −111.15 (d, J = 110.8 Hz). HRMS (ESI) (m/z) [M − H]− calculated for C14H9F2NO3P, 308.0294; found, 308.0298.
((3'-Cyano-[1,1'-biphenyl]-4-yl)difluoromethyl)phosphonic Acid (12b)
Compound 12b was prepared using general procedure C. Hence, using 100 mg (0.35 mmol) of 14 and 77 mg (0.52 mmol) of (3-cyanophenyl)boronic acid, the title compound was obtained in 35% yield (38 mg) as a white solid. 1H NMR (400 MHz, DMSO-d6) δ7.63–7.67 (m, 1H), 7.71–7.78 (m, 5H), 7.96–8.04 (m, 2H); 31P NMR (162 MHz, DMSO-d6) δ 5.81 (t, J = 112.2 Hz); 19F NMR (376 MHz, DMSO-d6) δ −111.49 (d, J = 112.0 Hz). HRMS (ESI) (m/z) [M − H]− calculated for C14H9F2NO3P, 308.0294; found, 308.0296.
((4'-Cyano-[1,1'-biphenyl]-4-yl)difluoromethyl)phosphonic Acid (12c)
Compound 12c was prepared using general procedure C. Hence, using 100 mg (0.35 mmol) of 14 and 77 mg (0.52 mmol) of (4-cyanophenyl)boronic acid, the title compound was obtained in 25% yield (27 mg) as a white solid. 1H NMR (400 MHz, DMSO-d6) δ 7.73 (d, J = 8.0 Hz, 2H), 7.78–7.87 (m, 6H); 31P NMR (162 MHz, DMSO-d6) δ 5.61 (t, J = 111.7 Hz); 19F NMR (376 MHz, DMSO-d6) δ −111.49 (d, J = 111.7 Hz). HRMS (ESI) (m/z) [M − H]− calculated for C14H9F2NO3P, 308.0294; found, 308.0297.
(Difluoro(3'-isopropoxy-[1,1'-biphenyl]-4-yl)methyl)phosphonic Acid (12d)
Compound 12d was prepared using general procedure C. Hence, using 100 mg (0.35 mmol) of 14 and 94 mg (0.52 mmol) of (3-isopropoxyphenyl)boronic acid, the title compound was obtained in 20% yield (24 mg) as a white solid.1H NMR (400 MHz, DMSO-d6) δ 1.33 (d, J = 6.0 Hz, 6H), 4.67 (sept, J = 6.0 Hz, 1H), 6.92 (ddd, J = 8.4 Hz, 2.4, 0.8 Hz, 1H), 7.13–7.19 (m, 2H), 7.34 (t, J = 8.4 Hz, 1H), 7.65–7.70 (m, 4H); 31P NMR (162 MHz, DMSO-d6) δ 5.88 (t, J = 112.9 Hz); 19F NMR (376 MHz, DMSO-d6) δ −111.15 (d, J = 112.9 Hz). HRMS (ESI) (m/z) [M − H]− calculated for C16H16F2O4P, 341.0760; found, 341.0760.
(Difluoro(4'-isopropoxy-[1,1'-biphenyl]-4-yl)methyl)phosphonic Acid (12e)
Compound 12e was prepared using general procedure C. Hence, using 100 mg (0.35 mmol) of 14 and 94 mg (0.52 mmol) of (4-isopropoxyphenyl)boronic acid, the title compound was obtained in 16% yield (19 mg) as a white solid. 1H NMR (400 MHz, DMSO-d6) δ 1.33 (d, J = 6.0 Hz, 6H), 4.65 (sept, J = 6.0 Hz, 1H), 6.98 (d, J = 8.8 Hz, 2H), 7.56 (d, J = 8.8 Hz, 2H), 7.63, 7.66 (ABq, J = 8.8 Hz, 4H); 31P NMR (162 MHz, DMSO-d6) δ 5.93 (t, J = 104.0 Hz); 19F NMR (376 MHz, DMSO-d6) δ −111.02 (d, J = 113.3 Hz). HRMS (ESI) (m/z) [M − H]− calculated for C16H16F2O4P, 341.0760; found, 341.0763.
(Difluoro(3'-isopropyl-[1,1'-biphenyl]-4-yl)methyl)phosphonic Acid (12f)
Compound 12f was prepared using general procedure C. Hence, using 80 mg (0.28 mmol) of 14 and 86 mg (0.52 mmol) of (3-isopropylphenyl)boronic acid, the title compound was obtained in 32% yield (29 mg) as a sticky oil. 1H NMR (400 MHz, DMSO-d6) δ 1.30 (d, J = 6.4 Hz, 6H), 2.98 (sept, J = 6.4 Hz, 1H), 7.24–7.27 (m, 1H), 7.37–7.45 (m, 3H), 7.66–7.72 (m, 4H); 31P NMR (162 MHz, DMSO-d6) δ 5.95 (t, J = 113.4 Hz); 19F NMR (376 MHz, DMSO-d6) δ −111.15 (d, J = 113.6 Hz). HRMS (ESI) (m/z) [M − H]− calculated for C16H16F2O3P, 325.0811; found, 325.08114.
(Difluoro(4'-isopropyl-[1,1'-biphenyl]-4-yl)methyl)phosphonic Acid (12g)
Compound 12g was prepared using general procedure C. Hence, using 80 mg (0.28 mmol) of 14 and 86 mg (0.52 mmol) of (4-isopropylphenyl)boronic acid, the title compound was obtained in 15 % yield (13 mg) as a white solid. 1H NMR (400 MHz, DMSO-d6) δ 1.28 (d, J = 6.4 Hz, 6H), 2.94 (sept, J = 6.4 Hz, 1H), 7.33 (d, J = 8.0 Hz, 2H), 7.58 (d, J = 8.0 Hz, 2H), 7.65 (d, J = 8.0 Hz, 2H), 7.69 (d, J = 8.0 Hz, 2H); 31P NMR (162 MHz, DMSO-d6) δ 5.99 (t, J = 113.9 Hz); 19F NMR (376 MHz, DMSO-d6) δ −111.19 (d, J = 114.0 Hz). HRMS (ESI) (m/z) [M − H]− calculated for C16H16F2O3P, 325.0811; found, 325.0815.
(Difluoro(2'-(methylsulfonamido)-[1,1'-biphenyl]-4-yl)methyl)phosphonic Acid (12h)
Compound 12h was prepared using general procedure D. Hence, using 80 mg (0.28 mmol) of 14 and 124 mg (0.42 mmol) of 2-(methanesulfonylamino)phenylboronic acid pinacol ester, the title compound was obtained in 12% yield (13 mg) as a white solid. 1H NMR (400 MHz, DMSO-d6) δ 2.74 (s, 3H), 7.29–7.46 (m, 4H), 7.53, 7.57 (ABq, J = 8.0 Hz, 4H), 9.01 (s, 1H); 31P NMR (162 MHz, DMSO-d6) δ 2.83 (t, J = 106.3 Hz); 19F NMR (376 MHz, DMSO-d6) δ −107.44 (d, J = 106.3 Hz). HRMS (ESI) (m/z) [M − H]− calculated for C14H13F2NO5PS, 376.0226; found, 376.0232.
(Difluoro(3'-(methylsulfonamido)-[1,1'-biphenyl]-4-yl)methyl)phosphonic Acid (12i)
Compound 12i was prepared using general procedure D. Hence, using 80 mg (0.28 mmol) of 14 and 90 mg (0.42 mmol) of 3-(methanesulfonylamino)phenylboronic acid, the title compound was obtained in 15% yield (16 mg) as a white solid. 1H NMR (400 MHz, DMSO-d6) δ 3.03 (s, 3H), 7.23–7.25 (m, 1H), 7.39–7.47 (m, 3H), 7.61 (d, J = 8.0 Hz, 2H), 7.70 (d, J = 8.0 Hz, 2H), 9.86 (s, 1H); 31P NMR (162 MHz, DMSO-d6) δ 2.86 (t, J = 106.6 Hz); 19F NMR (376 MHz, DMSO-d6) δ −108.08 (d, J = 106.7 Hz). HRMS (ESI) (m/z) [M − H]− calculated for C14H13F2NO5PS, 376.0226; found, 376.0230.
(Difluoro(3'-(1-hydroxyethyl)-[1,1'-biphenyl]-4-yl)methyl)phosphonic Acid (12j)
Compound 12j was prepared using general procedure D. Hence, using 80 mg (0.28 mmol) of 14 and 69 mg (0.42 mmol) of 3-(1-hydroxyethyl)phenylboronic acid, the title compound was obtained in 16% yield (22 mg) as a white solid. 1H NMR (400 MHz, DMSO-d6) δ 1.34 (d, J = 6.4 Hz, 3H), 4.77 (q, J = 6.4 Hz, 1H), 7.32–7.37 (m, 1H), 7.40 (t, J = 7.6 Hz, 1H), 7.48–7.53 (m, 1H), 7.56–7.65 (m, 3H), 7.73 (d, J = 8.4 Hz, 2H).; 31P NMR (162 MHz, DMSO-d6) δ 2.91 (t, J = 107.1 Hz); 19F NMR (376 MHz, DMSO-d6) δ −107.96 (d, J = 107.1 Hz). HRMS (ESI) (m/z) [M − H]− calculated for C15H14F2O4P, 327.0603; found, 327.0602.
