

Abstract
This report describes the discovery and initial characterization of the first positive allosteric modulator of muscarinic acetylcholine receptor subtype 5 (mAChR5 or M5). Functional HTS, identified VU0119498, which displayed micromolar potencies for potentiation of acetylcholine at M1, M3, and M5 receptors in cell-based Ca2+ mobilization assays. Subsequent optimization led to the discovery of VU0238429, which possessed an EC50 of approximately 1.16 µM at M5 with >30-fold selectivity versus M1 and M3, with no M2 or M4 potentiator activity.
The five cloned muscarinic acetylcholine receptor subtypes (mAChR1–5 or M1–5a) are known to play highly important and diverse roles in many basic physiological processes including gastrointestinal, cardiovascular, motor, attention, learning, memory, pain, sleep, and other functions.1–3 Correspondingly, muscarinic agonists and antagonists targeting one or more subtypes have been used preclinically and clinically for research and treatment of a wide range of pathologies.3,4 Given the high sequence homology of the mAChRs across subtypes and particularly within the orthosteric acetylcholine (ACh) binding site, discovery of truly subtype-selective compounds has proven historically difficult. Because of the paucity of selective compounds, a detailed understanding of the precise roles of each subtype in neurobiology and in various central nervous system (CNS) disorders has thus remained challenging.3,4
Recently, a number of novel highly subtype-selective allosteric ligands for M1 and M4 have emerged from functional cell-based screening efforts.5,6 However, no ligands have been reported to date as being highly M5-preferring or selective. Relative to the other mAChRs, little is known about M5, which is expressed at very low levels in the CNS and peripheral tissues.2–4
Interestingly, data from studies using mAChR5 knockout (M5-KO) mice suggest that M5 is the sole mediator of ACh-induced vasodilation in the cerebral vasculature and thereby may have therapeutic relevance for cerebrovascular diseases or acute ischemic stroke.7,8 M5-KO mice have also been found to exhibit deficits in long-term potentiation (LTP) at the hippocampal mossy fiber-CA3 synapse and show deficits in hippocampal-dependent behavioral cognitive tests.8
In light of these and related findings, activation of M5 has been suggested as a potential target for treatment of Alzheimer’s disease, perhaps in combination with M1 activation.9 Consistent with the putative postsynaptic localization of M5 in the ventral tegmental area (VTA), other M5-KO data suggest this subtype plays an important role in regulation of mesolimbic dopamine transmission.3,9 Indeed, M5-KO mice exhibit decreased reward responses to morphine, decreased self-administration of cocaine, and less pronounced drug withdrawal symptoms, suggesting that M5 antagonists or negative modulators may have therapeutic value in treatment of illicit drug addiction.9–11 Further pharmacological exploration of these and related hypotheses greatly depends on the discovery of novel M5-preferring or selective small molecule tools.
We recently reported on a diverse group of novel mAChR positive allosteric modulators (PAMs), some of which were highly selective for M1 or M4 (Chart 1, Figure 1).6,12,13 Other mAChR PAMs displayed mixed subtype-selectivity profiles.12 These compounds enhanced receptor activation in response to ACh in Ca2+ mobilization assays and did not compete with the orthosteric antagonist [3H]-N-methylscopolamine (NMS) in radioligand binding experiments performed with M1-CHO membranes, which strongly suggests an allosteric mechanism of modulation.12 Interestingly, the p-bromobenzyl-substituted isatin screening hit VU0119498 (113) was found to exhibit allosteric potentiator activity at the natively Gq-coupled M1, M3, and M5 receptors with comparable potency and efficacy in Ca2+ mobilization assays but lacked potentiator effects at the natively Gi-coupled M2 and M4 receptors in cells cotransfected with chimeric Gqi5 protein, which facilitates coupling to the PLC/ Ca2+ pathway (Figure 1).12 In similar assays, a high fixed 30 µM concentration of 113 induced a 14-fold leftward shift of a full ACh concentration response curve (CRC), signifying a robust potentiation effect. This hit was therefore chosen as an attractive starting point for subsequent optimization efforts directed toward increasing M5 selectivity and potency while simultaneously decreasing M1 and M3 activity.
