

Abstract
Background
Cardiac injury and dysfunction are contributors to disease progression and mortality in sepsis. This study evaluated the cardiovascular role of intrinsic A2A adenosine receptor (A2AAR) activity during lipopolysaccharide (LPS)-induced inflammation.
Methods
We assessed the impact of 24 h of LPS challenge (20 mg/kg, IP) on cardiac injury, coronary function and inflammatory mediator levels in Wild-Type (WT) mice and mice lacking functional A2AARs (A2AAR KO).
Results
Cardiac injury was evident in LPS-treated WTs, with ∼7-fold elevation in serum cardiac troponin I (cTnI), and significant ventricular and coronary dysfunction. Absence of A2AARs increased LPS-provoked cTnI release at 24 h by 3-fold without additional demise of contraction function. Importantly, A2AAR deletion per se emulated detrimental effects of LPS on coronary function, and LPS was without effect in coronary vessels lacking A2AARs. Effects of A2AAR KO were independent of major shifts in circulating C-reactive protein (CRP) and haptoglobin. Cytokine responses were largely insensitive to A2AAR deletion; substantial LPS-induced elevations (up to 100-fold) in IFN-γ and IL-10 were unaltered in A2AAR KO mice, as were levels of IL-4 and TNF-α. However, late elevations in IL-2 and IL-5 were differentially modulated by A2AAR KO (IL-2 reduced, IL-5 increased). Data demonstrate that in the context of LPS-triggered cardiac and coronary injury, A2AAR activity protects myocardial viability without modifying contractile dysfunction, and selectively modulates cytokine (IL-2, IL-5) release. A2AARs also appear to be targeted by LPS in the coronary vasculature.
Conclusions
These experimental data suggest that preservation of A2AAR functionality might provide therapeutic benefit in human sepsis.
Keywords: Adenosine receptors, Cardiac dysfunction, Cytokines, Inflammation, Troponin
1. Introduction
Sepsis is the chief cause of death in intensive care units, with associated mortality rates of 30–70% [1]. Trials of therapeutic interventions for sepsis have had limited success and a better understanding of mechanisms underlying disease progression is required [1–3]. Cardiac injury and dysfunction are key contributors to disease progression and mortality with sepsis [4]. Mechanisms underlying cardiovascular dysfunction in sepsis remain unclear [5].
Myocardial tolerance to injurious stressors and inflammation/ immune function are all sensitive to adenosine and its receptors [6,7]. Tissue adenosine levels rise with stress or inflammation, and mediate acute and longer-term benefit via one or more cell surface adenosine receptor (AR) subtypes. Adenosine is proposed to act as an endogenous ‘metabokine’ in an emerging paradigm of immunity, in which recognition of cell stress concomitantly instigates and inhibits inflammatory and immune responses [8]. Of the 4 AR subtypes (A1, A2A, A2B, and A3), the A2AAR possesses the greatest anti-inflammatory potential [8-10], regulating inflammation and injury in cardiac [11], renal [12], and hepatic systems [13]. Pharmacological manipulation of A2AAR activity is thus an attractive option for alleviating end-organ damage in sepsis [8]. However, controversy remains regarding the anti-inflammatory potential of adenosinergic therapy, and the intrinsic role of this receptor in determining cardiovascular resistance to inflammatory insult is not clear.
Despite support for adenosine-mediated protection against LPS-triggered inflammation and mortality in rodents [9,14], some human studies suggest limited anti-inflammatory potential for adenosine [15,16]. Moreover, effects of adenosinergic stimuli vary across experimental models. Augmented endogenous adenosine (via inhibition of cellular uptake) reduces TNF-α levels and leukopenia in an A2AR-dependent manner in male ddY mice [17]. In contrast, exogenous adenosine does not alter mortality or TNF-α levels in the LPS treated females of the CF1 mouse strain, though a related nucleoside analogue reduces both [18]. In humans, adenosine fails to substantially modify inflammation, TNF-α, IL-6 or IL-10 levels [15,16], but reduces potentially pro-inflammatory soluble receptor for advanced glycation end products [16].
More selective AR stimuli have been tested. A3AR agonism reduces LPS-triggered mortality, suppresses IL-12, IFN-γ, and IL-1α, and augments IL-10 [19]. Some evidence suggests that the effects of A2AAR agonism in LPS-dependent inflammation may be beneficial [14,20], however these findings are controversial. For example, Bonneau et al. [21] found that A2AAR agonism did not modify inflammatory cell influx, TNF-α, keratinocyte chemoattractant or MIP-2 levels in LPS-triggered lung inflammation, but did inhibit neutrophil activation. In contrast, another work indicates exogenous A2AAR activation does inhibit cytokine levels during LPS injury in lung [22]. However, the latter investigators also found that cytokine responses during lung inflammation were unaltered by A2AAR deletion. These disparities highlight differing responses to endogenous vs. exogenous AR ligands, and demonstrate that ARs are sub-maximally engaged by endogenous ligands (since they remain responsive to applied agonists).
The goal of this study was to examine the role of intrinsic A2AAR activity in regulating cardiac and systemic responses to inflammatory challenge. Since LPS triggers inflammation in the absence of infection, we evaluate effects of A2AARs on inflammatory and cardiac sequelae un-confounded by infection-mediated responses in A2AAR-deficient mice. We hypothesized that intrinsic A2AAR activity is cardioprotective against LPS challenge, and that deletion of this receptor would exacerbate inflammation-induced cardiac injury.
2. Materials and methods
2.1. Animals and LPS treatment
All animal procedures conformed to the Guide for the Care and Use of Laboratory Animals published by the NIH (publication number 85-25, revised 1996). Animals were cared for in accordance with protocols approved by the Animal Care and Use Committees of St. Jude Children's Research Hospital and the University of Tennessee Health Sciences Center. A2AAR KO mice and WT littermates were obtained from a sub-colony of the original lines generated and characterized by Ledent et al. [23]. Mice were bred and maintained on-site at St. Jude Children's Research Hospital, with standard laboratory food and water available ad libitum. Animal genotype was confirmed by PCR analysis of tail snips. All animals within experimental groups originated from the same breeding series and were matched for age and weight. Male and female mice were used with equal sex representation within each group. Both A2AAR KO and WT littermate mice received either a single intraperitoneal injection of 20mg/kg LPS isolated from Escherichia coli (Sigma-Aldrich, St. Louis, MO, catalog number L3129, lot number 78K4044), or an equal volume of sterile saline vehicle. Assessment of LPS (or vehicle) treatment effect was at 12 or 24 h as indicated.