(Difluoro(4'-(1-hydroxyethyl)-[1,1'-biphenyl]-4-yl)methyl)phosphonic Acid (12k)
Compound 12k was prepared from 14 in two steps. Suzuki-Miyaura cross coupling of 14 (100 mg, 0.35 mmol) and 4-acetylphenylboronic acid (86 mg, 0.52 mmol) using general procedure C afforded ((4'-acetyl-[1,1'-biphenyl]-4-yl)difluoromethyl)phosphonic acid (S1) as a yellow solid (18 mg, 20% yield). S1 (10 mg, 0.030 mmol) was dissolved in methanol (0.3 ml) and treated with NaBH4 (10 mg) at 0 °C. The reaction mixture was stirred at 0 °C for 20 min. More NaBH4 (10 mg) was added, and the reaction mixture was stirred for an additional 20 min. Acetic acid (0.1 ml) was added. The reaction mixture was purified by automated reversed-phase C-18 column chromatography (liquid injection, 50 gram C18 column, column volume = 42.6 mL, 40 mL/min, linear gradient of 5–100% acetonitrile/water with 0.1% trifluoroacetic acid over 20 min) to give the title compound as a white solid (5 mg, 50% yield). 1H NMR (400 MHz, DMSO-d6) δ 1.32 (d, J = 6.4 Hz, 3H). 4.73 (q, J = 6.4 Hz, 1H), 7.41(d, J = 8.0 Hz, 2H), 7.56 (d, J = 8.0 Hz, 2H), 7.61 (d, J = 8.0 Hz, 2H), 7.70 (d, J = 8.0 Hz, 2H); 31P NMR (162 MHz, DMSO-d6) δ 2.62 (t, J = 103.6 Hz); 19F NMR (376 MHz, DMSO-d6) δ −104.57 (d, J = 103.6 Hz). HRMS (ESI) (m/z) [M − H]− calculated for C15H14F2O4P, 327.0603; found, 327.0608.
(Difluoro(3'-(phenylamino)-[1,1'-biphenyl]-4-yl)methyl)phosphonic Acid (12l)
Compound 12l was prepared using general procedure D. Hence, using 80 mg (0.28 mmol) of 14 and 124 mg (0.42 mmol) of 3-(phenylamino)phenylboronic acid pinacol ester, the title compound was obtained in 10% yield (10 mg) as a white solid. 1H NMR (400 MHz, DMSO-d6) δ 6.82 (t, J = 7.2 Hz, 1H), 7.05–7.12 (m, 4H), 7.18–7.22 (m, 2H), 7.27–7.34 (m, 2H), 7.58 (d, J = 8.0 Hz, 2H), 7.68 (d, J = 8.0 Hz, 2H); 31P NMR (162 MHz, DMSO-d6) δ 2.91 (t, J = 107.3 Hz); 19F NMR (376 MHz, DMSO-d6) δ −108.01 (d, J = 107.3 Hz). HRMS (ESI) (m/z) [M − H]− calculated for C19H15F2NO3P, 374.0763; found, 374.0764.
((3'-Benzyl-[1,1'-biphenyl]-4-yl)difluoromethyl)phosphonic Acid (12m)
Compound 12m was prepared using general procedure D. Hence, using 80 mg (0.28 mmol) of 14 and 123 mg (0.42 mmol) of 22a, the title compound was obtained in 22% yield (25 mg) as a white solid. 1H NMR (400 MHz, DMSO-d6) δ 4.01 (s, 2H), 7.15–7.20 (m, 1H), 7.24–7.30 (m, 5H), 7.39 (t, J = 8.0 Hz, 1H), 7.51 (d, J = 8.0 Hz, 1H), 7.57–7.60 (m, 3H), 7.32 (d, J = 8.0 Hz, 2H); 31P NMR (162 MHz, DMSO-d6) δ 2.90 (t, J = 106.9 Hz); 19F NMR (376 MHz, DMSO-d6) δ −107.98 (d, J = 106.8 Hz). HRMS (ESI) (m/z) [M − H]− calculated for C20H16F2O3P, 373.0811; found, 373.0803.
(Difluoro(3'-(4-fluorobenzyl)-[1,1'-biphenyl]-4-yl)methyl)phosphonic Acid (12n)
Compound 12n was prepared using general procedure D. Hence, using 80 mg (0.28 mmol) of 14 and 131 mg (0.42 mmol) of 22b, the title compound was obtained in 34% yield (37 mg) as a white solid.1H NMR (400 MHz, DMSO-d6) δ 4.00 (s, 2H), 7.06–7.12 (m, 2H), 7.24 (d, J = 8.0 Hz, 1H), 7.29–7.34 (m, 2H), 7.39 (t, J = 8.0 Hz, 1H), 7.51 (d, J = 8.0 Hz, 1H), 7.51–7.60 (m, 3H), 7.72 (d, J = 8.0 Hz, 2H); 31P NMR (162 MHz, DMSO-d6) δ 2.90 (t, JFP = 106.92 Hz); 19F NMR (376 MHz, DMSO-d6) δ −107.78 (d, J = 106.7 Hz), −117.29 (m). HRMS (ESI) (m/z) [M − H]− calculated for C20H15F3O3P, 391.0716; found, 391.0719.
((3'-(4-Chlorobenzyl)-[1,1'-biphenyl]-4-yl)difluoromethyl)phosphonic Acid (12o)
Compound 12o was prepared using general procedure D. Hence, using 80 mg (0.28 mmol) of 14 and 138 mg (0.42 mmol) of 22c, the title compound was obtained in 24% yield (27 mg) as a white solid. 1H NMR (400 MHz, DMSO-d6) δ 3.99 (s, 2H), 7.22 (d, J = 7.6 Hz, 1H), 7.28–7.33 (m, 4H), 7.38 (t, J = 7.6 Hz, 1H), 7.51–7.53 (m, 1H), 7.56–7.58 (m, 3H), 7.69 (d, J = 8.0 Hz, 2H); 31P NMR (162 MHz, DMSO-d6) δ 2.59 (t, JFP = 103.7 Hz); 19F NMR (376 MHz, DMSO-d6) δ −108.01 (d, JFP = 105.3 Hz). HRMS (ESI) (m/z) [M –H]− calculated for C20H15F2ClO3P, 407.0421; found, 407.0418.
((3'-(3,4-Dichlorobenzyl)-[1,1'-biphenyl]-4-yl)difluoromethyl)phosphonic acid (12p)
Compound 12p was prepared using general procedure D. Hence, using 80 mg (0.28 mmol) of 14 and 152 mg (0.42 mmol) of 22d, the title compound was obtained in 28% yield (34 mg) as a white solid. 1H NMR (400 MHz, DMSO-d6) δ 4.01 (s, 2H), 7.25–7.29 (m, 2H), 7.40 (t, 8.0 Hz, 1H), 7.51–7.54 (m, 2H), 7.58–7.62 (m, 4H), 7.74 (d, J = 8.0 Hz, 2H). 31P NMR (162 MHz, DMSO-d6) δ 2.66 (t, JFP = 104.2 Hz). 19F NMR (376 MHz, DMSO-d6) δ −107.62 (d, JFP = 104.2 Hz). HRMS (ESI) (m/z) [M − H]− calculated for C20H14F2Cl2O3P, 441.0031; found, 441.0030.
((3'-((3,4-Dichlorophenyl)(hydroxy)methyl)-[1,1'-biphenyl]-4-yl)difluoromethyl)phosphonic Acid (12q)
Compound 12q was prepared using general procedure D. Hence, using 80 mg (0.28 mmol) of 14 and 124 mg (0.42 mmol) of 24, the title compound was obtained in 36% yield (46 mg) as a white solid. 1H NMR (400 MHz, DMSO-d6) δ 6.04 (s, 1H), 7.31 (d, J = 8.0 Hz, 1H), 7.42 (t, J = 8.0 Hz, 1H), 7.46 (dd, J = 8.0 Hz, 2.0 Hz, 1H), 7.53 (d, J = 2.0 Hz, 1H), 7.56 (d, J = 8.0 Hz, 1H), 7.60 (d, J = 8.0 Hz, 2H), 7.65 (s, 1H), 7.70 (d, J = 8.0 Hz, 2H), 7.74 (d, J = 8.0 Hz, 1H); 31P NMR (162 MHz, DMSO-d6) δ 2.91 (t, JFP = 106.9 Hz); 19F NMR (376 MHz, DMSO-d6) δ −108.09 (d, JFP = 107.0 Hz). HRMS (ESI) (m/z) [M –H]− calculated for C20H14Cl2F2O4P, 456.9980; found, 456.9979.
