

Abstract
Diverse eukaryotic hosts produce virus-derived small interfering RNAs (siRNAs) to direct antiviral immunity by RNA interference (RNAi). However, it remains unknown whether the mammalian RNAi pathway has a natural antiviral function. Here, we show that infection of hamster cells and suckling mice by Nodamura virus (NoV), a mosquito-transmissible RNA virus, requires RNAi suppression by its B2 protein. Loss of B2 expression or its suppressor activity leads to abundant production of viral siRNAs and rapid clearance of the mutant viruses in mice. However, viral small RNAs detected during virulent infection by NoV do not have the properties of canonical siRNAs. These findings have parallels with the induction and suppression of antiviral RNAi by the related Flock house virus in fruit flies and nematodes and reveal a mammalian antiviral immunity mechanism mediated by RNAi.
RNA interference (RNAi) acts as a natural antiviral defense in plants, insects, nematodes, and fungi; accordingly, virulent infection in these organisms requires suppression of antiviral RNAi by a virus-encoded suppressor of RNAi (VSR) (1–12). Induction of antiviral RNAi depends on the processing of virus-specific double-stranded RNA (dsRNA) by Dicer nuclease into 21- to 24-nucleotide (nt) small interfering RNAs (siRNAs), which are short dsRNAs with two unpaired nucleotides at the 3′ end of either strand (1–9). Mammalian viral mRNAs are as susceptible as cellular mRNAs to RNAi programmed by synthetic siRNAs, and virus-derived small RNAs (vsRNAs) are found in mammalian cells infected by RNA viruses (9, 13, 14). Mammalian viral proteins that can suppress insect and plant RNAi or artificially induced RNAi in mammalian cells have been identified, and the virulence function of one such protein can be complemented by distinct siRNA-sequestering plant VSRs (9, 15–19). However, it remains unknown whether virus infection triggers production of canonical viral siRNAs in mammals or if mammalian virus infections require specific suppression of an antiviral RNAi response (9).
Nodamura virus (NoV) is mosquito-transmissible, highly virulent to suckling mice and suckling hamsters, and belongs to the same bipartite positive-strand RNA virus genus as Flock house virus (FHV), an insect pathogen (20). FHV infection in Drosophila requires expression of its VSR protein B2, a dsRNA-binding protein, to inhibit Dicer processing of dsRNA viral replication intermediates into siRNAs (3, 12, 21–24). Clearance of a B2-deficient FHV mutant in cultured Drosophila cells is therefore associated with abundant accumulation of viral siRNAs (24). Because the B2 ortholog of NoV exhibits similar in vitro VSR activities and suppresses experimental RNAi in mammalian cells (15, 16, 24), we reasoned that use of NoVΔB2, a B2-deficient mutant of NoV (25), to challenge baby hamster kidney 21 (BHK-21) cells might facilitate detection of mammalian viral siRNAs. In two independent experiments, we compared deep sequencing profiles of 18- to 28-nt small RNAs from BHK-21 cells 2 or 3 days postinoculation (dpi) with either NoVor NoVΔB2. In cells infected by NoV, vsRNAs were highly abundant, but they displayed an overwhelming bias for positive strands (~97%), showed no size preference expected for Dicer products (Fig. 1A and table S1), and are likely breakdown products from the abundant positive-strand viral RNAs (9).
Fig. 1. siRNA properties of vsRNAs in BHK-21 cells.
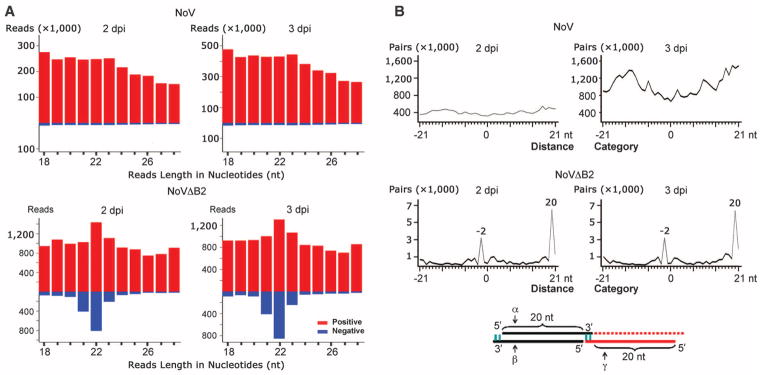
(A) Length distribution and abundance of positive- or negative-strand vsRNAs from cells 2 or 3 dpi with NoV or NoVΔB2. (B) Total counts of pairs of complementary 22-nt vsRNAs of NoV or NoVΔB2 in each distance category (in nucleotides) between 5′ and 3′ ends of a complementary vsRNA pair, defined as 0 for perfect base-paired 22-nt vsRNAs with blunt ends, –2 for pairs with 2-nt overhang at the 3′-end of each strand (α and β), or 20 for pairs with 20-nt overhang at the 5′-ends (α and γ).
