

Abstract
Ligand-independent, constitutive activation of Hedgehog signalling in mice expressing a mutant, activated SmoM2 allele results in the development of multifocal, highly differentiated tumours that express myogenic markers (including desmin, actin, MyoD and myogenin). The histopathology of these tumours, commonly classified as rhabdomyosarcomas, more closely resembles human fetal rhabdomyoma (FRM), a benign tumour that can be difficult to distinguish from highly differentiated rhabdomyosarcomas. We evaluated the spectrum of Hedgehog (HH) pathway gene mutations in a cohort of human FRM tumours by targeted Illumina sequencing and fluorescence in situ hybridization testing for PTCH1. Our studies identified functionally relevant aberrations at the PTCH1 locus in three of five FRM tumours surveyed, including a PTCH1 frameshift mutation in one tumour and homozygous deletions of PTCH1 in two tumours. These data suggest that activated Hedgehog signalling contributes to the biology of human FRM.
Keywords: fetal rhabdomyoma, hedgehog signalling, PTCH1
Introduction
Hedgehog (HH) signalling is essential in many developmental processes [1] and has been linked to the development of a variety of malignant neoplasms, including rhabdomyosarcoma [2,3]. HH binds to the transmembrane receptor Patched 1 (Ptch1) to relieve inhibition of the transmembrane signal transducer Smoothened (Smo). Activated Smo then mediates proteolytic processing of the Gli family of zinc finger transcription factors to drive transcription of downstream target genes, including Ptch1 and Gli1 (negative and positive components of the HH feedback system) [1,4] (Figure 1B). Mice that are haploinsufficient for the HH receptor Ptch1 [5] or for the HH binding protein Hip1 [6], both negative regulators of HH signalling, and mice that express an activated Smo allele (SmoM2; Figure 1A) [4] are prone to develop tumours with myogenic differentiation, commonly classified as embryonal rhabdomyosarcomas [4–6]. Our review of the histopathology of SmoM2 rhabdomyosarcomas revealed that these tumours show prominent cytodifferentiation, lack nuclear atypia and closer resemble human fetal rhabdomyomas (FRM). Human FRMs are rare benign tumours that typically arise in children and younger adults, have a predilection to occur at head/neck sites, occasionally infiltrate into adjacent normal skeletal muscle and can be complicated by local recurrence [7–9]. They do not cause regional or systemic metastases, and their ultimate outcome is benign [7]. Multifocal FRMs have been described in the context of nevoid basal cell carcinoma syndrome (NBCCS; a genetic disorder linked to PTCH1 mutations) [8,10].
Figure 1.

SmoM2 myogenic tumours in R26-SmoM2;CAGGS-CreER mice. (A) Constitutive activation of HH pathway signalling; Gli-R, Gli repressor; Gli-A, Gli-activating complex. (B) R26-SmoM2;CAGGS-CreER mice develop myogenic tumours with high penetrance within the first 3 months of life. (C) SmoM2 myogenic tumours are multifocal and reach a total tumour mass of 358 ± 67 mg/mouse. (D) Increased expression of HH target genes Ptch1 and Gli1 in SmoM2 tumours compared to wild-type (WT) mouse muscle (SM) reflects activated HH signalling in SmoM2 myogenic tumours (mean ± SD of six SmoM2 tumours and three muscle specimens). (E) Random muscle sections obtained from R26-SmoM2;CAGGS-CreER mice contain small tumour foci. Images were taken at ×10 and ×20 magnification.
FRMs typically consist of bland spindle cells and elongated muscle cells reminiscent of fetal myotubules in a fibromyxoid stroma (Figure 3A) [7], and their lack of nuclear atypia and low mitotic rate serves as a distinguishing feature between FRM and rhab-domyosarcoma. However, overlap in the histological presentation of human FRM and highly differentiated human rhabdomyosarcoma has been well documented in the literature, and the distinction of human FRM from highly differentiated RMS can sometimes be problematic [7,9,11]. As HH-driven tumours with myogenic differentiation in mice with hyperactive HH signalling resemble human FRMs, we sought to investigate the molecular underpinnings and, specifically, the frequency of HH pathway gene mutations in these rare tumours. Our studies identified functionally relevant aberrations at the PTCH1 locus in three of five human FRMs surveyed, including a frameshift mutation (g.98,220,305GC > G) in one tumour and homozygous deletions of PTCH1 in two tumours. The frequent detection of PTCH1 inactivating mutations in human FRMs suggests that hyperactive HH signalling contributes to the development or maintenance of human FRMs.
Figure 3.