Chart 1.

M1 and M4 Positive Allosteric Modulators
Figure 1.

HTS hit 113 potentiates responses in M1, M3, and M5-expressing CHO cells. In the presence of a fixed submaximal concentration of ACh (~EC20) in Ca2+, assays performed with CHO cells stably expressing each of the five mAChR subtypes (M2 and M4 cotransfected with Gqi5). M1 EC50 = 6.04 µM, %max = 82.4; M3 EC50 = 6.38 µM, %max = 65.5; M5 EC50 = 4.08 µM, %max = 74.4; inactive at M2 and M4 (data shown are means ± SEM, N ≥ 3).
To rapidly explore SAR around 113, we generated an analogue library in matrix format wherein each of eight commercially available isatins (1–8) was reacted with approximately 12 benzyl halides (9–20) under standard microwave alkylation conditions (Scheme 1).14 Resulting analogues (21–112) were then screened in a single point format at a 30 µM final concentration in Ca2+ mobilization assays using M5 and M1 cells receiving a fixed submaximal concentration (~EC20) of ACh (Figure 2, see full SAR table in Supporting Information).14 This method efficiently triaged analogues displaying high M1 vs M5 or M5 vs M1 preference. Interestingly, some analogues displayed robust potentiation effects at M5 (i.e., elevation of ACh ~EC20 to >50–60% of maximum ACh response) with absent or weak potentiation at M1, thus exhibiting strong preference for M5 versus M1 activity.
Scheme 1.

Library Synthesis of VU0119498 Analoguesa
aReagents and conditions: (a) K2CO3, KI, ACN, microwave 160°C, 10 min, 20–95%.
Figure 2.

Screen of analogue library (compounds 21–112) at 30 µM for potentiation of submaximal acetylcholine (~EC20) in M1 (A) and M5 (B) cells by Ca2+ assay (data shown are means ± SEM, N ≥3).14
In terms of maximal potentiation efficacy, SAR from the initial 30 µM screen of the entire library suggested that 5-OCF3 substitution of the isatin core (R1) was generally favored for increased M5 versus M1 activity, while halogen substitutions at the 7-position conferred a more dual M1/M5 activity (Table 1).14 Indeed, all 5-OCF3 substituted compounds chosen from the initial screen possessed 1–5 µM potencies at M5 and >30 µM potencies at M1, with the exception of compunds 63 and 112, which nonetheless possessed greater M5 versus M1 activities (Table 1).14 Numerous benzylic substitutions (R2) were tolerated for M1 and M5 activity, depending on the isatin core (R1).14 Methoxybenzyl was generally favored for M5 versus M1 activity, whereas trifluoromethylbenzyl was generally favored for dual M1/M5 potentiation activity (Table 1).14 Most other simple congeners, including those with various methyl substitutions at R1 or R2, displayed weak or no potentiation activity at either receptor.14 In light of the high M5 versus M1 potentiation preference displayed by analogue 42 (1.16 µM M5 EC50 and >30 µM M1 EC50) in these assays, the full subtype-selectivity profile of this compound was obtained in similar Ca2+ assays using M2, M3, and M4 cells. Likewise, full subtype-selectivity determination was also performed with compound 56, which was the most potent dual M1/M5 potentiator in this initial evaluation at both receptors (2.11 µM M5 EC50 and 3.20 µM M1 EC50).
Table 1.