((3'-(3,4-Dichlorobenzyl)-[1,1'-biphenyl]-4-yl)(hydroxy)methyl)phosphonic Acid (12r)
To a solution of 28 (35 mg, 0.083 mmol) in MeOH (0.2 ml) at 0 °C was added NaBH4 (16 mg, 0.41 mmol). The reaction mixture was stirred at 0 °C for 20 min. More NaBH4 (16 mg) was added, and the reaction mixture was stirred for an additional 20 min. Acetic acid (0.1 ml) was added. The reaction mixture was purified by automated reversed-phase C-18 column chromatography (liquid injection, 50 gram C18 column, column volume = 42.6 mL, 40 mL/min, linear gradient of 5–100% acetonitrile/water with 0.1% trifluoroacetic acid over 20 min) to give the title compound as a white solid (20 mg, 57% yield). 1H NMR (400 MHz, DMSO-d6) δ 4.00 (s, 2H), 4.70 (d, J = 14.0 Hz, 1H), 7.20 (d, J = 7.6 Hz, 1H), 7.28 (dd, J = 7.6, 2.0 Hz, 1H), 7.36 (t, J = 7.6 Hz, 1H), 7.45–7.58 (m, 8H). 31P NMR (162 MHz, DMSO-d6) δ 18.03. HRMS (ESI) (m/z) [M − H]− calculated for C20H16Cl2O4P, 421.0169; found, 421.0155.
((S)-(3'-((R)-(3,4-dichlorophenyl)(hydroxy)methyl)-[1,1'-biphenyl]-4-yl)(hydroxy)methyl)phosphonic Acid (12s)
Inhibitor 12s was prepared using general procedure D. Hence, using 80 mg (0.30 mmol) of 30b and 150 mg (0.40 mmol) of 32b, the title compound was obtained in 12% yield (16 mg) as a white solid. Reaction time = 4 h. 1H NMR (400 MHz, DMSO-d6) δ 4.70 (d, J = 14.0 Hz, 1H), 5.79 (s, 1H), 6.17 (s, 1H), 7.34 (d, J = 8.0 Hz, 1H), 7.36–7.41 (m, 2H), 7.45–7.51 (m, 3H), 7.54–7.56 (m, 3H), 7.67–7.69 (m, 2H).31P NMR (162 MHz, DMSO-d6) δ 17.97. HRMS (ESI) (m/z) [M − H]− calculated for C20H16Cl2O5P, 437.0118; found, 437.0110.
((S)-(3'-((S)-(3,4-dichlorophenyl)(hydroxy)methyl)-[1,1'-biphenyl]-4-yl)(hydroxy)methyl)phosphonic Acid (12t)
Inhibitor 12t was prepared using general procedure D. Hence, using 80 mg (0.30 mmol) of 30b and 150 mg (0.40 mmol) of 32a, the title compound was obtained in 10% yield (13 mg) as a white solid. Reaction time = 4 h. 1H NMR (400 MHz, DMSO-d6) δ 4.70 (d, J = 14.0 Hz, 1H), 5.79 (s, 1H), 6.17 (s, 1H), 7.34 (d, J = 8.0 Hz, 1H), 7.36–7.41 (m, 2H), 7.45–7.51 (m, 3H), 7.54–7.56 (m, 3H), 7.67–7.69 (m, 2H).31P NMR (162 MHz, DMSO-d6) δ 18.00. HRMS (ESI) (m/z) [M − H]− calculated for C20H16Cl2O5P, 437.0118; found, 437.0111.
((R)-(3'-((R)-(3,4-dichlorophenyl)(hydroxy)methyl)-[1,1'-biphenyl]-4-yl)(hydroxy)methyl)phosphonic Acid (12u)
Inhibitor 12u was prepared using general procedure D. Hence, using 80 mg (0.30 mmol) of 30a and 150 mg (0.40 mmol) of 32b, the title compound was obtained in 10% yield (13 mg) as a white solid. Reaction time = 4 h. The 1H NMR and 31P NMR spectra are the same as those reported for the enantiomer, 12t. HRMS (ESI) (m/z) [M − H]− calculated for C20H16Cl2O5P, 437.0118; found, 437.0111.
((R)-(3'-((S)-(3,4-dichlorophenyl)(hydroxy)methyl)-[1,1'-biphenyl]-4-yl)(hydroxy)methyl)phosphonic Acid (12v)
Inhibitor 12v was prepared using general procedure D. Hence, using 80 mg (0.30 mmol) of 30a and 150 mg (0.40 mmol) of 32a, the title compound was obtained in 15% yield (19 mg) as a white solid. Reaction time = 4 h. The 1H NMR and 31P NMR spectra are the same as those reported for the enantiomer, 12s. HRMS (ESI) (m/z) [M − H]− calculated for C20H16Cl2O5P, 437.0118; found, 437.0112.
((3'-((3,4-Dichlorophenyl)(hydroxy)methyl)-[1,1'-biphenyl]-4-yl)methyl)phosphonic Acid (12w)
Compound 12w was prepared using general procedure D. Hence, using 70 mg (0.28 mmol) of 4-bromobenzylphosphonic acid and 124 mg (0.42 mmol) of 24, the title compound was obtained in 14% yield (16 mg) as a white solid. 1H NMR (400 MHz, DMSO-d6) δ 2.97 (d, J = 21.2 Hz, 2H), 5.78 (s, 1H), 6.15 (br, 1H), 7.30–7.32 (m, 3H), 7.35–7.39 (m, 2H), 7.48 (d, J = 8.0 Hz, 1H), 7.50–7.55 (m, 3H), 7.65 (d, J = 4.0 Hz, 2H).31P NMR (162 MHz, DMSO-d6) δ 20.79 (s). HRMS (ESI) (m/z) [M − H]− calculated for C20H16Cl2O4P, 421.0169; found, 421.0162.
2-Bromo-6-cyclohexylphenol (16e)
General procedure A was followed for the ortho-bromination of 2-cyclohexylphenol starting with 880 mg of the phenol (5.0 mmol). The crude product was purified by column chromatography (9:1 hexanes/ethyl acetate; product Rf: 0.35) to afford the product as a yellow oil (1.16 g, 91%). 1H NMR (400 MHz, CDCl3) δ 1.19–1.52 (m, 5H), 1.71–1.80 (m, 1H), 1.80–1.93 (m, 4H), 2.86–3.04 (m, 1H), 5.56 (s, 1H), 6.77 (t, J = 7.8 Hz, 1H), 7.13 (dd, J = 7.7, 1.4 Hz, 1H), 7.29 (dd, J = 8.0, 1.5 Hz, 1H). MS (ESI) m/z 255 (M + H+).
Potassium ((2-Hydroxy-3-cyclohexyl)phenyl)trifluoroborate (17e)
Borylation of the phenol was accomplished by a modified version of general procedure B. Butyllitium (2.5 M in hexanes, 3.90 mL, 9.75 mmol, 2.15 equiv) was added dropwise over 5 min via syringe to a stirred solution of the ortho-bromophenol 16e (1.15 g, 4.5 mmol) in tetrahydrofuran, THF (25 mL, 0.18 M), in a dry ice-acetone cold temperature bath. The cold bath was removed and the reaction solution was allowed to warm to ambient temperature in air for 1.5 h. The reaction flask is resuspended in the dry ice-acetone cold temperature bath. After 10 min stirring in the cold bath, a solution of trimethyl borate (0.84 mL, 7.5 mmol, 1.67 equiv) in THF (5 mL, 1.5 M) was added via syringe. After stirring in the cold bath for 30 min, the reaction mixture was stirred at ambient temperature for 18 h. The reaction flask was placed in an ice water bath, and the reaction was quenched by addition of 7 mL of saturated potassium hydrogen difluoride (4.5 M, 7 equiv). The reaction mixture allowed to come to ambient temperature and stirred for 24 h. The volatile material was removed with a rotary evaporator under vacuum to afford the crude product. The crude products were dissolved in 150 mL of hot acetone, and the insoluble salts were filtered. The volatile components of the filtrate were removed with a rotary evaporator under vacuum until 5 mL of acetone was remaining. Approximately 30 mL of pentane was added to the acetone solution to precipitate the product. Filtration and washing with pentane afforded the purified product as a white solid (500 mg, 39%). 1H NMR (400 MHz, Acetone-d6) δ 1.18–1.46 (m, 5H), 1.67–1.75 (m, 1H), 1.75–1.86 (m, 4H), 2.86–2.96 (m, 1H), 6.57 (t, J = 7.3 Hz, 1H), 6.84 (dd, J = 7.5, 1.7 Hz, 1H), 7.11 (dd, J = 7.2, 1.9 Hz, 1H), 7.60 (q, J = 11.8 Hz, 1H). HRMS (ESI) m/z : calcd 219.1198 (boronic acid M − H+); found 219.1202.