Human fetal rhabdomyomas (FRMs). Six human FRM specimens were identified. (A) Representative images of H&E-stained sections of tumours FRM4, FRM5 and FRM6 are shown; images were taken at ×40 magnification. (B) All patients were male; age at diagnosis was 0–108 months, and four of six tumours were located in the head/neck region. Clinical and outcome data were available for three of six cases (FRM4–6). These three patients did not meet clinical criteria for NBCCS and were tumour-free 17 months–12 years post-diagnosis. No evidence of disease, NRD.
Materials and methods
Mice
R26-SmoM2 (mixed genetic background, including 129/Sv and Swiss Webster as main components) and CAGGS-CreER mice were purchased from the Jack-son Laboratory. The mice were bred and maintained at the Joslin Diabetes Center Animal Facility. Tamoxifen (1 mg/40 g body weight; Sigma, St. Louis, MO, USA) was injected intraperitoneally on postnatal day 10 (P10) to induce Cre expression. Animals were monitored once weekly for the onset of soft-tissue tumours or other health problems, and were sacrificed once they had palpable muscle tumours or were ill. All animal experiments were approved by the Joslin Diabetes Center Institutional Animal Care and Use Committee.
Human tissue specimens
Human FRM specimens (FRM1-6) and corresponding normal tissue were obtained from the pathology archives at Boston Children’s Hospital (FRM4-6) and the Brigham and Women’s Hospital, Boston, MA (FRM1-3). Accurate histopathological diagnosis was verified by LAT and CDF. Corresponding normal tissue was available for tumours FRM5-6 only (CONO5-6). Discarded normal human skeletal muscle obtained from donors without tumour history (NO1-3) was obtained from PJG (see supplementary material, Table S1). All studies involving human tissue samples were approved by the relevant institutional review boards (Boston Children’s Hospital, IRB-P00003845; Joslin Diabetes Center, CHS#06-42).
Histopathological evaluation of mouse SmoM2 tumours
Tumours harvested from mice were fixed in 4% v/v paraformaldehyde for 2 h, embedded in paraffin, sectioned and stained with haematoxylin and eosin (H&E). For immunohistochemistry, 4 μm sections of paraffin-embedded tissue were baked, deparaffinized and subjected to heat-induced antigen retrieval in target retrieval solution, pH 9 (Dako, Carpinteria, CA, USA). The sections were then treated with peroxidase and protein blockers (Dako) and incubated with antibodies against myogenin (1:100; M3559; Dako), MyoD1 (1:50; M3512; Dako), desmin (1:50; M0760; Dako), actin (1:200, Dako M0635) and Ki67 (1:250; VP-K451; Vector Laboratories). The detection system was mouse Envision (Dako) for 30 min, followed by development with a 3,3V-diaminobenzidine chromagen substrate for 5 min. The slides were lightly counter-stained with haematoxylin. Isotype-specific antibodies were used as negative controls.
RT–PCR
Total RNA was isolated by TRIzol extraction and reverse-transcribed using Superscript I First-Strand Synthesis System for RT–PCR (Invitrogen, Carlsbad, CA, USA). qRT–PCR was performed using an AV7900 PCR system (Invitrogen) with Taqman PCR reagents. The primers used were Taqman Gene Expression Gli1 (Mm00494654_m1), Ptch1 (Mm00436026_m1) and 18s (Mm03928990_g1).