2.2. Blood collection
At 12 or 24 h following LPS (or vehicle) injection, mice were anesthetized with sodium pentobarbital (50 mg/kg intraperitoneally). A 22 G needle was inserted into the inferior vena cava to inject 20IU of heparin in 0.2 mL normal saline, and this was allowed to circulate for ∼10 s prior to blood withdrawal. Whole blood was transferred to an EDTA vacutainer to prevent clotting. A 400 μL aliquot was used for complete blood count (CBC) analysis with the remainder centrifuged at 1500 RPM for 15 min. Plasma supernatant was removed and stored at —80 °C for analyses of cytokines, cTnI, CRP, and haptoglobin. Sufficient blood was not always available from each mouse to perform both CBC and cytokine analyses. In these cases, only CBC data were assessed.
2.3. Cytokine measurement
Four pro-inflammatory cytokines known to be modulated in response to A2AAR activation in inflammatory cells were measured: IL-2 [24,25], IL-5 [25], IFNγ [26,27] and TNFα [14,24]. The anti-inflammatory markers IL-4 [26] and IL-10, [14,24] are also known to be regulated by A2AARs and were assessed. Cytokine analyses were performed using a Bio-Rad BioPlex 100 system with Luminex xMAP technology (Bio-Rad Laboratories, Hercules, CA). Antibodies specific to the cytokines of interest (IL-2, IL-4, IL-5, IL-10, TNF-α, IFN-γ) were conjugated to carboxylated Luminex microspheres. Undiluted murine plasma samples were incubated on a 96 well plate with antibody-coupled microspheres for 1 h (at room temperature on an orbital shaker) to capture cytokines on the beads. Each well was washed with 100 μL PBS/ BSA. A solution of biotin and biotinylated antibodies against alternative epitopes on each cytokine of interest was placed in each well and similarly incubated for 1 h. After further washing, biotinylated cytokine conjugates were incubated for 30 min in streptavidin-phycoerythrin solution and detected by a dual-laser flow-based reader recognizing the internal bead signature and quantifying the cytokine bound to the bead. Cytokine concentration in each plasma sample was determined based on standards of known concentration. All samples were run in duplicate and reported (in pg/mL) as an average of two determinations.
2.4. CRP, haptoglobin, and cTnI assays
Inflammatory markers CRP and haptoglobin, were measured via solid phase ELISA kits (LifeDiagnostics, Inc.; West Chester, PA). High sensitivity murine cTnI was measured as a marker of myocardial cell damage using a similar ELISA kit (LifeDiagnostics, Inc.; West Chester, PA, USA). Each kit uses affinity purified anti-mouse antibodies against the biomarker of interest conjugated to microtiter wells for solid phase immobilization, and horseradish peroxidase conjugated anti-mouse antibodies for detection. Sample optical density was determined spectrophotometrically at 450 nM. Solutions of known concentration were used to develop standard curves for each biomarker and sample concentration was determined from these curves. All samples were run in duplicate and reported as an average of two determinations.
2.5. Left ventricular contractile function and coronary reactivity
At 24 h post-LPS treatment, mice were anesthetized with sodium pentobarbital (50 mg/kg intraperitoneally) and hearts rapidly removed into ice-cold perfusion fluid before cannulation and perfusion on a Langendorff apparatus, as detailed previously [28,29]. Hearts were perfused at a constant pressure of 80mm Hg with Krebs-Henseleit solution gassed with 95% O2, 5% CO2 and maintained at 37 °C. Left ventricular contractile function was assessed via a fluid-filled plastic film balloon introduced across the mitral valve and connected to a pressure transducer. The balloon was initially inflated to yield a diastolic pressure of 5 mm Hg. After a 10 min period of stabilization hearts were switched to electrical pacing at 420 beats/min. After 20 min of pacing, baseline functional parameters were assessed before testing for shifts in intrinsic vasoregulation through changes in the reactive hyperemic response [30]. Specifically, hearts were subjected to a single 30 s episode of total coronary occlusion and the subsequent hyperemia monitored over 5 min of reperfusion. We have previously identified the mechanistic components of this vasodilatatory response in the current murine model [30].
For more detailed analysis of contractile function, hearts were allowed to stabilize a further 5 min before baseline functional parameters were again determined. The ventricular balloon was then deflated to a volume yielding 0 mm Hg systolic pressure. This was defined as an arbitrary ‘zero’ volume, and the balloon was then incrementally inflated in 5 μL steps (remaining fixed at each volume for 30 s of measurement) until a plateau in systolic force occurred. Relationships between measures of ventricular function and balloon volume (normalized to cardiac mass) were assessed, and the slopes of the linear portions of individual ‘force-volume’ curves were calculated over a 10−40 μL volume range for systolic force, and 10−35 μL range for +dP/dt. These slopes are employed as surrogate measures of ventricular inotropic state normalized to heart mass.
2.6. Statistical analyses
Leukocyte count, platelet count, hemoglobin and hematocrit values were contrasted via factorial analysis of variance (ANOVA), functional parameters were contrasted via two-way ANOVA and coronary hyperemia via ANOVA with repeated measures. A least-significant differences (LSD) test was used to assess individual differences where ANOVA indicated significant effects. The presence of 0 values in some treatment groups in cytokine analyses caused variance heterogeneity which could not be rectified by data transformation for IL-4, IL-10, and IFN-γ (but not in the cases of TNF-α and IL-5). Data meeting assumptions of parametric tests were analyzed using a factorial ANOVA with a LSD post-hoc test determining the significance of individual comparisons. Where data did not meet assumptions of a parametric test, and in the absence of a factorial ANOVA equivalent, treatments were compared via a Kruskall-Wallis non-parametric one-way ANOVA. If differences were detected, a priori comparisons were used to detect differences between individual means using the Mann-Whitney test with a Bonferroni correction for repeated assessment of data.