By contrast, vsRNAs from NoVΔB2-infected cells were much less abundant and exhibited reduced positive-strand bias (~85%) (table S1). Notably, ~77% of the total negative-strand vsRNA reads in both libraries were in the 21- to 23-nt size range with a major 22-nt peak, similar to Dicer-dependent cellular microRNAs (Fig. 1A and fig. S1A). The unique negative-strand vsRNAs also had a dominant 22-nt peak (fig. S1B). Therefore, NoVΔB2 vsRNAs display patterns of length distribution and strand bias expected for Dicer products as found for plant and invertebrate viral siRNAs (9).
NoVΔB2 vsRNAs exhibited properties of canonical siRNAs (Fig. 1B and table S1). First, both NoVΔB2 libraries were enriched for a population of 22-nt vsRNAs that contained a 20-nt perfectly base-paired duplex region with 2-nt 3′ overhangs (Fig. 1B, peak “−2” and siRNAs α/β). Enrichment for 22-nt canonical siRNA pairs was not found for the comparably much more abundant vsRNAs of NoV (Fig. 1B). Second, we detected a more dominant population of complementary 22-nt vsRNA pairs with 20-nt 5′-end overhangs only for NoVΔB2 vsRNAs (Fig. 1B, peak “20” and siRNAs α/γ). These findings together suggest Dicer-dependent processing of the same viral dsRNA precursor into successive 22-nt viral siRNA duplexes in cells infected by NoVΔB2, but not by NoV.
In contrast to the efficient infection of BHK-21 cells by B2-expressing NoV (25), NoVΔB2 maintained infection only at low levels (Fig. 2). Higher accumulation levels of NoVΔB2 were restored (Fig. 2), however, in BHK-21 cells engineered with a stably expressed transgene encoding either NoV B2 or Ebola virus virion protein 35 (VP35), the latter of which suppresses experimental RNAi in mammalian cells by a distinct mechanism (26, 27). These results show that RNAi suppression by a cognate or heterologous VSR expressed from either the viral genome or an ectopic transgene is essential for robust virus infection in mammalian cells. We conclude therefore that NoVΔB2 is defective only in RNAi suppression, and the RNAi response induced by NoVΔB2, characterized by the production of viral siRNAs, has potent antiviral activity in BHK-21 cells. Consistently, rescue of NoVΔB2 infection was also observed in RNAi-defective mouse embryonic stem cells deleted of all Argonaute genes (28).
Fig. 2. NoV infection requires RNAi suppression.
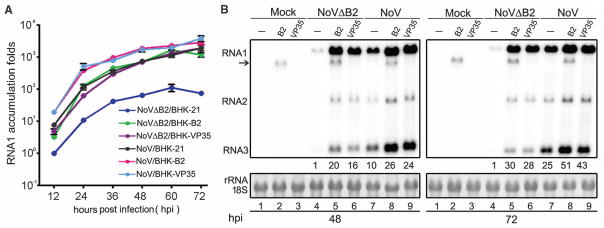
(A) BHK-21 cells or BHK cells stably expressing B2 or VP35 were mock-infected or infected by NoVΔB2 or NoV of the same titer. Every 12 hours postinfection (hpi), the viral genomic RNA1 levels were determined by quantitative RT-PCR with the accumulation level of NoVΔB2 in BHK-21 cells at 12 hpi set as 1. Error bars indicate standard deviation of three replicates. (B) Accumulation of NoV and NoVΔB2 RNAs 1 to 3 in the infected cells detected by Northern blotting. RNA1 signal quantified by phosphorimaging was shown with that of NoVΔB2 in BHK-21 cells (lanes 4) set as 1. Detection of B2 transgene mRNA (arrow) was visible. 18S rRNA staining served as loading control.