2-Cyclobutylphenol (15f)
Following a previous report, dilithiation of 2-bromophenol was accomplished.25 2-Bromophenol (3.5 g, 20 mmol, 1.0 equiv) was dissolved in diethyl ether (55 mL) and cooled in a dry ice-acetone cold bath. After stirring for 10 min, n-butyllithium (1.6 M in hexanes, 26.5 mL, 42 mmol, 2.1 equiv) was added dropwise by syringe. The cold bath was removed and the reaction mixture was stirred at ambient temperature for 2 h, resulting in the dilithiated reagent. LCMS of an acid quenched aliquot confirmed that there was no starting bromide remaining. 19 mL (approximately 5 mmol) of the above reagent was transferred to a reaction flask charged with a stir bar and was suspended in the dry ice-acetone cold bath. Next, 392 µL of cyclobutanone (5.5 mmol, 1.1 equiv) was added neat via syringe. The reaction mixture was allowed to warm to ambient temperature and was stirred for 14 h. The reaction was quenched by addition of 10 mL of saturated ammonium chloride. The layers were separated, and the water layer was washed with 15 mL of diethyl ether. The combined organic layers were dried over MgSO4, and the volatile material was removed with a rotary evaporator under vacuum to afford the crude benzyl alcohol product as a yellow liquid (880 mg). The crude product (~5 mmol) was dissolved in 5 mL of CH2Cl2 in a 20 mL scintillation vial open to atmosphere and triethylsilane (2.4 mL, 15 mmol, ~3 equiv) was added followed by 2.5 mL of trifluoroacetic acid (1:0.5 by volume). The reaction mixture was stirred at ambient temperature for 16 h. The volatile material was removed with a rotary evaporator under vacuum to afford the crude product. The crude product was purified via gradient column chromatography (2% to 20% ethyl acetate in hexanes) to afford the product as a clear oil (550 mg, 67%). 1H NMR (400 MHz, CDCl3) δ 1.80–1.95 (m, 1H), 2.00–2.12 (m, 1H), 2.12–2.26 (m, 2H), 2.30–2.46 (m, 2H), 3.58–3.75 (m, 1H), 4.63 (s, 1H), 6.76 (dd, J = 7.9, 1.2 Hz, 1H), 6.92 (td, J = 7.4, 1.1 Hz, 1H), 7.09 (td, J = 7.7, 1.7 Hz, 1H), 7.18 (dt, J = 7.6, 1.3 Hz, 1H). MS (ESI) m/z 149 (M + H+).
Potassium ((2-Hydroxy-3-cyclobutyl)phenyl)trifluoroborate (18a)
Borylation of 2-cyclobutylphenol, 15f, was accomplished by closely following a previous report.26 A 1 dram oven dried vial was charged with a stir bar and brought into an inert atmosphere glove box where it was charged with 2-cyclobutylphenol (222 mg, 1.5 mmol, 1.0 equiv), diethylsilane (290 µL, 2.25 mmol, 1.5 equiv), bis(1,5-cyclooctadiene)diiridium(I) dichloride (10 mg, 0.015 mmol, 0.01 equiv, 2 mol% Ir), and 3 mL of benzene (0.5 M). The vial was capped, removed from the inert atmosphere glove box, and the reaction mixture was stirred at ambient temperature for 1 h. Solvent was removed with a rotary evaporator under vacuum, and the reaction vial was brought back into the inert atmosphere glove box where it was charged with bis(pinacolato)diboron (381 mg, 1.5 mmol, 1.0 equiv), pinacolborane (21.8 µL, 0.15 mmol, 10 mol%), bis(1,5-cyclooctadiene)diiridium(I) dichloride (10 mg, 0.015 mmol, 0.01 equiv, 2 mol% Ir), 4,4-di-tert-butyl bipyridine (8 mg, 0.03 mmol, 2 mol%), and 3 mL of THF (0.5 M). The vial was capped, removed from the inert atmosphere glove box, placed in a heating block preheated to 80 °C, and the mixture was stirred vigorously for 2 h. The vial was cooled to ambient temperature and saturated potassium hydrogen difluoride (4.5 M, 3 mL, 13.5 mmol, 9 equiv) was added. The resulting reaction mixture was stirred vigorously for 18 h. The volatile material was removed with a rotary evaporator under vacuum to afford the crude product. The crude product was dissolved in 5 mL of hot acetone, and the insoluble salts were filtered. The volatile components of the filtrate were removed with a rotary evaporator under vacuum until 1 mL of acetone remained. The solution was transferred to a 15 mL conical centrifuge tube, and approximately 5 mL of pentane was added to the acetone solution to precipitate the product. The suspension was centrifuged to pellet the solid product and the black liquid was removed. Additional washes were accomplished by adding 5 mL of pentane, resuspending the solid followed by centrifugation to pellet the solid products for a total of three additional washes. Removal of residual solvent under vacuum afforded the product as a light gray solid (200 mg, 52%). 1H NMR (400 MHz, acetone-d6) δ 1.71–1.84 (m, 1H), 1.86–2.03 (m, 2H), 2.03–2.15 (m, 1H), 2.18–2.33 (m, 2H), 3.74 (p, J = 8.5 Hz, 1H), 6.60 (t, J = 7.3 Hz, 1H), 6.90 (dd, J = 7.5, 1.8 Hz, 1H), 7.15 (d, J = 7.0 Hz, 1H), 7.62 (q, J = 11.8 Hz, 1H). MS (ESI) m/z 191 (boronic acid M − H+).
2-Cycloheptylphenol (15g)
Synthesis was achieved analogously to that of 15f using 19 mL of the dilithiated reagent (approximately 5 mmol) and 650 µL of cycloheptanone (5.5 mmol, 1.1 equiv) to afford the crude benzyl alcohol product as a yellow liquid (1.072 g). The crude product (~5 mmol) was treated with triethylsilane (2.4 mL, 15 mmol, ~3 equiv) and trifluoroacetic acid and was purified via gradient column chromatography (2% to 20% ethyl acetate in hexanes) to afford the product as a clear oil (422 mg, 44%). 1H NMR (400 MHz, CDCl3) δ 1.48–1.77 (m, 8H), 1.76–1.86 (m, 2H), 1.87–1.99 (m, 2H), 2.92–3.02 (m, 1H), 4.71 (s, 1H), 6.74 (dd, J = 8.1, 1.2 Hz, 1H), 6.89 (td, J = 7.5, 1.2 Hz, 1H), 7.05 (td, J = 7.6, 1.7 Hz, 1H), 7.18 (dd, J = 7.7, 1.6 Hz, 1H). MS (ESI) m/z 191 (M + H+).
Potassium ((2-Hydroxy-3-cycloheptyl)phenyl)trifluoroborate (18b)
Borylation of 2-cycloheptylphenol, 15g, was accomplished in the same manner as 18a, starting with 285 mg of 2-cycloheptylphenol (1.5 mmol). The procedure yielded 90 mg of a gray solid (20%), which is taken directly into the next reaction without further purification.
(3-bromophenyl)(phenyl)methanol (20a)
To a solution of 3-bromobenzaldehyde (1.42 g, 7.67 mmol) in THF (15 mL) at −78 °C under nitrogen atmosphere was added PhMgBr (3.0 M in Et2O, 2.56 mL, 7.68 mmol) dropwise. The reaction solution was stirred at −78 °C for 2 h. Saturated aqueous NH4Cl solution (20 mL) and Et2O (40 mL) were added, and the reaction mixture was allowed to warm to ambient temperature. The layers were separated and the aqueous layer was extracted with Et2O (40 mL). The combined organic solution was dried over anhydrous MgSO4, concentrated, and the residue was purified by automated silica gel flash column chromatography (linear gradient of 0 to 70% ethyl acetate in hexanes over 25 min) to give 20a as a colorless oil (1.72 g, 85% yield). 1H NMR (400 MHz, CDCl3) δ 2.43 (d, J = 3.2 Hz, 1H), 5.76 (d, J = 3.2 Hz, 1H), 7.18–7.21 (m, 1H), 7.27–7.41 (m, 7 H), 7.56–7.57 (m, 1H). 13C NMR (101 MHz, CDCl3) δ 75.6, 122.6, 125.1, 126.5, 127.9, 128.6, 129.4, 130.0, 130.5, 143.1, 145.9.
1-Benzyl-3-bromobenzene (21a)
Compound 20a (1.70 g, 6.50 mmol) was dissolved in dichloromethane (6 mL). Et3SiH (TES, 3.0 mL, 18.7 mmol) was added at ambient temperature, followed by trifluoroacetic acid (TFA, 3.0 mL). The resulting solution was stirred at ambient temperature for 15 h. Volatiles were removed under reduced pressure. The residue was purified by automated silica gel flash column chromatography (80 gram flash column, column volume = 125 mL, 60 mL/min, linear gradient of 0 to 20% ether in hexanes over 25 min) to the title compound as a colorless oil (1.43 g, 89% yield). 1H NMR (400 MHz, CDCl3) δ 3.97 (s, 2H), 7.13–7.38 (m, 9H). 13C NMR (101 MHz, CDCl3) δ 41.6, 122.6, 126.4, 127.6, 128.6, 128.9, 129.2, 130.0, 131.9, 140.2, 143.5.