DNA isolation and sequencing analysis of human tissue specimen
Genomic DNA was isolated from 10–15 punches (diameter 0.6 mm) obtained from formalin-fixed paraffin-embedded (FFPE) tissue blocks or from five unstained sections/specimen, using the DNeasy Blood and Tissue kit (Qiagen, Hilden, Germany). DNA was analysed using PICOgreen fluorescence to quantify double-stranded DNA. Total DNA yield was 61–816 and 51–2327 ng for FRM and normal tissue specimens, respectively. Of note, all FRM tissue samples were older than 10 years. Sequencing was performed using OncoPanel v.1, a targeted Illumina sequencing strategy of the coding regions of 645 genes previously linked to human cancer, including HH pathway genes PTCH1, SMO, suppressor of fused homologue (SUFU; acts as a negative regulator of HH signalling by downregulating GLI1 -mediated transactivation of target genes) and HIP1 [1]. Prior to library preparation, 51–200 ng DNA was fragmented by Covaris sonication to 150 bp and further purified using Agentcourt AMPure XP beads. 50 ng size-selected DNA was then ligated to sequencing adaptors with sample-specific barcodes during library preparation (Illumina TruSeq) and quantified by qPCR. The yields of the libraries were in the range 83–2308 ng. The yield of the library obtained from sample FRM1 was only 3 ng, and this sample was discarded from further analysis. The remaining samples were pooled in equimolar concentrations to a total of 500 ng for targeted exon enrichment, using the OncoPanel v.1 gene set with the Agilent SureSelect hybrid capture kit, and sequenced in one lane for 100 bp paired-end reads/fragment on an Illumina Hiseq 2000. Pooled sample reads were deconvoluted (demultiplexed) and sorted using the Picard tools (for details, see: http://picard.sourceforge.net/command-line-overview.shtml). Reads were aligned to the reference sequence b37 edition from the Human Genome Reference Consortium, using bwa (http://bio-bwa.sourceforge.net/bwa.shtml) and the parameters ‘-q 5 -l 32 -k 2 -o 1’. Duplicate reads were identified and removed using the Picard tools. The alignments were further refined using the GATK tool for localized realignment around indel sites (http://www.broadinstitute.org/gsa/wiki/index.php/Local_realignment_around_indels). Recalibration of the quality scores was performed using GATK tools (http://www.broadinstitute.org/gsa/wiki/index.php/Base_quality_score_recalibration). The sequence generated a total of 482 330 586 reads and 43 ± 5 × 106 reads/sample. Samples NO1-3 had high mean target coverages of ×232–284, while the tumour and matched normal samples only reached a mean target coverage of ×44–66. However, in all cases taken together, 63.4–90.2% of target bases were covered at least ×30, including 79% of Hedgehog (HH) gene-coding regions (see supplementary material, Table S4). Mutation analysis for single nucleotide variants (SNVs) was performed using MuTect v. 1 0.27200 (https://confluence.broadinstitute.org/display/CGATools/MuTect) and annotated by Oncotator (http://www.broadinstitute.org/oncotator), developed by the Cancer Biology Group at the Broad Institute. MuTect was made available through the generosity of Kristian Cibulskis and the Cancer Genome Analysis Program at the Broad Institute, Inc. Insertions and deletions (indels) were called using Indel Locator (https://confluence.broadinstitute.org/display/CGATools/Indelocator). We considered only those SNVs that were non-synonymous, detected in a tumour fraction > 30% (total pairs > 30, MQ0 < 10), corresponding to a 60% tumour purity, absent in normal muscle specimens NO1-3 and listed in the Exome Variant Server, NHLBI Exome Sequencing Project (ESP; Seattle, WA, USA) with a population frequency < 1%. We considered those indels found in an allele fraction > 25%.
Fluorescence in situ hybridization (FISH)
A two-colour FISH probe set was hybridized to 5 μm sections of seven paraffin-embedded tissue samples (FRM4-6, NO1-3, CONO5), including a custom-made probe for PTCH1 (RP11-435O5, labelled with spectrum orange) and a commercial probe for CEP9 (labelled with spectrum green). For tumours FRM2-3 and corresponding normal tissue CONO6, no remaining paraffin-embedded tissue was available for FISH testing.
Results
HH-driven myogenic tumours in SmoM2 mice resemble human FRM
In R26-SmoM2;CAGGS-CreER mice, expression of a mutant, activated Smo allele (SmoM2) via an ubiquitously expressed transgene encoding a tamoxifen-inducible Cre recombinase (Figure 1A; CAGGS-CreER) resulted in hyperactivity of HH pathway signalling (Figure 1B) [4]. Tamoxifen-induced mice developed a spectrum of neoplasms, including tumours with myogenic differentiation previously classified as rhabdomyosarcomas (Figure 2) [4]. These tumours developed with high penetrance within the first 3 months of life (Figure 1C; 32 mice evaluated). The majority of tumours arose in the rear thigh, hindlimb and chest wall muscles, as previously described [4]. They were typically multifocal (average 4 ± 1 tumours/mouse detected; Figure 1D), and random skeletal muscle sections revealed numerous microscopic tumour foci (Figure 1F). Tumours reached considerable size (average 358 ± 67 mg total tumour mass/mouse; Figure 1D), but did not metastasize to the lungs of tumour-bearing animals. No metastatic foci were detected in random lung sections of 10 tumour-bearing mice. Expression of HH target genes Ptch1 and Gli1 was evaluated by qRT–PCR in six primary tumours obtained from SmoM2 mice compared to wild-type mouse muscle, and up-regulation of Ptch1 and Gli1 in tumours supported activation of HH signalling consistent with activation of SmoM2 (Figure 1E).
Figure 2.

Morphological and immunohistochemical (IHC) evaluation of SmoM2 tumours: H&E and IHC staining against Actin, Desmin, MyoD, Myogenin and Ki67. Representative images are shown for tumours originating in 56 day-old (A) and 27 day-old (B) R26-SmoM2;CAGGS-CreER mice; images were taken at ×20 magnification.