NoV is lethal to 7-day-old mice when injected intraperitoneally (29). Quantitative reverse transcription polymerase chain reaction (RT-PCR) analysis validated the spread of both NoV and NoVΔB2 from the injected abdominal cavity to the fore- and hindlimb tissues 24 hours after inoculation (Fig. 3A). The difference between NoV and7 NoVΔB2 accumulation levels was small at 1 dpi, although higher doses of NoVΔB2 were inoculated into each mouse (supplementary materials), but became progressively more pronounced at later infection times. By 4 dpi, NoV RNA levels were comparable to those of ribosomal RNAs (rRNAs), whereas the accumulation of NoV was more than 1000 times that of NoVΔB2 (Fig. 3, A and B). Accordingly, unlike the 100% mortality observed 5 days post–NoV infection, suckling mice challenged by NoVΔB2 remained healthy for the duration of the experiment, up to 4 weeks postinoculation (Fig. 3C). Our quantitative RT-PCR analysis on the expression of 84 key genes from the known innate antiviral pathways (30) in suckling mice at 4 dpi detected no major differences between infection by NoVΔB2 and NoV (table S2). This suggested that rapid in vivo clearance of NoVΔB2 was not mediated by one of the known innate antiviral pathways. Moreover, we found that a NoV mutant (NoVmB2) carrying a single Arg to Gln mutation at position 59 of B2, known to abolish B2’s VSR activity in vitro (3, 24), was as nonvirulent as NoVΔB2 in suckling mice and was also progressively cleared from 4 dpi (Fig. 3A). Thus, in vivo infection and virulence of NoV require the RNAi suppressor activity of B2.
Fig. 3. In vivo virus clearance associated with production of viral siRNAs.

(A and B) Accumulation of NoV, NoVΔB2, and NoVmB2 in mouse fore- (F) and hind- (H) limb tissues detected by quantitative RT-PCR of the viral RNA1 and Northern blotting, respectively. NoVΔB2 level in hind limb at 1 dpi was set as 1, and error bars indicate standard deviation of three replicates (A). NoV RNAs 1 and 2 (arrows) were visible after rRNA staining to show equal loading (B). (C) Suckling mice remained as healthy 4 weeks post-infection with either NoVΔB2 (right) or NoVmB2 (not shown) as mock-inoculated mice (left), whereas all of the five NoV-inoculated mice died by 5 dpi (not shown). (D) Northern blot detection of negative-strand viral siRNAs in mice infected with NoVΔB2 (middle) or NoVmB2 (left) and of vsRNAs from NoV-infected mice (right). The hybridizing positions of four siRNA probes were given in Fig. 4B, and size markers were synthetic 21- and 25-nt RNAs. The same filters were probed for mouse microRNA 127 (miR-127) and U6 RNA as loading controls. At least three independent repeats with reproducible results were performed with each experiment.
Northern blot hybridization detected accumulation of discrete species of viral siRNAs in NoVΔB2-inoculated suckling mice (Fig. 3D, right panel), as found in plant and invertebrate hosts after virus infection (1, 2, 9). The mouse viral siRNAs migrated as a dominant 22-nt band alongside a weaker, 21-nt signal and became detectable at 2 dpi and remained so up to 7 dpi even through the accumulation of NoVΔB2 was low at both 2 and 7 dpi (Fig. 3, A and B). In contrast, vsRNAs from NoV-infected mice appeared as bands of heterogeneous sizes (Fig. 3D, right panel). Notably, the 22-nt viral siRNAs became readily detectable in suckling mice inoculated with NoVmB2 (Fig. 3D, left panel). Therefore, rapid virus clearance resulting from loss of viral suppression of RNAi in NoVΔB2-and NoVmB2-infected mice was consistently accompanied with abundant production of the 22-nt viral siRNAs.