2-(3-Benzylphenyl)-4,4,5,5-tetramethyl-1,3,2-dioxaborolane (22a)
To a Schlenk flask equipped with a stirring bar was added 21a (0.80 g, 3.23 mmol), bis(pinacolato)diboron (B2Pin2, 1.64 g, 6.48 mmol), KOAc (1.27 g, 12.95 mmol), and DMSO (13 mL). Nitrogen was bubbled through the reaction mixture for 0.5 h. Pd(dppf)Cl2 (0.13 g, 0.16 mmol) was added. Nitrogen was bubbled through the reaction mixture for an additional 0.5 h. The reaction mixture was then stirred at 80 °C for 6 h, cooled down to ambient temperature and poured into water (130 mL). The resulting suspension was extracted with Et2O (2 × 100 mL). The combined organic extracts was dried over anhydrous MgSO4, concentrated, and the residue was purified by automated silica gel flash column chromatography (80 gram flash column, column volume = 125 mL, 60 mL/min, linear gradient of 0 to 20% ether in hexanes over 25 min) to give the title compound as a colorless oil (0.75 g, 79% yield). 1H NMR (400 MHz, CD3OD) δ 1.32 (s, 12H), 3.94 (s, 2H), 7.12–7.17 (m, 3H), 7.22–7.29 (m, 4H), 7.56 (d, J = 8.0 Hz, 1H), 7.59 (s, 1H). 13C NMR (101 MHz, CD3OD) δ 23.8, 41.4, 83.6, 125.6, 127.5, 128.0, 128.4, 131.6, 132.0, 134.7, 140.6, 141.2.
(3-Bromophenyl)(4-fluorophenyl)methanol (20b)
To a solution of 3-bromoiodobenzene (2.0 g, 7.1 mmol) in THF (15 mL) at −78 °C under nitrogen atmosphere was added dropwise n-BuLi (2.5 M in hexanes, 2.8 mL, 7.0 mmol). The reaction mixture was stirred at the same temperature for 0.5 h. 4-Fluorobenzaldehyde (0.75 mL, 7.1 mmol) was added. The reaction mixture was kept at −78 °C for 2 h. Saturated NH4Cl (50 mL) and ether (50 mL) were added, and the reaction mixture was allowed to warm to ambient temperature. The phases were separated. The aqueous phase was extracted with ether (100 mL). The combined organic solution was dried over anhydrous MgSO4, filtered, and concentrated to give a viscous oil, which was used without purification and characterization.
1-Bromo-3-(4-fluorobenzyl)benzene (21b)
The crude product of 20b was dissolved in dichloromethane (7 mL). Et3SiH (TES, 3.3 mL, 20.6 mmol) and trifluoroacetic acid (TFA, 3.5 mL) were added. The resulting solution was stirred at ambient temperature for 15 h. Volatiles were removed under reduced pressure. The residue was purified by automated silica gel flash column chromatography (80 gram flash column, column volume = 125 mL, 60 mL/min, linear gradient of 0 to 20% ether in hexanes over 25 min) to give the title compound as a colorless oil (1.18 g, 64% yield for 2 steps). 1H NMR (400 MHz, CDCl3) δ 3.92 (s, 2H), 6.97–7.02 (m, 2H), 7.08–7.18 (m, 4H), 7.31–7.36 (m, 2H).
2-(3-(4-Fluorobenzyl)phenyl)-4,4,5,5-tetramethyl-1,3,2-dioxaborolane (22b)
Compound 22b (0.58 g) was obtained as a colorless oil in 67% yield starting from 21b (0.75 g, 2.8 mmol) by following the procedure described for the preparation of 22a. 1H NMR (400 MHz, CDCl3) δ 1.35 (s, 12H), 3.95 (s, 2H), 6.93–6.98 (m, 2H), 7.12–7.15 (m, 2H), 7.23 (d, J = 8.0 Hz, 1H), 7.30 (t, J = 8.0 Hz, 1H), 7.66–7.68 (m, 2H). 13C NMR (101 MHz, CDCl3) δ 24.8, 41.0, 83.8, 115.0 (d), 128.0, 130.2(d), 131.8, 132.7, 135.1, 136.9 (d), 140.1, 160.1, 162.6. 19F NMR (376 MHz, CDCl3) δ −117.58 (m).
(3-Bromophenyl)(4-chlorophenyl)methanol (20c)
Compound 20c was prepared from4-chlorobenzaldehyde (1.09 g, 7.78 mmol) by following the procedure described for the preparation of 20b. The crude material was used in the following step without purification and characterization.
1-Bromo-3-(4-chlorobenzyl)benzene (21c)
Compound 21c (1.22 g) was obtained as a colorless oil in 56% yield starting from 4-chlorobenzaldehyde (1.09 g, 7.78 mmol) by following the procedure described for the preparation of 21b. 1H NMR (400 MHz, DMSO) δ 3.92 (s, 2H), 7.21–7.27 (m, 4H), 7.31–7.37 (m, 3H), 7.43–7.44 (m, 1H).
2-(3-(4-Chlorobenzyl)phenyl)-4,4,5,5-tetramethyl-1,3,2-dioxaborolane (22c)
The title compound (1.26 g) was obtained as a white solid in 72% yield starting from 21c (1.50 g, 5.33 mmol) by following the procedure described for the preparation of 22a. 1H NMR (400 MHz, CDCl3) δ 1.34 (s, 12H), 3.95 (s, 2H), 7.10 (d, J = 8.0 Hz, 2H), 7.21–7.26 (m, 3H), 7.30 (t, J = 8.0 Hz, 1H), 7.66–7.68 (m, 2H).
(3-Bromophenyl)(3,4-dichlorophenyl)methanol (20d)
The title compound (1.85 g) was obtained as a viscous oil in 72% yield starting from 3,4-dichlorobenaldehyde (1.36 g, 7.77 mmol) by following the procedure described for the preparation of 20b. 1H NMR (400 MHz, CDCl3) δ 2.31 (d, J = 3.2 Hz, 1H), 5.74 (d, J = 3.2 Hz, 1H), 7.16–7.27 (m, 3H), 7.40–7.44 (m, 2H), 7.48 (m, 1H), 7.51 (m, 1H).
4-(3-bromobenzyl)-1,2-dichlorobenzene (21d)
The title compound (1.57 g) was obtained as a white solid in 92% yield starting from 20d (1.80 g, 5.42 mmol) by following the procedure described for the synthesis of 21a. 1H NMR (400 MHz, CDCl3) δ 3.89 (s, 2H), 7.00 (dd, J = 8.0, 2.0 Hz, 1H), 7.06–7.09 (m, 1H), 7.17 (t, J = 8.0 Hz, 1H), 7.25 (d, J = 2.0 Hz, 1H), 7.30–7.31 (m, 1H), 7.35–7.38 (m, 2H).
2-(3-(3,4-Dichlorobenzyl)phenyl)-4,4,5,5-tetramethyl-1,3,2-dioxaborolane (22d)
The title compound (1.60 g) was obtained as a white solid in 82% yield starting from 21d (1.70 g, 5.38 mmol) by following the procedure described for the synthesis of 22a. 1H NMR (400 MHz, CDCl3) δ 1.35 (s, 12H), 3.92 (s, 2H), 7.0 (d, J = 8.0 Hz, 1H), 7.22–7.26 (m, 2H), 7.32 (t, J = 8.0 Hz, 2H), 7.64 (s, 1H), 7.69 (d, J = 8.0 Hz, 1H).
(3-((3,4-Dichlorophenyl)(hydroxy)methyl)phenyl)boronic Acid (24)
To a solution of compound 23 (1.5 g, 5.3 mmol) in THF (45 mL) was added n-BuLi (2.5 M in hexanes, 2.1 mL, 5.3 mmol) dropwise at −78 °C under nitrogen atmosphere. The resulting solution was stirred at the same temperature for 0.5 h. 3,4-Dichlorobenzaldehyde (0.93 g, 5.3 mmol in THF (3 mL) was added. The reaction mixture was stirred at −78 °C for 1 h. 1 N HCl (16.5 mL) and ether (100 mL) were added. The reaction mixture was allowed to warm to ambient temperature. The layers were separated, and the aqueous layer was extracted with diethyl ether (2 × 100 mL). The combined organic solution was dried over anhydrous MgSO4 and concentrated under reduced pressure to give the title compound, which was used in the following step without purification and characterization.
(3'-(3,4-dichlorobenzyl)-[1,1'-biphenyl]-4-carbonyl)phosphonic Acid (28)
Compound 28 was prepared using general procedure D. Hence, using 80 mg (0.28 mmol) of 27 and 152 mg (0.42 mmol) of 22d, the title compound was obtained in 42% yield (49 mg) as a white solid. 1H NMR (400 MHz, DMSO-d6) δ 4.03 (s, 2H), 7.28–7.32 (m, 2H), 7.43 (t, J = 8.0 Hz, 1H), 7.52 (d, J = 8.0 Hz, 1H), 7.58–7.60 (m, 2H), 7.70 (s, 1H), 7.84 (d, J = 8.0 Hz, 2H), 8.26 (d, J = 8.0 Hz, 2H). 31P NMR (162 MHz, DMSO-d6) δ −3.07. HRMS (ESI) (m/z) [M − H]− calculated for C20H14Cl2O4P, 419.0012; found, 419.0020.