Histopathologically, SmoM2 tumours exhibited a highly differentiated myogenic phenotype and lacked cellular atypia (Figure 2). They contained numerous ‘myofibre-like’, multinucleated, elongated cells as well as small round cells with uniform appearance, ovoid nuclei and evenly distributed chromatin. Tumour cells expressed myogenic markers, including the myogenic regulator factors myogenin and MyoD (normally expressed in activated satellite cells and proliferating myoblasts [12]), and exhibited uniformly high levels of the late myogenic differentiation markers desmin and actin (Figure 2). Ki67 indices were variable, including extremely low Ki67 indices in many tumours (19.1 ± 15.9%; range 3.4–41.8%). When we reviewed SmoM2 tumour histology (n = 15 tumours) and myogenic marker staining (n = 8 tumours) in the context of human pathology conventions, SmoM2 tumours consistently showed more prominent cytodifferentiation and less nuclear atypia than expected in embryonal rhabdomyosarcomas. It was also noted that SmoM2 tumours bear close resemblance to human FRMs, rare benign tumours with muscle differentiation.
Human FRMs
Six FFPE FRMs were identified in the pathology archives at Boston Children’s Hospital and the Brigham and Women’s Hospital, Boston, MA (Figure 3; see also supplementary material, Table S1). All six patients were male, age at diagnosis was 0–108 months and four of six tumours were located at head/neck sites. Notably, two tumours arose in the postauricular area, which is a frequent site of origin of FRMs (Figure 3B). For FRM4, a previously described germline polymorphism at the PTCH1 locus had been detected [Thr1195Ser, present in > 1% of the population, according to the Exome Variant Server, NHLBI Exome Sequencing Project (ESP), Seattle, WA, USA; accessed August 2012]. No information on germline mutational status was available for the other tumours. Clinical and outcome data were available for three of the six cases (FRM4-6, Figure 3B). These three patients had no family history of nevoid basal cell carcinoma syndrome (NBCCS), did not meet clinical criteria for NBCCS, had their tumours removed surgically, did not receive chemotherapy and/or radiation and were disease-free at 17 months–12 years after initial diagnosis. One patient (FRM4) had two local recurrences, both of which were resected (Figure 3B).
Mutational spectrum of human FRMs
We used hybrid-capture and targeted Illumina sequencing (OncoPanel v. 1) to investigate the molecular underpinnings of human FRMs, using DNA obtained from five FFPE tumour specimens (FRM2-6, Figure 3B). FRM1 yielded insufficient amounts of DNA and was discarded from the sequencing analysis. Normal corresponding tissue was available for tumours FRM5 (CONO5, muscle) and FRM6 (CONO6, lymph node). Three normal human muscle specimens obtained from donors without tumour history were also sequenced (NO1-3). We identified 10–17 SNVs in each of tumours FRM2–6 [non-synonymous, not present in normal muscle samples NO1-3, listed in the Exome Variant Server, NHLBI Exome Sequencing Project (ESP), Seattle, WA, USA, with a population frequency < 1%] (see supplementary material, Table S2). All 10–13 SNVs detected in tumours FRM5–6 were present in corresponding normal tissue (germline). All 15–17 SNVs detected in tumours FRM2–4 had indeterminate germline status. We also identified one to three insertions/deletions (indels; detected in > 25% of alleles) in tumours FRM2, FRM3, FRM4 (germline status indeterminate) and FRM6 (germline status somatic) (see supplementary material, Table S3). In addition to SNVs and indels, there was clear indication of deletion of PTCH1 and FANCC, which are co-located on chromosome 9, in samples FRM4 and FRM5 (Figure 4A, B, deleted regions marked by a red circle). For FRM5, the deleted region spanned Chr9, 95178901–98244322. Within this region, our targeted sequencing approach covered 1 exon of CENPP, all exons of FANCC and all exons of PTCH1 except exon 3. All exons within this region were different from the genome mean by four or five standard deviations (SDs; in log2 space, except PTCH1 exon 18). For FRM4, the deleted region spanned Chr9, 97873745–98248156. Within this region, OncoPanel v. 1 covered all exons of FANCC and PTCH1. All exons differed from the genome mean by six to eight SDs (in log2 space).
Figure 4.