3. Results
3.1. Cardiac injury as evidenced by cTnI release
Release of cTnI was assayed as a specific marker of myocardial damage/cell death. Serum cTnI was low and stable at 12 and 24 h in vehicle-treated animals, and was unaltered by A2AAR deletion (Fig. 1). In comparison with vehicle, LPS treatment in WT mice caused a significant 7.3-fold elevation in cTnI at 12 h, and a 2.7-fold elevation at 24 h (Fig. 1). The observation of a marked increase in cTnI levels between 12 and 24 h in A2AAR KO mice vs. a decline in cTnI levels at 24 h in WTs suggests a shift towards greater or more sustained injury (trending upwards in KO mice vs. falling in WT mice) in the absence of the receptor. The summed mean cTnI release for both time points (different animals were assessed at 12 vs. 24 h, limiting ability to calculate a true ‘area under the curve’) indicates that total cTnI release is ∼35% higher in A2AAR KO mice.
Fig. 1.
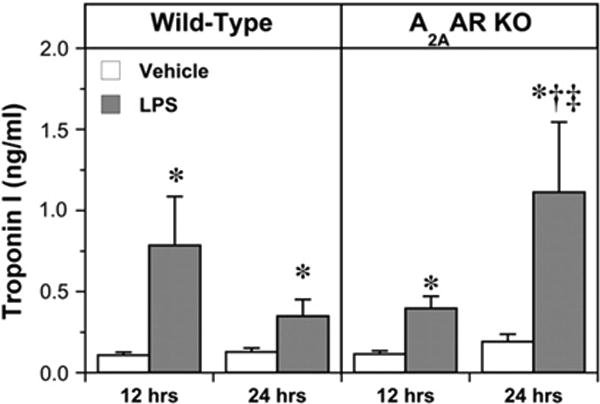
Serum levels of cardiac troponin I in the following groups: Wild-Type, vehicle-treated 12 h (n = 7); Wild-Type, LPS-treated, 12 h (n = 8); Wild-Type vehicle-treated 24 h (n = 11); Wild-Type, LPS-treated, 24 h (n = 11); A2AAR KO, vehicle-treated, 12 h (n = 11); A2AAR KO, LPS-treated, 12 h (n = 11); A2AAR KO, vehicle-treated, 24 h (n = 11); A2AAR KO, LPS-treated, 24 h (n = 10). Data are means±S.E.M. * P<0.05 vs. corresponding vehicle value; † P<0.05 vs. corresponding value at 12 h; ‡ P<0.05 vs. corresponding Wild-Type.
3.2. Cardiac contractile dysfunction
Baseline functional parameters for ex vivo Langendorff perfused hearts are presented in Table 1. Deletion of A2AARs did not modify baseline contractile function or coronary perfusion. LPS treatment did not modify coronary perfusion, but did significantly reduce measures of ventricular contractility by 25–30% (Table 1). The negative inotropic and lusitropic responses to LPS was unaltered by A2AAR deletion.
Table 1.
Age, weight, and baseline cardiac parameters from vehicle- and LPS-treated Wild-Type and A2AAR KO animals.
| Wild-Type | A2AAR KO | |||
|---|---|---|---|---|
|
|
|
|||
| Vehicle (n = 8) | LPS (n = 9) | Vehicle (n = 8) | LPS (n = 9) | |
| Age (weeks) | 14.1 ±0.3 | 13.9 ±0.4 | 14.2 ±0.3 | 14.3 ±0.4 |
| Body weight (g) | 27.1 ±0.4 | 26.4 ± 0.9 | 28.5 ±1.2 | 29.4 ±1.2 |
| Heart weight (mg) | 110 ± 4.0 | 109 ± 4.0 | 105 ± 4.0 | 109 ±5.0 |
| Heart rate (beats/min) | 341 ± 6.9 | 352 ±9.1 | 341 ±6.7 | 354 ± 9.9 |
| Absolute coronary flow (mL/min) | 2.02 ±0.11 | 1.95 ±0.24 | 1.78 ±0.14 | 1.77 ±0.12 |
| Normalized coronary flow (mL/min/g) | 18.3 ±0.78 | 17.9 ±2.3 | 17.0 ±1.2 | 16.4 ±0.82 |
| LV developed pressure (mm Hg) | 126 ±7 | 85 ±9* | 128 ±6 | 89 ±3* |
| Positive dP/dt (mm Hg/s) | 6493 ±251 | 4531 ±508* | 6420 ±191 | 4761 ± 223* |
| Negative dP/dt (mm Hs/s) | 3804 ±137 | 2832 ±239* | 3831 ±116 | 2951 ± 98* |
All data are means ± S.E.M (n = number of mice or hearts). Functional parameters were measured after 30 min of normothermic aerobic Langendorff perfusion of hearts. Intrinsic heart rate was recorded immediately prior to pacing.
P<0.05 vs. corresponding values in vehicle-treated hearts. There were no significant effects of A2AAR KO on these parameters. LV, left ventricular; dP/dt, first derivative of pressure over time.
As ventricular force development is volume-dependent, the relationship between ventricular balloon volume and contractile function was examined. Data in Fig. 2A and B demonstrate that LPS depressed the relationship between ventricular function and volume, triggering a 20–25% reduction in contractile parameters at a fixed (and near optimal) intraventricular balloon volume of 50 μL (∼420 μL/g) independently of A2AARs. Inotropic state, estimated from slopes of relationships between ventricular volume and either systolic force (Fig. 2C) or + dP/dt (Fig. 2D), was also reduced 25–30% with LPS and was insensitive to A2AAR deletion.
Fig. 2.