Bis((1R,2S,5R)-2-isopropyl-5-methylcyclohexyl) ((R)-(4-bromophenyl)(hydroxy)methyl)phosphonate (29a) and Bis((1R,2S,5R)-2-isopropyl-5-methylcyclohexyl) ((S)-(4-bromophenyl)(hydroxy)methyl)phosphonate (29b)
Tris[(1R,2S,5R)-menth-2-yl]phosphite is prepared according to literature procedure.30 Hence, to a solution of phosphorus trichloride (1.7 mL, 20.0 mmol) in anhydrous toluene (30 mL) at −20 °C was added slowly a solution of D-menthol (9.4 g, 60.0 mmol) and triethylamine (10 mL) in toluene (50 mL). Upon complete addition, the cooling bath was removed and the reaction mixture was stirred at ambient temperature for 5 h. The white precipitate was filtered off and the filtrate was concentrated under reduced pressure to give tris[(1R,2S,5R)-menth-2-yl]phosphite (9.2 g), which was treated with 4-bromobenzaldehyde (3.4 g, 18.4 mmol) and chlorotrimethylsilane (5.4 mL, 42.6 mmol) at 0 °C. The suspension was stirred at 0 °C for 2 h and then at ambient temperature for an additional 1 h. Volatiles were removed under reduced pressure. The residue was purified by automatic silica gel column chromatography (220 gram flash column, column volume = 334 mL, 150 mL/min, linear gradient of 0–10% hexane/ethyl acetate over 35 min) to give the product as a mixture of diastereoisomers in a ratio of 1:0.7 (8.8 g, 88%). The diastereoisomeric mixture was dissolved in acetonitrile (1 g/100 mL) at 60 °C and cooled to room temperature. The solid was collected by filtration and recrystallized in acetonitrile one more time to give diastereomerically pure 29b. The filtrate solution was cooled to 0 °C. The solid was collected and recrystallized in acetonitrile one more time to give diastereomerically pure 29a. Compound 29a : 1H NMR (400 MHz, CDCl3) δ 0.69 (d, J = 7.2 Hz, 3H), 0.76 (d, J = 6.8 Hz, 3H), 0.80 (d, J = 7.2 Hz, 3H), 0.85–1.41 (m, 19 H), 1.59–1.65 (m, 4H), 1.77–1.81 (m, 1H), 1.97–2.06 (m, 2H), 2.14–2.19 (m, 1H), 3.42 (br, 1H), 4.15–4.24 (m, 2H), 4.92 (dd, J = 11.2, 5.2 Hz, 1H), 7.37 (dd, J = 8.0, 4.0 Hz, 2H), 7.45 (d, J = 8.0 Hz, 2H). 31P NMR (162 MHz, CDCl3) δ 18.75. Compound 29b : 1H NMR (400 MHz, CDCl3) δ 0.72 (d, J = 6.8 Hz, 3H), 0.76 (d, J = 7.2 Hz, 3H), 0.79–1.42 (m, 22H), 1.59–1.70 (m, 4H), 1.91–1.99 (m, 3H), 2.13–2.16 (m, 1H), 3.36 (dd, J = 9.2, 4.8 Hz, 1H), 4.16–4.26 (m, 2H), 4.91 (dd, J = 11.2, 5.2 Hz, 1H), 7.35 (dd, J = 8.0, 4.0 Hz, 2H), 7.45 (d, J = 8.0 Hz, 2H). 31P NMR (162 MHz, CDCl3) δ 19.06.
(S)-((4-bromophenyl)(hydroxy)methyl)phosphonic acid (30b)
To a suspension of 29b (1.8 g, 3.3 mmol) and NaI (2.0 g, 13.3 mmol) in acetonitrile (20 mL) under nitrogen atmosphere was added chlorotrimethylsilane (1.7 mL, 13.5 mmol). The reaction mixture was refluxed for 10 h and then cooled down to ambient temperature. The precipitates were filtered off. The filtrate was concentrated under reduced pressure. The residue was purified by automated reversed-phase C-18 column chromatography (150 gram C18 column, column volume = 130 mL, 85 mL/min, liquid injection, linear gradient of 5–30% acetonitrile in water with 0.1% trifluoroacetic acid over 25 min) to give the title compound as a white solid (0.52 g, 59%). 1H NMR (400 MHz, DMSO-d6) δ 4.62 (d, J = 12.0 Hz, 1H), 7.33 (dd, J = 8.0, 4.0 Hz, 2H), 7.48 (d, J = 8.0 Hz, 2H).31P NMR (162 MHz, DMSO-d6) δ 17.43.
(S)-(Hydroxy(phenyl)methyl)phosphonic Acid
A mixture of 30b (43 mg, 0.16 mmol) and 10% Pd/C (dry, 16 mg) in MeOH (4 mL) was stirred under hydrogen atmosphere (1 atm) for 3 h. The reaction mixture was filtered through a syringe filter (0.2 µm). The filtrate was concentrated to give the title compound (28 mg, 93%). 1H NMR (400 MHz, DMSO-d6) δ 4.64 (d, J = 14.0 Hz, 1H), 7.17–7.39 (m, 5H). 31P NMR (162 MHz, DMSO-d6) δ 18.14. The phosphonic acid was dissolved in ethanol (0.5 mL) and excess of cyclohexylamine (100 µL, 0.87 mmol) was added. The white precipitate was collected by filtration. [α]D −10.6 (c 0.77, 50% MeOH/H2O at 20 °C) (lit.31a −13.8, c 0.77, 50% MeOH/H2O).
(R)-((4-bromophenyl)(hydroxy)methyl)phosphonic Acid (30a)
Compound 30a (0.22 g) was obtained as a white solid in 45% yield starting from 29a (1.0 g, 1.8 mmol) by following the procedure described for the synthesis of compound 30b. The 1H NMR and 31P NMR spectra are the same as those of 30b.
(R)-(3-bromophenyl)(3,4-dichlorophenyl)methanol (31a)
Compound 31a was prepared by adapting a literature procedure.32a To a solution of 3-bromo-1,2-dichlorobenzene (1.00 g, 4.42 mmol) in tert-butyl methyl ether (5.50 mL) at −78 °C was added dropwise n-BuLi (2.5 M in hexanes, 1.77, 4.42 mmol). The reaction mixture was stirred at −78 °C for 1 h. The dry ice/acetone cooling bath was replaced with an ice/water cooling bath. Anhydrous ZnCl2 (0.63 g, 4.64 mmol) was added. The resulting suspension was stirred at 0 °C for 1 h. Additional n-BuLi (2.5 M in hexanes, 1.77 mL, 4.42 mmol) was added. The cooling bath was removed and the reaction mixture was stirred at ambient temperature for 4.5 h. Toluene (anhydrous, 22.5 mL) and tetraethylethylenediamine (0.37 mL, 1.77 mmol) was added. The reaction mixture was stirred at ambient temperature for an additional 1 h. (+)-3-exo-(morpholino)isoborneol ((+)-MIB32, 27 mg, 0.11 mmol) was added. The reaction mixture was cooled to 0 °C and stirred for 0.5 h. 3-Bromobenzaldehyde (0.78 g, 2.21 mmol) was added, and the reaction mixture was stirred at 0 °C for 17 h. Water (30 mL) and ether (30 mL) were added. The phases were separated, and the aqueous layer was extracted with ether (2 × 50 mL). The combined organic solution was dried over anhydrous MgSO4, filtered, and concentrated. The residue was purified by automated silica gel column chromatography (80 gram flash column, column volume = 125 mL, 60 mL/min, linear gradient of 0–50% ethyl acetate in hexane over 25 min) to afford the title compound as a colorless oil (0.51 g, 70%). The enantiomeric excess was determined by HPLC (AS-H column, isopropanol/hexane = 5:95, flow rate = 0.5 mL/min, tr, major = 32.3 min, minor enantiomer tr, minor = 35.4 min, 90% ee). 1H NMR (400 MHz, CDCl3) δ 2.30 (d, J = 3.2 Hz, 1H), 5.75 (d, J = 3.2 Hz, 1H), 7.18 (dd, J = 8.4. 2.0 Hz, 1H), 7.20–7.27 (m, 2H), 7.40–7.44 (m, 2H), 7.48 (d, J = 2.0 Hz, 1H), 7.52 (s, 1H).
(S)-(3-Bromophenyl)(3,4-dichlorophenyl)methanol (31b)
Compound 31b (0.49 g) was obtained as a colorless oil in 67% yield starting from 3-bromo-1,2-dichlorobenzene (1.00 g, 4.42 mmol) by following the procedure used for the synthesis of 31a but replacing the catalyst (+)-MIB with (−)-MIB. The enantiomeric excess was determined by HPLC (AS-H column, isopropanol/hexane = 5:95, flow rate = 0.5 mL/min, tr, major = 35.4 min, tr, minor = 32.3 min, 90% ee). The 1H NMR spectrum is the same as that of 31a.