Structures and Activities at M1 and M5 of Analogues of Compound 113
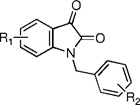 | ||||
|---|---|---|---|---|
| compd | R1 | R2 | M1 EC50 (µM) | M5 EC50 (µM) |
| 22 | H | 4-OMe | >10 | >30 |
| 25 | 7-F | 4-Br | 3.99 | 1.93 |
| 33 | 7-Cl | 4-OMe | 5.38 | 3.96 |
| 38 | 4,7-Cl | 3-OMe | >30 | 7.15 |
| 41 | 5-OCF3 | 3-OMe | >30 | 1.70 |
| 42 | 5-OCF3 | 4-OMe | >30 | 1.16 |
| 44 | H | 4-CF3 | 5.88 | 3.19 |
| 46 | 5-F | 2-CF3 | 6.21 | 4.09 |
| 47 | 5-F | 4-CF3 | 8.55 | 7.13 |
| 49 | 7-F | 2-CF3 | >30 | >30 |
| 50 | 7-F | 4-CF3 | 5.18 | 3.26 |
| 56 | 7-Cl | 4-CF3 | 3.20 | 2.11 |
| 63 | 5-OCF3 | 4-CF3 | 7.09 | 1.85 |
| 64 | 5-OCF3 | 2-Cl | >30 | 4.07 |
| 103 | 7-Cl | 2-F, 4-Br | 7.14 | 4.66 |
| 110 | 5-OCF3 | 4-OCF3 | >30 | 1.86 |
| 111 | 5-OCF3 | 2-Me | >30 | 3.28 |
| 112 | 5-OCF3 | 2-F, 4-Br | >30 | >10 |
Remarkably, 42 (VU0238429) had similarly poor potency at M3 as at M1, with potentiation of the ACh ~EC20 beginning to emerge only in the double-digit micromolar range (Figure 3). Gratifyingly, this compound maintained its lack of potentiation activity at M2 and M4 in cells cotransfected with Gqi5 at up to 30 µM (Figure 3). Given these data, the approximately 1.16 µM M5 potency of compound 42 provides >30× selectivity for M5 versus the other four subtypes, thus representing the first highly M5-preferring muscarinic receptor ligand ever reported. Conversely, compound 56 (VU0238441) was found to retain the M1/M3/M5 potentiation profile of the parent HTS hit 113 and interestingly possessed modest potentiation at M2 and M4 at high concentrations (Figure 4).
Figure 3.

Concentration response curve for 42 performed in the presence of a fixed ACh ~EC20 in Ca2+ assays performed with CHO cells stably expressing each of the five mAChR subtypes (M2 and M4 cotransfected with Gqi5). M1 EC50 = >30 µM; M3 EC50 = >30 µM; M5 EC50 = 1.16 µM, %max = 91; inactive at M2 and M4 (data shown are means ± SEM, N ≥ 3).
Figure 4.

Concentration response curve for 56 performed in the presence of a fixed ACh ~EC20 in Ca2+ assays performed with CHO cells stably expressing each of the five mAChR subtypes (M2 and M4 cotransfected with Gqi5). M1 EC50 = 3.20 µM, %max = 87; M3 EC50 = 2.22 µM, %max = 78; M5 EC50 = 2.11 µM, %max = 86; M2 EC50 = 2.84 µM, %max = 55; M4 EC50 = >10 µM (data shown are means ± SEM, N ≥ 3).
To further evaluate the ability of 42 to potentiate ACh-induced activation of mAChR5, we obtained full CRCs for ACh in M5 cells in the presence of either vehicle or a fixed (30 µM) concentration of 42 (Figure 5, top right).
Figure 5.

Compound 42 CRC alone or in the presence of fixed ACh EC20 demonstrating an absence of intrinsic agonist activity at M5 by Ca2+ assay (top left), effect of fixed 30 µM 42 on an ACh CRC demonstrating a 14-fold increase in ACh potency at M5 by Ca2+ assay (top right), lack of competition with [3H-NMS] binding to M5 by 42 versus atropine control (Ki = 0.21 nM, bottom left), and the effect of fixed 30 µM 42 on ACh competition with [3H-NMS] binding demonstrating a 10-fold increase in ACh affinity for M5 (ACh + vehicle Ki = 1.61 µM; ACh + 42 Ki = 159 nM; data shown are means ± SEM, N ≥ 3).