PTCH1 copy number variations in human fetal rhabdomyomas (FRMs). Ratio of sequence coverage (log2 scale) of tumours FRM4 (A) and FRM5 (B) against CONO5, showing a deletion of FANCC and PTCH1 on chromosome 9 (marked by the red circle). For FRM5, the deleted region spanned Chr9, 95178901–98244322. Within this region, OncoPanel v. 1 covered one exon of CENPP, all exons of FANCC and all exons of PTCH1 except exon 3. All the exons except PTCH1 exon 18 were four to five SDs different from the genome mean (in log2 space). For FRM4, the deleted region spanned Chr9, 97873745–98248156. Within this region, OncoPanel v. 1 covered all exons of FANCC and PTCH1. All the exons were six to eight SDs different from the genome mean (in log2 space).
Several abnormalities were seen in more than one of the FRM specimens surveyed: First, indels in epithelial splicing regulatory protein 1 (ESPR1) were detected in three tumours, resulting in codon deletions in two tumours (FRM2, FRM4) and a frameshift mutation in one tumour (FRM3) (Table S3). Second, abnormalities in Fanconi anaemia complementation group C (FANCC) were present in three tumours, including two tumours with FANCC deletions (FRM4, FRM5) and one tumour with a FANCC SNV (FRM3) (Table S2, Figure 4). Third, missense mutations in the nuclear membrane protein spectrin repeat-containing nuclear envelope 1 (SYNE1) were detected in three tumours (FRM3, FRM5, FRM6) (Table S2). Finally, aberrations in HH pathway genes were found in three of five tumours, including two tumours with large deletions at the PTCH1 locus (FRM4, FRM5) and one tumour with a PTCH1 indel and a SMO SNV (FRM3) (Table 1, Figure 4; see also supplementary material, Tables S2, S3). The SMO mutation was previously found in 0.9% of the general population (according to the Exome Variant Server, NHLBI Exome Sequencing Project (ESP), Seattle, WA, USA). The PTCH1 Indel caused a frameshift and thus represented a high-impact aberration. No SUFU or HIP1 mutations were detected.
Table 1.
Aberrations in Hedgehog pathway genes in fetal rhabdomyomas
| PTCH1 | SMO | SUFU | HIP1 | |
|---|---|---|---|---|
| FRM2 | – | – | – | g.75172270G > A (p.P930P) |
| FRM3 | g.98,220,305 GC > G | g.128845511G > A (p.V270I) | – | – |
| FRM4 | Homozygous deletion* | – | – | – |
| FRM5 | Homozygous deletion* | – | – | – |
| FRM6 | –* | – | – | – |
Functionally relevant aberrations are marked in bold. PTCH1 indel 98,220,305 GC > G represents a frameshift mutation. SMO p.V270I is present in > 1% of the population. HIP1 p.P930P was predicted to be silent.
Presence or lack of deletion was confirmed by FISH (Figure 5).
HH pathway gene aberrations in human FRMs
Our sequencing studies identified aberrations in PTCH1 and SMO in three of five human FRMs sequenced (Table 1). FISH testing of PTCH1 was performed on FFPE sections obtained from tumours FRM4–6 and normal tissue specimens (NO1-3), using a custom-made probe for PTCH1 (labelled with spectrum orange) and a commercial probe for CEP9 (labelled with spectrum green) (Figure 5). For tumour FRM3, there was no remaining tissue or DNA available to verify the abnormalities noted by OncoPanel v. 1 sequencing. The frequency of hybridization signal patterns in the PTCH1 region was first analysed in the three normal tissue specimens (NO1-3). Heterozygous deletion patterns were observed in 11–37 of 100 cells, with a cut-off of 49 of 100 (determined as mean ± two SDs); homozygous deletion patterns (zero red and one green signal, or zero red and two green signals) were not observed in normal tissues. For all three tumours, heterozygous deletion patterns were within the range observed in normal tissues and did not exceed the cutoff. No homozygous deletion patterns were observed in tumour FRM6. However, homozygous deletion of PTCH1 was noted in tumours FRM4 and FRM5 (homozygous deletion patterns in 73/100 and 76/100 cells, respectively) (Figure 5). This is consistent with the results of our sequencing studies, demonstrating deletions at the PTCH1 locus in FRM4 and FRM5.
Figure 5.
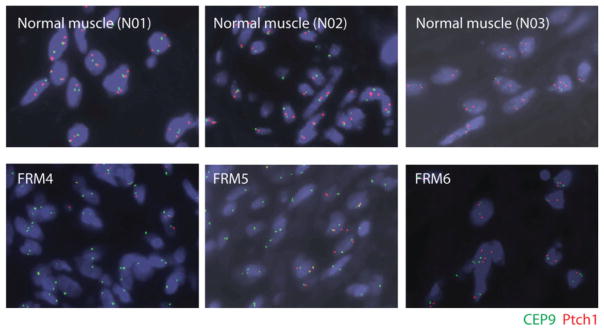
PTCH1 deletion in human fetal rhabdomyomas (FRM). Fluorescence in situ hybridization testing of PTCH1 revealed homozygous deletion of PTCH1 in tumours FRM4 (lower left panel) and FRM5 (lower middle panel), consistent with large deletions at the PTCH1 locus noted by sequencing. PTCH1 expression in tumour FRM6 (lower right panel) was normal, without evidence of homozygous deletion patterns. Heterozygous deletion patterns in tumours FRM4–6 were within the range observed in normal muscle specimens (upper panels).