(S)-(3,4-Dichlorophenyl)(3-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)phenyl)methanol (32a)
Compound 32a (0.28 g) was obtained as a colorless oil in 64% yield starting from 31a (0.38 g, 1.14 mmol) by following the procedure described for the synthesis of 22a. 1H NMR (400 MHz, CDCl3) δ 1.35 (s, 12H), 2.23 (d, J = 3.2 Hz, 1H), 5.80 (d, J = 3.2 Hz, 1H), 7.20 (ddd, J = 8.4, 2.4, 0.8 Hz, 1H), 7.33–7.44 (m, 3H), 7.51 (dd, J = 2.4, 0.8 Hz, 1H), 7.75 (dt, J = 6.8, 1.2 Hz, 1H), 7.79–7.80 (m, 1H).
(S)-(3,4-dichlorophenyl)(3-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)phenyl)methanol (32b)
Compound 32b was prepared in 58% yield starting from 31b (0.50 g, 1.51 mmol) by following the procedure described the synthesis of 32a. The 1H NMR spectrum of 32b is the same as that of 32a.
Supplementary Material
Acknowledgments
Funding Sources
JAE gratefully acknowledges the support of Yale University Set Up Funds and NIH R01 GM054051; PJL acknowledges support from NIMH R01s MH052711 and MH091037.
ABBREVIATIONS
- AD
Alzheimer’s disease
- B2pin2
bis(pinacolato)diboron
- BBB
blood-brain barrier
- cod
1,5-cyclooctadiene
- dppf
1,1’-bis(diphenylphosphino)ferrocene
- LAR
leukocyte common antigen-related
- DFMP
difluoromethylphosphonic acid
- dtbpy
4,4’-di-tert-butyl-2,2’-bipyridine
- HBpin
pinacolborane
- MKP5
mitogen-activated protein (MAP) kinase phosphatase 5
- MIB
3-exo-(morpholino)isoborneol
- PTP
protein tyrosine phosphatase
- STEP
striatal-enriched protein tyrosine phosphatase
- TC-Ptp
T-cell protein tyrosine phosphatase
Footnotes
Supporting Information Available: NMR Spectra of compounds 12s and 12t, enzyme assay curves for the inhibitors, table of antibodies used for Western blotting, and BBB PAMPA results. This material is available free of charge via the Internet at http://pubs.acs.org.
Author Contributions
The manuscript was written with contributions from all authors. All authors have given approval to the final version of the manuscript.
REFERENCES
- 1.For a general overview of synaptic plasticity in neuropsychiatric disorders, see: Lüscher C, Issac JT. The synapse: center stage for many brain diseases. J. Physiol. 2009;587:727–729. doi: 10.1113/jphysiol.2008.167742. Palop JJ, Chin J, Mucke L. A network dysfunction perspective on neurodegenerative diseases. Nature. 2006;443:768–773. doi: 10.1038/nature05289.
- 2.(a) Selkoe DJ. Alzheimer's disease is a synaptic failure. Science. 2002;298:789–791. doi: 10.1126/science.1074069. [DOI] [PubMed] [Google Scholar]; (b) Burke SN, Barnes CA. Neural plasticity in the ageing brain. Nat. Rev. Neurosci. 2006;7:30–40. doi: 10.1038/nrn1809. [DOI] [PubMed] [Google Scholar]
- 3.(a) Stephan KE, Friston KJ, Frith CD. Dysconnection in Schizophrenia: from abnormal synaptic plasticity to failures of self-monitoring. Schizophrenia Bull. 2009;35:509–527. doi: 10.1093/schbul/sbn176. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Stephan KE, Baldeweg T, Friston KJ. Synaptic plasticity and dysconnection in Schizophrenia. Biol. Psychiatry. 2006;59:929–939. doi: 10.1016/j.biopsych.2005.10.005. [DOI] [PubMed] [Google Scholar]
- 4.Duman RS. Pathophysiology of depression: the concept of synaptic plasticity. Eur. Psychiatry. 2002;17:306–310. doi: 10.1016/s0924-9338(02)00654-5. [DOI] [PubMed] [Google Scholar]
- 5.(a) Garner CC, Wetmore DZ. Synaptic pathology of Down syndrome. Adv. Exp. Med. Biol. 2012;970:451–468. doi: 10.1007/978-3-7091-0932-8_20. [DOI] [PubMed] [Google Scholar]; (b) Huttenlocher PR. Dendritic and synaptic pathology in mental retardation. Pediatr. Neurol. 1991;7:79–85. doi: 10.1016/0887-8994(91)90001-2. [DOI] [PubMed] [Google Scholar]; (c) O’Donnell WT, Warren ST. A decade of molecular studies of Fragile X syndrome. Annu. Rev. Neurosci. 2002;25:315–338. doi: 10.1146/annurev.neuro.25.112701.142909. [DOI] [PubMed] [Google Scholar]
- 6.Gerdeman GL, Partridge JG, Lupica CR, Lovinger DM. It could be habit forming: drugs of abuse and striatal synaptic plasticity. Trends Neurosci. 2003;26:184–192. doi: 10.1016/S0166-2236(03)00065-1. [DOI] [PubMed] [Google Scholar]
- 7.Paul S, Nairn A, Wang P, Lombroso PJ. NMDA-mediated activation of the protein tyrosine phosphatase, STEP, regulates the duration of ERK signaling. Nat. Neurosci. 2003;6:34–42. doi: 10.1038/nn989. [DOI] [PubMed] [Google Scholar]
- 8.Xu J, Kurup P, Bartos JA, Hell JW, Lombroso PJ. STriatal-Enriched protein tyrosine Phosphatase (STEP) regulates Pyk2 activity. J. Biol. Chem. 2012;287:20942–20956. doi: 10.1074/jbc.M112.368654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Munoz JJ, Tarrega C, BlancoAparicio C, Pulido R. Differential interaction of the tyrosine phosphatases PTP-SL, STEP and HePTP with the mitogen-activated protein kinases ERK1/2 and p38α is determined by a kinase specificity sequence and influenced by reducing agents. Biochem. J. 2003;372:193–201. doi: 10.1042/BJ20021941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Snyder EM, Nong Y, Almeida CG, Paul S, Moran T, Choi EY, Nairn AC, Salter MW, Lombroso PJ, Gouras GK, Greengard P. Regulation of NMDA receptor trafficking by amyloid-β. Nat. Neurosci. 2005;8:1051–1058. doi: 10.1038/nn1503. [DOI] [PubMed] [Google Scholar]
- 11.(a) Zhang YF, Kurup P, Xu J, Carty N, Fernandez S, Nygaard HB, Pittenger C, Greengard P, Strittmatter S, Nairn AC, Lombroso PJ. Genetic reduction of the tyrosine phosphatase STEP reverses cognitive and cellular deficits in a mouse model of Alzheimer’s disease. Proc. Nat. Acad. Sci. 2010;107:19014–19019. doi: 10.1073/pnas.1013543107. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Zhang YF, Kurup P, Xu J, Anderson G, Greengard P, Nairn AC, Lombroso PJ. Reduced levels of the tyrosine phosphatase STEP block beta amyloid-mediated GluA1/GluA2 receptor internalization. J. Neurochem. 2011;119:664–672. doi: 10.1111/j.1471-4159.2011.07450.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.(a) Soellner MB, Rawls KA, Grundner C, Alber T, Ellman JA. Fragment-based substrate activity screening method for the identification of potent inhibitors of the Mycobacterium tuberculosis phosphatase PtpB. J. Am. Chem. Soc. 2007;129:9613–9615. doi: 10.1021/ja0727520. [DOI] [PubMed] [Google Scholar]; (b) Rawls KA, Lang PT, Takeuchi J, Imamura S, Baguley TD, Grundner C, Alber T, Ellman JA. Fragment-based discovery of selective inhibitors of the Mycobacterium tuberculosis protein tyrosine phosphatse PtpA. Bioorg. Med. Chem. Lett. 2009;19:6851–6854. doi: 10.1016/j.bmcl.2009.10.090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sahn JJ, Martin SF. Expedient synthesis of norbenzomorphan library via multicomponent assembly process coupled with ring-closing reactions. ACS Comb. Sci. 2012;14:496–502. doi: 10.1021/co300068a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.For a discussion of the high incidence of false positives in phosphatase, see: Moretto AF, Kirincich SJ, Xu WX, Smith MJ, Wan ZK, Wilson DP, Follows BC, Binnun E, Joseph-McCarthy D, Foreman K, Erbe DV, Zhang YL, Tam SK, Tam SY, Lee J. Bicyclic and tricyclic thiophenes as protein tyrosine phosphatase 1B inhibitors. Bioorg. Med. Chem. 2006;14:2162–2177. doi: 10.1016/j.bmc.2005.11.005.
- 15.For recent reviews and leading references on the development of phosphatase inhibitors, see: Vintonyak VV, Waldmann H, Rauh D. Inhibitors of protein tyrosine phosphatases: next-generation drugs? Bioorg. Med. Chem. 2011;19:2145–2155. doi: 10.1016/j.bmc.2011.02.047. Combs AP. Recent advances in the discovery of competitive protein tyrosine phosphatase 1B inhibitors for the treatment of diabetes, obesity, and cancer. J. Med. Chem. 2010;53:2333–2344. doi: 10.1021/jm901090b. Blaskovich MAT. Drug discovery and protein tyrosine phosphatases. Curr. Med. Chem. 2009;16:2095–2176. doi: 10.2174/092986709788612693.