In these assays, compound 42 induced a robust 14-fold left-shift of the ACh CRC, which was virtually identical to that found with the parent HTS hit 113 when evaluated in a similar assay at M1, as previously reported.12 Moreover, 42 lacked intrinsic agonist activity at M5 when tested up to 30 µM in the absence of ACh (Figure 5, top left).
We also evaluated HTS hit 113 and the highly M5-preferring analogue 42 in competition-binding experiments with M5-CHO membrane preparations using the orthosteric radioligand [3H]-N-methylscopolamine ([3H]-NMS), a potent pan-mAChR antagonist. At up to 30 µM, 42 lacked competition with [3H]-NMS, consistent with an allosteric mode of binding to M5 (Figure 5, bottom left). By contrast, 113 exhibited approximately 50% inhibition of radioligand binding at 30 µM (Figure 5, bottom left). Compound 42 also caused a 10-fold increase in ACh affinity for M5 at 30 µM, indicating that its mechanism of potentiation is in part due to enhancement of ACh binding to M5.
We then evaluated pharmacokinetics and brain penetration for the M5 PAM to evaluate its ability to serve as an in vivo probe. Unfortunately, 42 is characterized by poor systemic absorption after intraperitoneal administration with maximum concentration in plasma (161.7 ng/mL) being achieved within 1 h.14 However, it is slowly eliminated from systemic circulation and has elimination half-life of 4.7 h.14 Although quickly taken up in the brain, it exhibits poor brain penetration with AUCbrain/ AUCplasma value of 0.25.14 Future lead optimization will focus on improving brain penetration.
Although a growing number of allosteric modulators have been recently documented for mAChR1 and mAChR4, to date no selective mAChR5 ligand has been reported. Compound 42, a congener of 113 identified from an analogue library synthesis approach, serves as the first highly mAChR5-preferring small molecule. Despite the relatively limited scope of this analogue library in terms of structural modification, we found clear SAR associated with this novel mAChR chemotype, with 5-OCF3 modification of the isatin core and 3- or 4-position OMe substitutions of the benzyl ring conferring strong M5-preference versus M1–M4. In contrast, virtually all modifications conferring increased M1 activity simultaneously increased M5 activity.14 As exemplified in the case of 56, 7-Cl substitution of the isatin core carried similar and increased M1/M5 potency and efficacy but also caused degeneration into a panmuscarinic potentiator when profiled at all five subtypes. Further chemical modification of compound 42 to improve M5 PAM potency and characterization in electrophysiology and in vivo experiments are underway and will be reported in due course.
Supplementary Material
Acknowledgment
We thank the NIH and NIMH for support of our programs. We specifically acknowledge the support of the Alzheimer’s Association (IIRG-07-57131) and NIMH (1RO1MH082867-01 and 5R01MH073673). T.M.B. acknowledges an ITTD (T90-DA022873) predoctoral training grant.
Footnotes
Abbreviations: mAChR, muscarinic acetylcholine receptor; ACh, acetylcholine; CNS, central nervous system; KO, knockout; LTP, long-term potentiation; VTA, ventral tegmental area; PAM, positive allosteric modulator; NMS, N-methylscopolamine; AUC, area under curve; CRC, concentration–response curve.