We further deep sequenced small RNAs from suckling mice 4 days after NoV inoculation and from those 1 or 2 days after NoVΔB2 inoculation. NoV vsRNAs showed no size preference, and the 22-nt vsRNAs of NoV were not enriched for canonical siRNAs (Fig. 4, A and B), which suggested B2 suppression of viral siRNA biogenesis in NoV-infected mice. We noted that NoV vsRNAs cloned from mice contained more abundant negative strands (16%) than those from cell culture (3%) (table S1), which might indicate weak in vivo dicing of NoV dsRNA in the presence of B2. In contrast, ~85% of NoVΔB2 small RNAs from mice were 21- to 23-nt long, with 22 nt as the predominant size for both strands (Fig. 4A and fig. S1B). A higher density of viral siRNAs was found to target the RNA3-transcribing region of RNA1 and the 5′-terminal region of RNAs 1 and 2 in NoVΔB2-infected mice and BHK-21 cells (Fig. 4C). The relative abundance of viral siRNAs in NoVΔB2-infected mice (0.3%) (table S1) was similar to that found in fruit flies (0.5 to 0.9%) undergoing FHV B2 clearance (12). NoVΔB2 siRNAs from mice at 2 dpi were divided approximately equally into positive and negative strands (Fig. 4A), and 65% of the 22-nt viral siRNAs in both NoVΔB2 libraries could form canonical siRNA duplexes with 2-nt 3′ overhang (Fig. 4B and table S1). The 22-nt viral siRNAs of NoVΔB2 detected by Northern blotting therefore have the properties of canonical viral siRNAs processed from dsRNA viral replication intermediates, which demonstrates induction of a typical antiviral RNAi response in mice by NoVΔB2 infection. Together, our findings reveal that, without viral suppression of RNAi, mice are able to launch a potent antiviral RNAi response sufficiently effective to provide full protection from lethal viral infection.
Fig. 4. Properties of mouse viral siRNAs produced in vivo.

(A) Length distribution and abundance of positive- or negative-strand vsRNAs from mice 1 or 2 dpi with NoVΔB2 or with NoV at 4 dpi. (B) Total counts of pairs of complementary 22-nt vsRNAs of NoVΔB2 and NoV in each distance category as defined in Fig. 1B. (C) Virus genome distribution of 21-to 23-nt viral siRNAs sequenced from either sucking mice (top two panels) or BHK-21 cells (bottom two panels) after infection by NoVΔB2. The functional proteins encoded by the viral bipartite RNA genome and transcription of B2 mRNA (RNA3) from RNA1 are shown. Arrows indicate the positions of the four locked nucleic acid probes used to detect negative-strand viral siRNAs in mice.
Here, we have found that an RNA virus infection in cultured hamster cells and suckling mice induces a typical antiviral RNAi response, characterized by the production of viral siRNAs with clearly defined properties of canonical siRNAs. Our findings and those of Maillard et al. (28) illustrate that Dicer-dependent processing of dsRNA viral replication intermediates into successive siRNAs is a conserved mammalian immune response to infection by two distinct positive-strand RNA viruses (table S3). Consistent with the known in vitro activity of the B2 protein to inhibit the processing of long dsRNA into siRNAs (3, 16, 21, 24), however, viral small RNAs detected by either deep sequencing or Northern blotting during wild-type NoV infection do not have the properties of canonical siRNAs. Northern blot detection of viral siRNAs in NoVΔB2-infected mice suggests that the use of in vivo infection models and/or viruses incapable of inhibiting siRNA biogenesis may facilitate detection of siRNAs targeting other mammalian viruses. Moreover, NoV infection both in vitro and in vivo requires the RNAi suppressor activity of its B2 protein. In particular, suckling mice produced abundant viral siRNAs and became completely resistant to the lethal infection by NoV after substitution of a single amino acid in B2 that eliminates its RNAi suppressor activity. Thus, the typical RNAi response induced by virus infection in mammals has potent antiviral activity. The striking similarities in the induction and suppression of antiviral RNAi by the closely related FHVand NoVin fruit flies, nematodes, and mammals (2, 3, 8, 12, 21–24) highlight an evolutionary conserved role of RNAi in antiviral defense within the animal kingdom. Compared with the antiviral immunity mechanisms reported to date in mammals, virus clearance by antiviral RNAi has a distinct effector mechanism and does not require cell death (9, 30). Nevertheless, this mammalian immunity mechanism exhibits properties known to be associated with innate and adaptive immunity because it involves rapid host recognition of a microbe-associated molecular pattern dsRNA and a mechanism of specificity determined by pathogen-derived siRNAs (9). Discovery of mammalian antiviral RNAi provides a framework to investigate the innate and adaptive control of important human viral pathogens.
Supplementary Material
Acknowledgments
We thank J. Jovel, J. Mai, X, Wang, Q. Wu, W.-X. Li, I.-C. Huang, G. Wang, and J. Pedra for technical assistance or advice; K. Johnson, A. Ball, and C. Basler for cDNA clones; and O. Voinnet for comments on the manuscript. X.F. was supported by China Scholarship Council. This study was supported by NIH grants AI52447 and GM94396 and College of Natural and Agricultural Sciences, University of California, Riverside (to S.-W.D.).