- 16.(a) Hajduk PJ, Bures M, Praesstgaard J, Fesik SW. Privileged molecules for protein binding identified from NMR-based screening. J. Med. Chem. 2000;43:3443–3447. doi: 10.1021/jm000164q. [DOI] [PubMed] [Google Scholar]; (b) Horton DA, Bourne GT, Smythe ML. The combinatorial synthesis of bicyclic privileged structures or privileged substructures. Chem. Rev. 2003;103:893–930. doi: 10.1021/cr020033s. [DOI] [PubMed] [Google Scholar]
- 17.Burke TR, Kole HK, Roller PP. Potent inhibition of insulin receptor dephosphorylation by a hexamer peptide containing the phosphotyrosyl mimetic F2Pmp. Biochem. Biophys. Res. Commun. 1994;204:129–134. doi: 10.1006/bbrc.1994.2435. [DOI] [PubMed] [Google Scholar]
- 18.Montalibet J, Skorey KI, Kennedy BP. Protein tyrosine phosphatase: enzymatic assays. Methods. 2005;35:2–8. doi: 10.1016/j.ymeth.2004.07.002. [DOI] [PubMed] [Google Scholar]
- 19.For reviews and general references, see: McGovern SL, Caselli E, Grigorieff N, Shoichet BK. A common mechanism underlying promiscuous inhibitors from virtual and high-throughput screening. J. Med. Chem. 2002;45:1712–1722. doi: 10.1021/jm010533y. McGovern SL, Helfand BT, Feng B, Shoichet BK. A specific mechanism of nonspecific inhibition. J. Med. Chem. 2003;46:4265–4272. doi: 10.1021/jm030266r. Shoichet BK. Screening in a spirit haunted world. Drug Discovery Today. 2006;11:607–615. doi: 10.1016/j.drudis.2006.05.014.
- 20.Murano T, Takechi H, Yuasa Y, Yokomatsu T, Umesue I, Soeda S, Shimeno H, Shibuya S. The difluoromethylenephosphonothioic acid group as a phosphate surrogate for obtaining PTP-1B inhibitors. Transformation of difluoromethyenephosphonic acids into difluoromethylenephosphonothioic acids. ARKIVOC. 2003;8:256–266. [Google Scholar]
- 21.(a) Montserat J, Chen L, Lawrence DS, Zhang Z-Y. Potent low molecular weight substrates for protein-tyrosine phosphatase. J. Biol. Chem. 1996;271:7868–7872. doi: 10.1074/jbc.271.13.7868. [DOI] [PubMed] [Google Scholar]; (b) Bahta M, Lountos GT, Dyas B, Kim S-E, Ulrich RG, Waugh DS, Burke TR., Jr Utilization of nitrophenylphosphates and oxime-based ligation for the development of nanomolar affinity inhibitors of the Yersinia pestis outer protein H (YopH) phosphatase. J. Med. Chem. 2011;54:2933–2943. doi: 10.1021/jm200022g. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Chen L, Montserat J, Lawrence DS, Zhang Z-Y. VHR and PTP1 protein phosphatases exhibit remarkably different active site specificities toward low molecular weight nonpeptidic substrates. Biochemistry. 1996;35:9349–9354. doi: 10.1021/bi960700+. [DOI] [PubMed] [Google Scholar]
- 22.(a) Di L, Kerns EH, Bezar IF, Petusky SL, Huang Y. Comparison of blood-brain barrier permeability assays: in situ brain perfusion, MDR1-MDKII and PAMPA-BBB. J. Pharm. Sci. 2009;98:1980–1991. doi: 10.1002/jps.21580. [DOI] [PubMed] [Google Scholar]; (b) Mensch J, Melis A, Mackie C, Verreck G, Brewster ME, Augustijns P. Evaluation of various PAMPA models to identify the most discriminating method for the prediction of BBB permeability. Eur. J. Pharm. Biopharm. 2010;74:495–502. doi: 10.1016/j.ejpb.2010.01.003. [DOI] [PubMed] [Google Scholar]
- 23.(a) Rautio J, Laine K, Gynther M, Savolainen J. Prodrug Approaches for CNS Delivery. AAPS J. 2008;10:92–102. doi: 10.1208/s12248-008-9009-8. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Pavan B, Dalpiaz A, Ciliberti N, Biondi C, Manfredini S, Vertuani S. Progress in drug discovery to the central nervous system by the prodrug approach. Molecules. 2008;13:1035–1065. doi: 10.3390/molecules13051035. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Prokai L, Prokai-Tatrai K, Bodor N. Targeting Drugs to the Brain by Redox Chemical Delivery Systems. Med. Res. Rev. 2000;20:367–416. doi: 10.1002/1098-1128(200009)20:5<367::aid-med3>3.0.co;2-p. [DOI] [PubMed] [Google Scholar]
- 24.Fujisaki S, Eguchi H, Omura A, Okamato A, Nishida A. Halogenation using N-halogenocompounds. I. Effect of amines on ortho-bromination of phenols with NBS. Bull. Chem. Soc. Jpn. 1993;66:1576–1579. [Google Scholar]
- 25.Konakahara T, Kiran YB, Okuno Y, Ikeda R, Sakai N. An expedient synthesis of ellipticine via Suzuki-Miyaura coupling. Tetrahedron Lett. 2010;51:2335–2338. [Google Scholar]
- 26.Boebel TA, Hartwig JF. Silyl-Directed, Iridium-catalyzed ortho-borylation of arenes. A one-pot ortho-borylation of phenols, arylamines, and alkylarenes. J. Am. Chem. Soc. 2008;130:7534–7535. doi: 10.1021/ja8015878. [DOI] [PubMed] [Google Scholar]
- 27.Ishiyama T, Murata M, Miyaura N. Palladium(0)-catalyzed cross-coupling reaction of alkoxydiboron with haloarenes: a direct procedure for arylboronic esters. J. Org. Chem. 1995;60:7508–7510. [Google Scholar]
- 28.Vedso P, Olesen PH, Hoeg-Jensen T. Synthesis of sulfonyl chlorides of phenyl-boronic acids. Synlett. 2004;5:892–894. [Google Scholar]
- 29.Li X, Szardenings AK, Holmes CP, Wang L, Bhandari A, Shi L, Navre M, Jang L, Grove JR. A new and direct approach to functionalized biaryl α-ketophosphonic acids via aqueous Suzuki coupling on solid support. Tetrahedron Lett. 2006;47:19–22. [Google Scholar]
- 30.(a) Kolodiazhnyi OI, Grishkun EV, Sheiko S, Demchuk O, Thoennessen H, Jones P, Schmutzler R. Asymmetric synthesis of α-substituted alkylphosphonates based on symmetrical dialkyl phosphites. Russ. Chem. Bull. 1999;48:1568–1573. [Google Scholar]; (b) Kolodiazhnyi OI, Sheiko S, Grishkun EV. C3-symmetric trialkyl phosphites as starting compounds of asymmetric synthesis. Heteroatom. Chem. 2000;11:138–143. [Google Scholar]
- 31.(a) Blazis VJ, Koeller KJ, Spilling CD. Reactions of chiral phosphorous acid diamides: the asymmetric synthesis of chiral α-hydroxy phosphonamides, phosphonates, and phosphonic acids. J. Org. Chem. 1995;60:931–940. [Google Scholar]; (b) Smaardijk AA, Noorda S, Van Bolhuis F, Wynberg H. The absolute configuration of α-hydroxyphosphonates. Tetrahedron Lett. 1985;26:493–496. [Google Scholar]
- 32.(a) Salvi L, Kim JG, Walsh PJ. Practical catalytic asymmetric synthesis of diaryl-, aryl heteroaryl-, and diheteroarylmethanols. J. Am. Chem. Soc. 2009;131:12483–12493. doi: 10.1021/ja9046747. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Nugent WA. MIB: an advantageous alternative to DAIB for the addition of organozinc reagents to aldehydes. Chem. Commun. 1999:1369–1370. [Google Scholar]
- 33.Ishiyama T, Murata M, Miyaura N. Palladium(0)-catalyzed cross-coupling reaction of alkoxydiboron with haloarenes: a direct procedure for arylboronic esters. J. Org. Chem. 1995;60:7508–7510. [Google Scholar]
- 34.Xu J, Kurup P, Zhang YF, Goebel-Goody S, Wu P, Hawasli AH, Baum M, Bibb J, Lombroso PJ. Extrasynaptic NMDA receptors couple preferentially to excitotoxicity via calpain-mediated cleavage of STEP. J. Neuroscience. 2009;29:9330–9343. doi: 10.1523/JNEUROSCI.2212-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Li X, Szardenings AK, Holmes CP, Wang L, Bhandari A, Shi L, Navre M, Jang L, Grove JR. A new and direct approach to functionalized biaryl α-ketophosphonic acids via aqueous Suzuki coupling on solid support. Tetrahedron Lett. 2006;47:19–22. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.