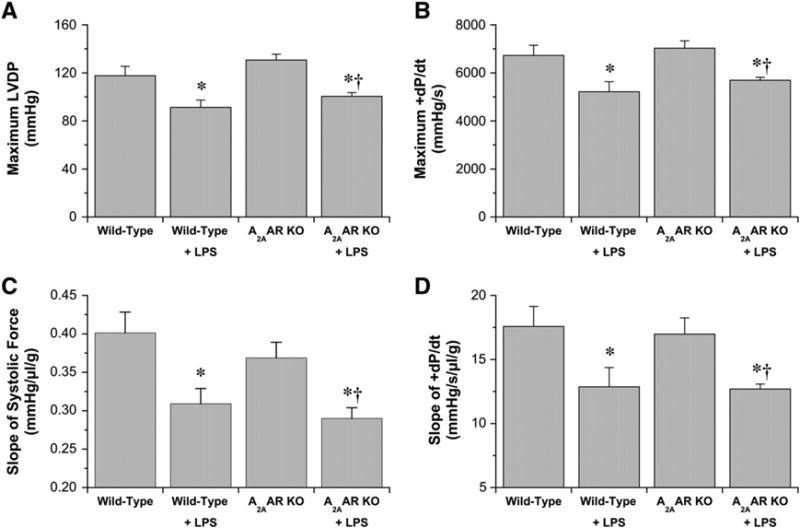
Contractile parameters in hearts from vehicle- and LPS-treated Wild-Type and A2AAR KO mice (24 h post-injection). Maximal values for left ventricular developed pressure (A) and +dP/dt (B) achieved at optimal ventricular balloon volumes of 50 μL. Inotropic state is reflected in the linear slopes of the relationships between ventricular contractile function and ventricular balloon volume (normalized to heart mass), as outlined in the Materials and methods. Slopes are shown for systolic pressure (C), and+dP/dt (D). Data are shown for Wild-Type, vehicle-treated (n = 8); Wild-Type, LPS treated (n = 9); A2AAR KO, vehicle-treated (n = 8); A2AAR KO, LPS-treated (n = 9). Data are means±S.E.M. * P<0.05 vs. corresponding vehicle value; † P<0.05 for A2AAR KO values vs. vehicle-treated Wild-Type.
3.3. Coronary dysfunction
Baseline coronary flow was not modified by A2AAR deletion (Table 1), whereas intrinsic vasodilatation during reactive hyperemia was modified (Fig. 3). Peak hyperemic flow in response to 30 s occlusion increased to ∼250% above baseline in the initial 5 s of reperfusion, and was similar across all groups. However, subsequent hyperemia between 12and 30 s of reperfusion was significantly reduced in A2AAR KO hearts (Fig. 3A). Effects of LPS on vasoregulation were almost identical to those of A2AAR deletion (Fig. 3A and B), with no impact on baseline or peak hyperemic flows but significant inhibition of dilatation between 10 and 30 s of reperfusion. Importantly, absence of A2AARs eliminated coronary vascular sensitivity to LPS (Fig. 3C), with hyperemic responses coincident for A2AAR KO hearts±LPS (and also LPS-treated Wild-Type hearts, shown in Fig. 3C for comparison). Collectively these observations support: 1) a role for A2AARs in mediating hyperemic flow subsequent to peak dilatation (between 10 and 30 s of reperfusion), in agreement with prior data [30]; 2) a select (near identical) effect of LPS on this phase of hyperemia; and 3) elimination of the coronary response to LPS in hearts lacking functional A2AARs. Thus, LPS eliminates A2AAR-dependent hyperemia.
Fig. 3.
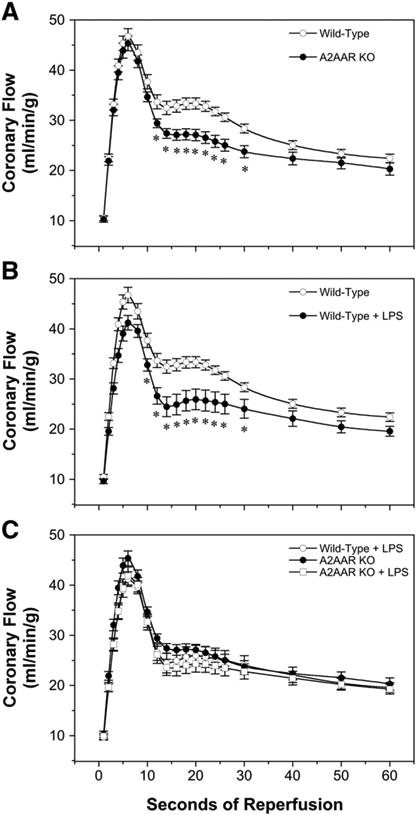
Coronary reactive hyperemic responses to 30 s occlusion in hearts from vehicle-and LPS-treated Wild-Type and A2AAR KO mice (24 h post-injection). For purposes of comparison, responses are shown for: effects of A2AAR KO in vehicle-treated hearts (A), effects of LPS treatment in Wild-Type hearts (B), and effects of LPS treatment in A2AAR KO hearts, together with LPS treatment in Wild-Type hearts (C). Data are means±S.E.M for the following groups: Wild-Type, vehicle-treated (n = 8); Wild-Type, LPS-treated (n = 9); A2AAR KO, vehicle-treated (n = 8); A2AAR KO, LPS-treated (n = 9). * P<0.05 vs. corresponding vehicle value.
3.4. Complete blood count profile
All mice received an injection of either LPS or sterile saline vehicle. Complete blood count profiles of vehicle- and LPS-treated mice are provided in Fig. 4. Leukocyte and platelet counts (Fig. 4A and B, respectively) did differ with LPS vs. vehicle treatment, while hematocrit and hemoglobin content was unaltered by LPS or vehicle (Fig. 4C and D, respectively).
Fig. 4.