Supporting Information Available: Experimental procedures and analytical data for compounds 113, 42, and 56 are provided as kinetics study. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Bonner TI, Buckley NJ, Young AC, Brann MR. Identification of a Family of Muscarinic Acetylcholine Receptor Genes. Science. 1987;237:527–532. doi: 10.1126/science.3037705. [DOI] [PubMed] [Google Scholar]
- 2.Bonner TI, Young AC, Brann MR, Buckley NJ. Cloning and Expression of the Human and Rat m5 Muscarinic Acetylcholine Receptor Genes. Neuron. 1988;1:403–410. doi: 10.1016/0896-6273(88)90190-0. [DOI] [PubMed] [Google Scholar]
- 3.Wess J. Muscarinic Acetylcholine Receptor Knockout Mice: Novel Phenotypes and Clinical Implications. Annu. ReV. Pharmacol. Toxicol. 2004;44:423–450. doi: 10.1146/annurev.pharmtox.44.101802.121622. [DOI] [PubMed] [Google Scholar]
- 4.Langmead CJ, Watson J, Reavill C. Muscarinic acetylcholine receptors as CNS drug targets. Pharmacol. Ther. 2008;117:232–243. doi: 10.1016/j.pharmthera.2007.09.009. [DOI] [PubMed] [Google Scholar]
- 5.Birdsall NJM, Lazareno S. Allosterism at Muscarinic Receptors: Ligands and Mechanisms. Mini. ReV. Med. Chem. 2005;5:523–543. doi: 10.2174/1389557054023251. [DOI] [PubMed] [Google Scholar]
- 6.Conn PJ, Christopoulos A, Lindsley CW. Allosteric modulators of GPCRs: a novel approach for the treatment of CNS disorders. Nat. ReV. Drug DiscoVery. 2009;8:41–54. doi: 10.1038/nrd2760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Yamada M, Lamping KG, Duttaroy A, Zhang W, Cui Y, Bymaster FP, McKinzie DL, Felder CC, Deng C, Faraci FM, Wess J. Cholinergic dilation of cerebral blood vessels is abolished in M5 muscarinic acetylcholine receptor knockout mice. Proc. Natl. Acad. Sci. U.S.A. 2001;98:14096–14101. doi: 10.1073/pnas.251542998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Araya R, Noguchi T, Yuhki M, Kitamura N, Higuchi M, Saido TC, Seki K, Itohara S, Kawano M, Tanemura K, Takashima A, Yamada K, Kondoh Y, Kanno I, Wess J, Yamada M. Loss of M5 muscarinic acetylcholine receptors leads to cerebrovascular and neuronal abnormalities and cognitive deficits in mice. Neurobiol. Dis. 2006;24:334–344. doi: 10.1016/j.nbd.2006.07.010. [DOI] [PubMed] [Google Scholar]
- 9.Wess J, Eglen RM, Gautam D. Muscarinic acetylcholine receptors: mutant mice provide new insights for drug development. Nat. ReV. Drug DiscoVery. 2007;6:721–733. doi: 10.1038/nrd2379. [DOI] [PubMed] [Google Scholar]
- 10.Basile AS, Fedorova I, Zapata A, Liu X, Shippenberg T, Duttaroy A, Yamada M, Wess J. Deletion of the M5 muscarinic acetylcholine receptor attenuates morphine reinforcement and withdrawal but not morphine analgesia. Proc. Natl. Acad. Sci. U.S.A. 2002;99:11452–11457. doi: 10.1073/pnas.162371899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Thomsen M, Woldbye DPD, Wortwein G, Fink-Jensen A, Wess J, Caine SB. Reduced Cocaine Self-Administration in Muscarinic M5 Acetylcholine Receptor-Deficient Mice. J. Neurosci. 2005;25:8141–8149. doi: 10.1523/JNEUROSCI.2077-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Marlo JE, Niswender CM, Days EL, Bridges TM, Xiang Y, Rodriguez AL, Shirey JK, Brady AE, Nalywajko T, Luo Q, Austin CA, Williams MB, Kim K, Williams R, Orton D, Brown HA, Lindsley CW, Weaver CD, Conn PJ. Discovery and characterization of novel allosteric potentiators of m1 muscarinic receptors reveals multiple modes of activity. Mol. Pharmacol. 2009;75:577–588. doi: 10.1124/mol.108.052886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Brady AE, Jones CK, Bridges TM, Kennedy JP, Thompson AD, Heiman JU, Breininger ML, Gentry PR, Yin H, Jadhav SB, Shirey JK, Conn PJ, Lindsley CW. Centrally active allosteric potentiators of the M4 muscarinic acetylcholine receptor reverse amphetamine-induced hyperlocomotor activity in rats. J. Pharmacol. Exp. Ther. 2008;327:941–953. doi: 10.1124/jpet.108.140350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.See Supporting Information for details.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.