Discussion
This study was motivated by our observation that HH-driven myogenic tumours in mice with hyperactive HH signalling due to expression of an activated SmoM2 allele resemble human FRMs. We evaluated the mutational spectrum of five human FRMs and noted functionally relevant aberrations in three of five human FRMs. Of note, Tostar et al previously reported a germline frameshift mutation and a somatic in-frame deletion at the PTCH1 locus in a FRM diagnosed in a woman with NBCCS [13]. These findings suggest that activated HH signalling is a driving force behind the development of human FRMs.
Two of three FRMs with PTCH1 aberrations carried mutations of indeterminate germline status (FRM3, FRM5). For FRM4, a germline PTCH1 SNV had been detected. This SNV was previously linked to NBCCS [14], but subsequently found to be present in > 1% of the general population and reclassified as a non-pathogenic polymorphism. Patient FRM4 did not meet clinical criteria for NBCCS at the age of 10 years [15] but, interestingly, he experienced two local recurrences of his FRM. As multifocal rhabdomyomas were previously described in NBCCS [8], it is conceivable that the germline PTCH1 variant in patient FRM4 contributed to the development of these tumours. We also note that our FISH studies demonstrated homozygous PTCH1 loss in FRM4 and FRM5. This could be consistent with tumour development as a consequence of the combined effects of a somatic mutation in one allele and a germline lesion in the other.
We did not detect abnormalities in HH pathway genes in two of five tumours evaluated. It is possible that certain mutations in HH pathway genes were missed, due to the limited quality of the DNA obtained from > 10 year-old FFPE specimens or due to our sequencing approach (covering coding regions only). Alternatively, activated HH signalling may only contribute to a subset of human FRMs. For example, we also noted mutations in ESPR1, FANCC and SYNE1 in three FRMs each. ESPR1 encodes a splicing factor that regulates the formation of epithelial cell-specific isoforms and has been linked to epithelial–mesenchymal transition [16]. FANCC encodes a DNA repair protein [17]. SYNE1 encodes a nuclear membrane protein that has intracellular scaffold and linker functions [18]. Neither of these genes was previously linked to the biology of rhabdomyomas or rhabdomyosarcomas.
Multifocal muscle tumours in SmoM2 mice do not metastasize to the lungs of tumour-bearing animals and have a highly differentiated myogenic phenotype. Similarly, low Ki67 indices and highly myogenic transcriptional profiles were previously observed in Ptch+/− mouse rhabdomyosarcomas [19]. We here note marked similarities in the histopathological presentation of SmoM2 tumours and human FRM. Human FRMs are highly differentiated myogenic tumours that do not cause regional and/or systemic metastases. However, they can be difficult to distinguish from highly differentiated non-alveolar rhabdomyosarcoma [7]; Kodet et al previously remarked that it might be difficult to draw a sharp line between highly differentiated rhabdomyosarcomas and FRMs [9,11]. Interestingly, two of 15 tumours in a previously published case series of embryonal rhabdomyosarcomas with advanced differentiation resembled FRM; they consisted of almost entirely mature rhabdomyoblasts, interspersed with clusters of undifferentiated cells [20]. The question arises as to whether highly differentiated, rhabdomyoma-like non-alveolar rhabdomyosarcomas and human FRMs represent a biologically distinct category of myogenic tumours resulting from hyperactive HH signalling, or whether they are separate and distinct entities.
The presence of mutations in HH pathway genes in rhabdomyosarcoma tissue has been investigated previously. Somatic mutations in PTCH1 were detected in seven non-alveolar rhabdomyosarcomas by FISH [13,21]. However, published studies aimed at determining the frequency of HH pathway gene mutations in rhabdomyosarcoma did not consider the degree of morphological differentiation within the spectrum of non-alveolar rhabdomyosarcoma and did not detect pathogenic mutations in PTCH1. It will be interesting to evaluate the presence of HH pathway gene mutations and expression patterns (including PTCH1) in highly differentiated, ‘rhabdomyoma-like’ rhabdomyosarcomas in order to directly address the question of whether highly differentiated RMS and FRM represent a biologically distinct category and/or a spectrum of tumours with inactivating mutations in PTCH1 (which could serve as a biomarker to facilitate diagnosis).