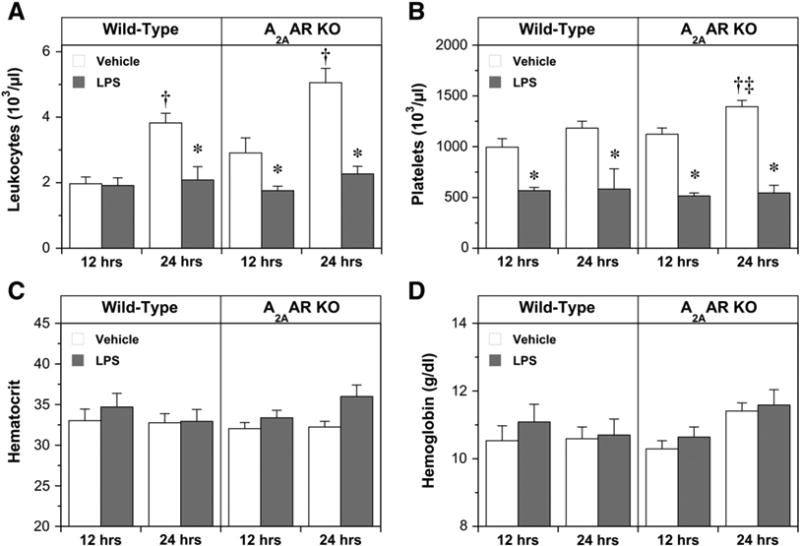
Complete blood count parameters at 12 and 24 h following either vehicle or LPS injection in the following treatment groups: Wild-Type, vehicle, 12 h (n = 9); Wild-Type, LPS, 12 h (n = 8); Wild-Type, vehicle, 24 h (n = 8); Wild-Type, LPS, 24 h (n = 6); A2AAR KO, vehicle,12 h(n = 11); A2AAR KO, LPS, 12 h (n = 11); A2AAR KO, vehicle, 24 h (n = 10); A2AAR KO, LPS, 24 h (n = 10). Data are means±S.E.M. * P<0.05 vs. corresponding vehicle value; † P<0.05 vs. corresponding value at 12 h; ‡ P<0.05 vs. corresponding Wild-Type value.
Interestingly, normal saline vehicle injection triggered a rise in leukocyte numbers at 24 h in WT mice, an effect accelerated (evident at 12 h) and augmented by A2AAR KO (Fig. 4A). This provides evidence of depression of leukocyte count with LPS vs. vehicle, and of A2AAR dependence of the response to vehicle (but not LPS). LPS also triggered a decline in platelets relative to vehicle treatment (Fig. 4B), although in this instance there was no apparent impact of A2AAR deletion. Overall, blood cell and hemoglobin responses to LPS are largely A2AAR independent.
3.5. Circulating cytokines
Circulating cytokine levels at 12 and 24 h are depicted in Fig. 5. In vehicle-treated mice, IL-2, IL-5, IL-10 and IFN-γlevels were very low. Overall, LPS treatment generated substantial elevations (vs. vehicle) in IL-2, IL-5, IL-10, and IFN-γ (Fig. 5). Deletion of A2AARs exerted modest and very select effects on cytokine release, limited to augmentation of IL-5 at 24 h, modulation of IL-2 release (eliminating the late rise at 24 h while inducing an early elevation at 12 h), and a shift in the pattern of IL-10 levels (further elevating IL-10 at 24 vs. 12 h, not observed in WT mice). While changes in IL-4 did not achieve statistical significance (Fig. 5B), there was evidence of early induction by LPS in WTs whereas IL-4 was unresponsive to LPS in A2AAR KO mice. Levels of IFN-γ were low and unaltered by vehicle, but increased markedly at 12 h of LPS treatment (with a subsequent decline to levels still above vehicle at 24 h; Fig. 5E). IFN- levels were unaltered by A2AAR KO. Changes in TNF-α exhibited some A2AAR dependence: TNF-α levels with vehicle were markedly reduced by A2AAR KO at 12 h and rose moderately after 24 h, contrasting WT mice which exhibited stable or slightly falling TNF-α levels to 24 h (Fig. 5F). In terms of response to LPS, there was no significant change in TNF-α levels relative to vehicle in WT mice, while a significant elevation was evident in A2AAR KO mice at 12 h (Fig. 5F). Despite this difference in response pro-file in KO mice, levels of TNF-α did not differ between WT and A2AAR KO mice at 12 or 24 h post-LPS.
Fig. 5.
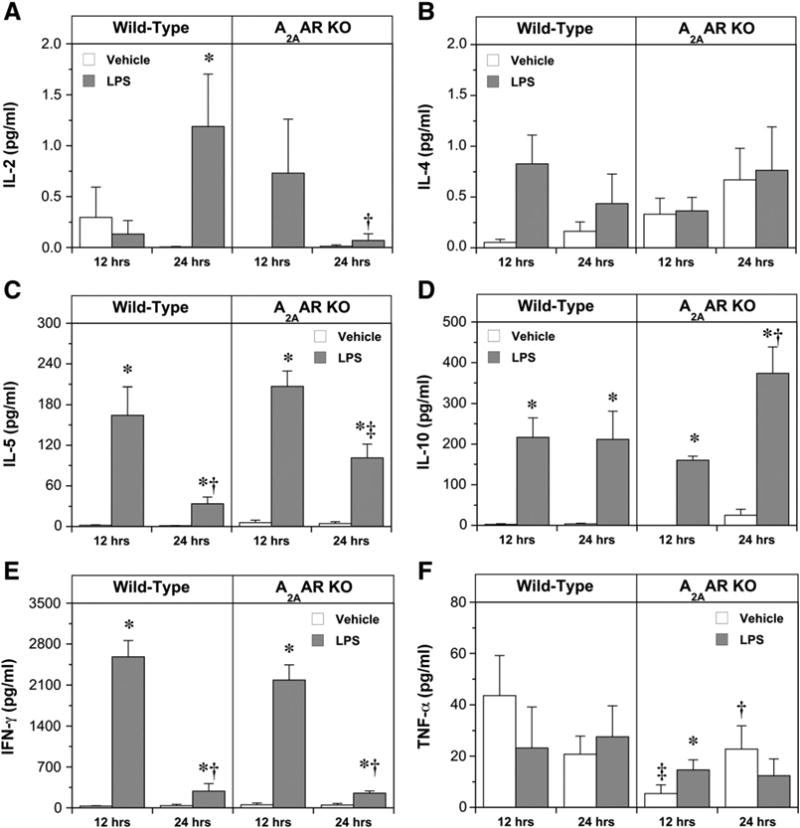
Serum levels of IL-2 (A), IL-4 (B), IL-5 (C), IL-10 (D), IFN-γ (E) and TNF-α (F) in the following treatment groups: Wild-Type, vehicle, 12 h (n = 9); Wild-Type, LPS, 12 h (n = 8); Wild-Type, vehicle, 24 h (n = 8); Wild-Type, LPS, 24 h (n = 6); A2AAR KO, vehicle, 12 h (n = 11); A2AAR KO, LPS, 12 h (n = 11); A2AAR KO, vehicle, 24 h (n = 10); A2AAR KO, LPS, 24 h (n = 10). Data are means±S.E.M. * P<0.05 vs. corresponding vehicle value; † P<0.05 vs. corresponding value at 12 h; ‡ P<0.05 vs. corresponding Wild-Type.