Footnotes
References and Notes
- 1.Hamilton AJ, Baulcombe DC. Science. 1999;286:950–952. doi: 10.1126/science.286.5441.950. [DOI] [PubMed] [Google Scholar]
- 2.Li HW, Li WX, Ding SW. Science. 2002;296:1319–1321. doi: 10.1126/science.1070948. [DOI] [PubMed] [Google Scholar]
- 3.Lu R, et al. Nature. 2005;436:1040–1043. doi: 10.1038/nature03870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wilkins C, et al. Nature. 2005;436:1044–1047. doi: 10.1038/nature03957. [DOI] [PubMed] [Google Scholar]
- 5.Segers GC, Zhang X, Deng F, Sun Q, Nuss DL. Proc Natl Acad Sci USA. 2007;104:12902–12906. doi: 10.1073/pnas.0702500104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Drinnenberg IA, Fink GR, Bartel DP. Science. 2011;333:1592. doi: 10.1126/science.1209575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Félix MA, et al. PLOS Biol. 2011;9:e1000586. doi: 10.1371/journal.pbio.1000586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Guo X, Li WX, Lu R. J Virol. 2012;86:11645–11653. doi: 10.1128/JVI.01501-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ding SW. Nat Rev Immunol. 2010;10:632–644. doi: 10.1038/nri2824. [DOI] [PubMed] [Google Scholar]
- 10.Deleris A, et al. Science. 2006;313:68–71. doi: 10.1126/science.1128214. [DOI] [PubMed] [Google Scholar]
- 11.Diaz-Pendon JA, Li F, Li WX, Ding SW. Plant Cell. 2007;19:2053–2063. doi: 10.1105/tpc.106.047449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Han YH, et al. J Virol. 2011;85:13153–13163. doi: 10.1128/JVI.05518-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bitko V, Barik S. BMC Microbiol. 2001;1:34. doi: 10.1186/1471-2180-1-34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Parameswaran P, et al. PLOS Pathog. 2010;6:e1000764. doi: 10.1371/journal.ppat.1000764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Li WX, et al. Proc Natl Acad Sci USA. 2004;101:1350–1355. doi: 10.1073/pnas.0308308100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sullivan CS, Ganem D. J Virol. 2005;79:7371–7379. doi: 10.1128/JVI.79.12.7371-7379.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bennasser Y, Le SY, Benkirane M, Jeang KT. Immunity. 2005;22:607–619. doi: 10.1016/j.immuni.2005.03.010. [DOI] [PubMed] [Google Scholar]
- 18.Qian S, et al. Proc Natl Acad Sci USA. 2009;106:605–610. doi: 10.1073/pnas.0806822106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Schnettler E, et al. EMBO Rep. 2009;10:258–263. doi: 10.1038/embor.2009.6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Schneemann A, Ball LA, Delsert C, Johnson JE, Nishizawa T. In: Virus Taxonomy—Eighth Report of the International Committee on Taxonomy of Viruses. Fauquet CM, et al., editors. Elsevier; San Diego, CA: 2005. pp. 865–872. [Google Scholar]
- 21.Chao JA, et al. Nat Struct Mol Biol. 2005;12:952–957. doi: 10.1038/nsmb1005. [DOI] [PubMed] [Google Scholar]
- 22.Wang XH, et al. Science. 2006;312:452–454. doi: 10.1126/science.1125694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Galiana-Arnoux D, Dostert C, Schneemann A, Hoffmann JA, Imler JL. Nat Immunol. 2006;7:590–597. doi: 10.1038/ni1335. [DOI] [PubMed] [Google Scholar]
- 24.Aliyari R, et al. Cell Host Microbe. 2008;4:387–397. doi: 10.1016/j.chom.2008.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Johnson KL, Price BD, Ball LA. Virology. 2003;305:436–451. doi: 10.1006/viro.2002.1769. [DOI] [PubMed] [Google Scholar]
- 26.Haasnoot J, et al. PLOS Pathog. 2007;3:e86. doi: 10.1371/journal.ppat.0030086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Fabozzi G, Nabel CS, Dolan MA, Sullivan NJ. J Virol. 2011;85:2512–2523. doi: 10.1128/JVI.01160-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Maillard PV, et al. Science. 2013;342:235–238. doi: 10.1126/science.1241930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ball LA, Amann JM, Garrett BK. J Virol. 1992;66:2326–2334. doi: 10.1128/jvi.66.4.2326-2334.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Goubau D, Deddouche S, Reis C, Sousa E. Immunity. 2013;38:855–869. doi: 10.1016/j.immuni.2013.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.