Supplementary Material
Acknowledgments
We thank C Namgyal, T Bowman and C Unitt in the DF/HCC research pathology core for help with DNA isolation and IHC; C Zhang and R Lee in the DF/HCC cytogenetics core for PTCH1 FISH testing; R Ehrlich, M Ducar, A Sunkavalli and M Hanna in the DFCI Center for Cancer Genome Discovery (CCGD) for assistance with OncoPanel v. 1 sequencing; and D Tchessalova for animal colony maintenance/technical assistance. MuTect was made available through the generosity of Kristian Cibulskis and the Cancer Genome Analysis Program at the Broad Institute, Inc. This work was funded in part by the Harvard Stem Cell Institute, the National Institutes of Health (NIH; Grant Nos NIH DP2 OD004345 to AJW and NIH NS033642 to APM) and PALS Bermuda/St. Baldrick’s and the Alex’s Lemonade Stand Foundation (to SH). Content is solely the responsibility of the authors and does not necessarily represent the official views of the NIH or other funding agencies.
Abbreviations
- ESP
Exome Sequencing Project
- ESPR1
epithelial splicing regulatory protein 1 (human)
- FANCC
Fanconi anaemia complementation group C (human)
- FFPE
formalin-fixed, paraffin-embedded
- FISH
fluorescence in situ hybridization
- FRM
fetal rhabdomyoma
- HH
Hedgehog
- HIP1
Hedgehog interacting protein 1 (human)
- Hip1
Hedgehog interacting protein 1 (mouse)
- indel
insertion/deletion
- NED
no evidence of disease
- NBCCS
nevoid basal cell carcinoma syndrome
- NHLBI
National Heart, Lung, and Blood Institute
- PCR
polymerase chain reaction
- Ptch1
Patched 1 (mouse)
- PTCH1
PATCHED1 (human)
- SMO
SMOOTHENED (human)
- Smo
Smoothened (mouse)
- SNV
single nucleotide variant
- SUFU
suppressor of fused (human)
- SYNE1
spectrin repeat containing, nuclear envelope 1 (human)
Footnotes
No conflicts of interest were declared.
Author contributions
SH, CDF and AJW conceived experiments; SH and PVH carried out experiments; SH, LAT, PVH, RTB and CDF analysed data; and SH, LAT, PVH, LM, RTB, CD, JM, AM, PJG, HEG, CRG, CDF and AJW interpreted data. All authors were involved in the writing of the manuscript and had final approval of the submitted manuscript.
SUPPORTING INFORMATION ON THE INTERNET
The following supplementary material may be found in the online version of this article:
Table S1. Age of human tissue specimens surveyed, quality control measures and sequencing metrics
Table S2. List of non-synonymous SNVs in human FRMs
Table S3. List of indels observed in human FRMs
Table S4. OncoPanel coverage of Hedgehog pathway genes PTCH1, SMO, SUFU and HIP1
References
- 1.Ingham PW, McMahon AP. Hedgehog signaling in animal development: paradigms and principles. Genes Dev. 2001;15:3059–3087. doi: 10.1101/gad.938601. [DOI] [PubMed] [Google Scholar]
- 2.Chari NS, McDonnell TJ. The sonic hedgehog signaling network in development and neoplasia. Adv Anat Pathol. 2007;14:344–352. doi: 10.1097/PAP.0b013e3180ca8a1d. [DOI] [PubMed] [Google Scholar]
- 3.Corcoran RB, Scott MP. A mouse model for medulloblastoma and basal cell nevus syndrome. J Neurooncol. 2001;53:307–318. doi: 10.1023/a:1012260318979. [DOI] [PubMed] [Google Scholar]
- 4.Mao J, Ligon KL, Rakhlin EY, et al. A novel somatic mouse model to survey tumorigenic potential applied to the Hedgehog pathway. Cancer Res. 2006;66:10171–10178. doi: 10.1158/0008-5472.CAN-06-0657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hahn H, Wojnowski L, Zimmer AM, et al. Rhabdomyosarcomas and radiation hypersensitivity in a mouse model of Gorlin syndrome. Nat Med. 1998;4:619–622. doi: 10.1038/nm0598-619. [DOI] [PubMed] [Google Scholar]