A limitation relevant to all in vivo studies of inflammation is that experimental intervention and surgery can themselves invoke significant inflammatory responses. It is thus critical to employ vehicle groups in studying inflammatory processes in vivo. This is exemplified in our data for blood cell counts and cytokine levels: the process of handling and saline injection increases leukocyte numbers at 24 h (Fig. 4), and may also specifically modify levels of TNF-α in the A2AAR KO group (Fig. 5). The detectable response to injection thus appears sensitive to A2AAR deletion and interpretation of the data in this context is necessary.
Summarizing these cytokine responses, in WT mice LPS triggered early elevations (by 12 h) in IL-4, IL-5, IL-10, and IFN-γ, with levels subsequently stabilizing (IL-10) or gradually falling (IL-4, IL-5, and IFN-γ). IL-2 appears to take longer (24 h) to rise in response to LPS. Deletion of A2AARs failed to significantly modify changes in IFN-γ, TNF-α and IL-4 yet selectively modified patterns of change for IL-2 and IL-5. We detected a difference in IL-10 between 12 and 24 h in A2AAR KO animals that was not present in WT animals. However, there was no statistically significant difference between WT and KO animals at either time point.
3.6. Changes in inflammatory biomarkers
As positive acute phase reactants, CRP and haptoglobin were both elevated by LPS in WT and A2AAR KO mice (Fig. 6). Levels of CRP in vehicle-treated mice were stable at 12 and 24 h, and tended to be lower in A2AAR KO animals. A transient peak in CRP was observed at 12 h in both groups, with A2AAR KO modestly reducing the level achieved (though not achieving significance). In contrast, haptoglobin levels were significantly elevated in A2AAR KO vs. WT animals treated with vehicle, and were enhanced by LPS at 12 h and further at 24 h in both groups. Overall, A2AAR deletion appears to reduce baseline CRP and augment baseline haptoglobin, with no major effects on accumulation of these markers during LPS challenge (Fig. 6).
Fig. 6.
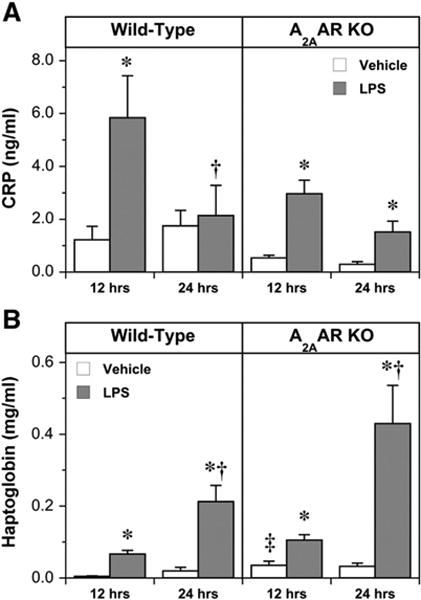
Serum levels of CRP (A) and haptoglobin (B) in the following treatment groups: Wild-Type, vehicle, 12 h (n = 9); Wild-Type, LPS, 12 h (n = 8); Wild-Type, vehicle, 24 h (n = 8); Wild-Type, LPS, 24 h (n = 6); A2AAR KO, vehicle, 12 h (n = 11); A2AAR KO, LPS, 12 h (n = 11); A2AAR KO, vehicle, 24 h (n = 10); A2AAR KO, LPS, 24 h (n = 10). Data are means ± S.E.M. * P<0.05 vs. corresponding vehicle value; † P<0.05 vs. corresponding value at 12 h; ‡ P<0.05 vs. corresponding Wild-Type.
4. Discussion
Our primary aim was to examine the role of intrinsic A2AAR activity in modifying cardiac injury, coronary vascular function, and circulating cytokines/inflammatory markers following LPS challenge. Effects of A2AAR deletion provide evidence that this receptor does mediate intrinsic protection, which reduces the overall extent of cardiac injury (Fig. 1). Interestingly, salutary effects occur independently of changes in contractile or coronary dysfunction (Figs. 2–3), and with minor and select changes in release patterns for cytokines and acute phase reactants (Figs. 5 and 6).
4.1. LPS-dependant cardiac injury but not dysfunction is exaggerated by A2AAR deletion
The LPS-induced rise in cTnI is consistent with sepsis-dependent TnI elevations in humans [31]. We note, however, that the basis for elevated markers of cardiac injury with inflammation/sepsis is not clear and it has also been suggested that factors released with sepsis/ inflammation may trigger degradation of cell troponin to low molecular weight fragments that can be released upon increases in membrane permeability [32]. It is thus unclear whether cardiac damage evidenced by cTnI is permanent or not, though reversibility is supported by clinical observations of resolution of sepsis-dependent cardiac depression in surviving patients [33]. With A2AAR deletion in the KO, the temporal pattern of TnI release differs to WT. In WT, from 12 to 24 h the decrease in TnI levels indicates injury resolution, while over the same time in the A2AAR KO, the injury response is increased. This raises the possibility that A2AARs play an important role in promoting recovery from endotoxemia, though perhaps not reducing the initial injury.
Sepsis triggers cardiac contractile dysfunction, which in vivo would contribute to hypotension/hypoperfusion and potentially to increased mortality. Here we show LPS mediates 20–30% reductions in ventricular force and contractility (Fig. 2). This dysfunction is unaltered by A2AAR deletion, despite augmentation of cTnI release (Fig. 1). Thus, while A2AARs may be engaged to reduce cardiac protein efflux during endotoxemia, their actions do not extend to protection against contractile dysfunction. This tends to counter cardiac injury (reflected in cTnI release) as a determinant of contractile changes observed with LPS.
4.2. LPS-triggered coronary dysfunction is mimicked by A2AAR deletion
Detrimental effects of sepsis have been linked to impaired vascular control in key organs [34,35]. Multiple mechanisms may contribute, including shifts in vascular reactivity, endothelial/nitric oxide (NO) control. By assessing reactive hyperemia following brief occlusion the effects on intrinsic control mechanisms can be examined. Most prior studies focus on responses of isolated vessel segments to applied dilators and constrictors (with a focus on NO bioavailability and actions), while very few examine intrinsic coronary vasoregulation. We have shown previously that the reactive coronary hyperemic response studied here involves contributions from A2AARs, NO, KATP channels and other mediators [30]. The current data support specific impairment of A2AAR-mediated coronary control by LPS (Fig. 3).
No reduction in baseline coronary flow following LPS is detected (Table 1), in agreement with reports of unchanged or increased flow with sepsis in human hearts [36], and with LPS treatment in rat hearts [37]. Maintenance of basal perfusion argues against an ischemic basis to cardiac depression, although highly focal ischemia/ hypoxia cannot be excluded. Despite unaltered baseline flow, coronary reactivity was impaired by LPS (Fig. 3): hyperemic flow subsequent to initial peak dilatation (which was unchanged across groups) was significantly depressed by LPS. Avontuur et al. [37] observed reduced coronary hyperemia in hearts from LPS treated rats, with unchanged peak flow but reduced hyperemic duration and flow repayment (consistent with our data). In contrast, Buffington et al. found no effect of endotoxin on coronary reactive hyperemia in dogs [38], although this latter study assessed responses at 4 h whereas our data and those of Avontuur et al. document later changes at 12–24 h.
Importantly, the inhibitory effect of LPS was remarkably similar to that of A2AAR deletion (Fig. 3), which also reduced hyperemia between 10 and 30 s of reperfusion without altering peak flow. Moreover, LPS failed to further modify hyperemia in hearts lacking A2AARs. Prior analysis of reactive hyperemia in this same model [39] supports a non-redundant role for the A2AAR in mediating dilatation subsequent to peak hyperemia (in an NO and KATP channel dependent manner). This previously documented A2AAR-sensitive component of hyperemia coincides precisely with both A2AAR KO- and LPS-dependent components of hyperemia here (Fig. 3). Near identical effects of LPS and A2AAR KO on hyperemia, and elimination of coronary sensitivity to LPS in A2AAR KO mice, suggests coronary dysfunction with LPS involves select targeting of A2AAR-dependent vascular control.
4.3. A2AAR deletion selectively modifies cytokine responses to LPS
The mechanistic basis of A2AAR-dependent cytoprotection during sepsis is not fully elucidated. A central idea is that the release and impact of inflammatory cytokines is selectively modulated, though this remains controversial. Nemeth et al. report A2AAR deletion augments LPS-dependent changes in TNF-α and IL-6, but not IL-10 [40]. This contrasts with the data of Reutershan et al. that indicate that A2AAR deletion does not modify cytokine responses during LPS-triggered lung inflammation [22]. In a different inflammatory model A2AAR-mediated protection is linked to repression of IFN-γ, TNF-α, and IL-4 [41]. Csóka et al. [42] recently showed that macrophages lacking A2AARs release less of the anti-inflammatory IL-10 when challenged, implicating A2AARs as an essential synergistic trigger for IL-10 production in response to infection. In contrast to in vivo evidence of reduced IL-6 and MIP-2 in A2AAR KO mice [40], macrophages from A2AAR KO animals display enhanced IL-6 and MIP-2 production [42]. This highlights that multiple cell types contribute to cytokine levels, and responses in different populations may be opposed, generating complex interactions in vivo.
Here we detect no significant impact of A2AAR deletion on patterns of IFN-γ, IL-4 or IL-10 release with LPS (Fig. 5). The pattern of change in circulating TNF-α was problematic to interpret, with minimal changes likely reflecting recovery of TNF-α by 12–24 h following earlier (and transient) release [43–45]. Importantly, there were no substantial differences or trends in TNF-α levels between WT and A2AAR KO mice (Fig. 5F). Our data thus do not support A2AAR-mediated protection via shifts in IFN-γ, TNF-α, IL-4, or IL-10 at 12–24 h, though it remains possible that earlier differences in circulating levels (not detected here) may play a role.
In contrast, responses of IL-2 and IL-5 were significantly modified by A2AAR KO, with augmented IL-5 and reduced IL-2 at 24 h (Fig. 5). IL-2 has both pro- and anti-inflammatory functions, promoting inflammation/immunity through proliferation and differentiation of T-cells, B-cells and natural killer cells while at the same time IL-2 (released from other cell types) is critical in development and maintenance of regulatory T-cells, and their immunosuppressive function [46]. Deficiency in IL-2 leads to multi-organ inflammation and fatal autoimmune/inflammatory disease. Enhanced IL-2 levels in animals possessing functional A2AARs may serve a protective function through regulatory T-cell suppression of inflammation. Evidence also supports A2AR mediated inhibition of IL-2 stimulated T-cell proliferation [47]. Thus, active A2AARs may not only augment IL-2 to suppress inflammation, but inhibit the proliferative effects of IL-2 to dampen further cellular responses. IL-2 may also induce IL-5, which we show was augmented modestly by A2AAR KO (Fig. 5). It is interesting that IL-5 was augmented at a time when IL-2 was reduced suggesting other important (and A2AAR sensitive) determinants of circulating IL-5. This cytokine induces differentiation of activated B-cells, and promotes proliferation, differentiation, and release of eosinophils. Reduced levels in the presence of functional A2AARs may thus limit eosinophil-mediated inflammation. Moreover, adenosine acting via the A3AR sub-type may additionally inhibit eosinophil degranulation and release of reactive oxygen species [48].
Shifts in acute phase proteins CRP and haptoglobin, were also assessed and results were discrepant (Fig. 6). In humans, CRP activates complement in response to binding of ligands from injured tissue, invading pathogens, or antigen–antibody complexes. CRP has predictive power for cardiovascular events [49], and elevated CRP may be injurious, as implicated by infarct amelioration or expansion upon CRP manipulation [50]. Significant extracellular accumulation of CRP was observed at 12 h following LPS treatment, with a tendency to reduced levels with A2AAR KO (Fig. 6). The pattern of change in haptoglobin differed, progressively rising at 12 and 24 h post-LPS, and exaggerated (albeit insignificantly) by A2AAR KO. Haptoglobin is produced in response to IL-6 and TNF-α, and scavenges hemoglobin during intra- or extravascular hemolysis, modulates immune function, and plays a role in major inflammatory disorders. The significant elevation in baseline haptoglobin in A2AAR KO animals was not associated with relevant change in TNF-α, which was paradoxically depressed in vehicle treated A2AAR KO animals. Data for haptoglobin are consistent with a beneficial role for intrinsic A2AAR activity during LPS treatment.