- 6.Gerber AN, Wilson CW, Li YJ, et al. The hedgehog regulated oncogenes Gli1 and Gli2 block myoblast differentiation by inhibiting MyoD-mediated transcriptional activation. Oncogene. 2007;26:1122–1136. doi: 10.1038/sj.onc.1209891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kapadia SB, Meis JM, Frisman DM, et al. Fetal rhabdomyoma of the head and neck: a clinicopathologic and immunophenotypic study of 24 cases. Hum Pathol. 1993;24:754–765. doi: 10.1016/0046-8177(93)90013-7. [DOI] [PubMed] [Google Scholar]
- 8.Klijanienko J, Caillaud JM, Micheau C, et al. Basal-cell nevomatosis associated with multifocal fetal rhabdomyoma. A case. Presse Med. 1988;17:2247–2250. [PubMed] [Google Scholar]
- 9.Kodet R, Fajstavr J, Kabelka Z, et al. Is fetal cellular rhabdomyoma an entity or a differentiated rhabdomyosarcoma? A study of patients with rhabdomyoma of the tongue and sarcoma of the tongue enrolled in the intergroup rhabdomyosarcoma studies I, II, and III. Cancer. 1991;67:2907–2913. doi: 10.1002/1097-0142(19910601)67:11<2907::aid-cncr2820671133>3.0.co;2-b. [DOI] [PubMed] [Google Scholar]
- 10.Dahl I, Angervall L, Save-Soderbergh J. Foetal rhabdomyoma. Case report of a patient with two tumours. Acta Pathol Microbiol Scand A. 1976;84:107–112. [PubMed] [Google Scholar]
- 11.Jacques SM, Lawrence WD, Malviya VK. Uterine mixed embryonal rhabdomyosarcoma and fetal rhabdomyoma. Gynecol Oncol. 1993;48:272–276. doi: 10.1006/gyno.1993.1047. [DOI] [PubMed] [Google Scholar]
- 12.Rudnicki MA, Le Grand F, McKinnell I, et al. The molecular regulation of muscle stem cell function. Cold Spring Harb Symp Quant Biol. 2008;73:323–331. doi: 10.1101/sqb.2008.73.064. [DOI] [PubMed] [Google Scholar]
- 13.Tostar U, Malm CJ, Meis-Kindblom JM, et al. Deregulation of the hedgehog signalling pathway: a possible role for the PTCH and SUFU genes in human rhabdomyoma and rhabdomyosarcoma development. J Pathol. 2006;208:17–25. doi: 10.1002/path.1882. [DOI] [PubMed] [Google Scholar]
- 14.Boutet N, Bignon YJ, Drouin-Garraud V, et al. Spectrum of PTCH1 mutations in French patients with Gorlin syndrome. J Invest Dermatol. 2003;121:478–481. doi: 10.1046/j.1523-1747.2003.12423.x. [DOI] [PubMed] [Google Scholar]
- 15.Lo Muzio L. Nevoid basal cell carcinoma syndrome (Gorlin syndrome) Orphanet J Rare Dis. 2008;3:32. doi: 10.1186/1750-1172-3-32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Reinke LM, Xu Y, Cheng C. Snail represses the splicing regulator epithelial splicing regulatory protein 1 to promote epithelial–mesenchymal transition. J Biol Chem. 2012;287:36435–36442. doi: 10.1074/jbc.M112.397125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Yuan F, Song L, Qian L, et al. Assembling an orchestra: Fanconi anemia pathway of DNA repair. Front Biosci. 2010;15:1131–1149. doi: 10.2741/3666. [DOI] [PubMed] [Google Scholar]
- 18.Rajgor D, Mellad JA, Autore F, et al. Multiple novel nesprin-1 and nesprin-2 variants act as versatile tissue-specific intracellular scaffolds. PLoS One. 2012;7:e40098. doi: 10.1371/journal.pone.0040098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kappler R, Bauer R, Calzada-Wack J, et al. Profiling the molecular difference between Patched- and p53-dependent rhabdomyosarcoma. Oncogene. 2004;23:8785–8795. doi: 10.1038/sj.onc.1208133. [DOI] [PubMed] [Google Scholar]
- 20.Kodet R, Newton WA, Jr, Hamoudi AB, et al. Orbital rhabdomyosarcomas and related tumors in childhood: relationship of morphology to prognosis—an Intergroup Rhabdomyosarcoma study. Med Pediatr Oncol. 1997;29:51–60. doi: 10.1002/(sici)1096-911x(199707)29:1<51::aid-mpo10>3.0.co;2-7. [DOI] [PubMed] [Google Scholar]
- 21.Bridge JA, Liu J, Weibolt V, et al. Novel genomic imbalances in embryonal rhabdomyosarcoma revealed by comparative genomic hybridization and fluorescence in situ hybridization: an intergroup rhabdomyosarcoma study. Genes Chromosomes Cancer. 2000;27:337–344. doi: 10.1002/(sici)1098-2264(200004)27:4<337::aid-gcc1>3.0.co;2-1. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.