4.4. Conclusions
In summary, the current study reveals that endogenous A2AAR activity significantly modifies injury in the context of endotoxemia. Challenge with LPS induces marked cardiac injury, with contractile and coronary dysfunction. Intrinsic A2AAR activity reduces the severity of cardiac injury (assessed from cTnI release). This A2AAR-mediated suppression of cardiac injury occurs even though ventricular dysfunction is not acutely remediated. The findings indicate that coronary dysfunction with LPS challenge may actually involve specific inhibition of vascular A2AAR responses. Benefit from intrinsic A2AAR activity does not involve major shifts in levels of IFN-γ, TNF-α, IL-4 or IL-10, whereas changes in IL-2 and IL-5 may play some role.
We thus conclude that the A2AAR is concomitantly a protectant and target in the context of sepsis-induced cardiac injury – a protectant that reduces cardiac cell death, and an intrinsic mediator of coronary vascular function, which is a target of LPS-induced sepsis. Interventions aimed at restoring/optimizing cardiovascular A2AAR responses could provide therapeutic benefit in the context of sepsis.
Acknowledgments
This work was supported by the National Heart, Lung and Blood Institute grants HL-74001 (RRM), HL-027339 and HL-094447 (SJM) and HL-48839 (PAH), an American Heart Association Southeast Affiliate Grant-in-Aid (PAH), a grant from the National Health and Medical Research Council of Australia (JPH), and by the American Lebanese Syrian Associated Charities. We gratefully acknowledge the technical assistance of Ms. Mary A. Cerniway. The authors of this manuscript have certified that they comply with the Principles of Ethical Publishing in the International Journal of Cardiology.
Footnotes
Disclosures: None of the authors have anything to disclose.
References
- 1.Riedemann NC, Guo RF, Ward PA. Novel strategies for the treatment of sepsis. Nat Med. 2003;9:517–24. doi: 10.1038/nm0503-517. [DOI] [PubMed] [Google Scholar]
- 2.Flynn A, Chokkalingam Mani B, Mather PJ. Sepsis-induced cardiomyopathy: a review of pathophysiologic mechanisms. Heart Fail Rev. 2010;15:605–11. doi: 10.1007/s10741-010-9176-4. [DOI] [PubMed] [Google Scholar]
- 3.Daniels R. Surviving the first hours in sepsis: getting the basics right (an intensivist's perspective) J Antimicrob Chemother. 2011;66(Suppl 2):ii11–ii23. doi: 10.1093/jac/dkq515. [DOI] [PubMed] [Google Scholar]
- 4.Merx MW, Weber C. Sepsis and the heart. Circulation. 2007;116:793–802. doi: 10.1161/CIRCULATIONAHA.106.678359. [DOI] [PubMed] [Google Scholar]
- 5.Balija TM, Lowry SF. Lipopolysaccharide and sepsis-associated myocardial dysfunction. Curr Opin Infect Dis. 2011;24:248–253. doi: 10.1097/QCO.0b013e32834536ce. [DOI] [PubMed] [Google Scholar]
- 6.Peart JN, Headrick JP. Adenosinergic cardioprotection: multiple receptors, multiple pathways. Pharmacol Ther. 2007;114:208–21. doi: 10.1016/j.pharmthera.2007.02.004. [DOI] [PubMed] [Google Scholar]
- 7.Blackburn MR, Vance CO, Morschl E, Wilson CN. Adenosine receptors and inflammation. Handb Exp Pharmacol. 2009:215–69. doi: 10.1007/978-3-540-89615-9_8. [DOI] [PubMed] [Google Scholar]
- 8.Sitkovsky MV, Ohta A. The ‘danger’ sensors that STOP the immune response: the A2 adenosine receptors? Trends Immunol. 2005;26:299–304. doi: 10.1016/j.it.2005.04.004. [DOI] [PubMed] [Google Scholar]
- 9.Hasko G, Linden J, Cronstein B, Pacher P. Adenosine receptors: therapeutic aspects for inflammatory and immune diseases. Nat Rev Drug Discov. 2008;7:759–70. doi: 10.1038/nrd2638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sitkovsky MV, Lukashev D, Apasov S, et al. Physiological control of immune response and inflammatory tissue damage by hypoxia-inducible factors and adenosine A2A receptors. Annu Rev Immunol. 2004;22:657–82. doi: 10.1146/annurev.immunol.22.012703.104731. [DOI] [PubMed] [Google Scholar]
- 11.Yang Z, Day YJ, Toufektsian MC, et al. Myocardial infarct-sparing effect of adenosine A2A receptor activation is due to its action on CD4+ T lymphocytes. Circulation. 2006;114:2056–64. doi: 10.1161/CIRCULATIONAHA.106.649244. [DOI] [PubMed] [Google Scholar]
- 12.Day YJ, Huang L, Ye H, Li L, Linden J, Okusa MD. Renal ischemia-reperfusion injury and adenosine 2A receptor-mediated tissue protection: the role of CD4+ T cells and IFN-gamma. J Immunol. 2006;176:3108–14. doi: 10.4049/jimmunol.176.5.3108. [DOI] [PubMed] [Google Scholar]
- 13.Lappas CM, Day YJ, Marshall MA, Engelhard VH, Linden J. Adenosine A2A receptor activation reduces hepatic ischemia reperfusion injury by inhibiting CD1d- dependent NKT cell activation. J Exp Med. 2006;203:2639–48. doi: 10.1084/jem.20061097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Moore CC, Martin EN, Lee GH, Obrig T, Linden J, Scheld WM. An A2A adenosine receptor agonist, ATL313, reduces inflammation and improves survival in murine sepsis models. BMC Infect Dis. 2008;8:141. doi: 10.1186/1471-2334-8-141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Soop A, Johansson C, Hjemdahl P, et al. Adenosine treatment attenuates cytokine interleukin-6 responses to endotoxin challenge in healthy volunteers. Shock. 2003;19:503–7. doi: 10.1097/01.shk.0000051756.08171.11. [DOI] [PubMed] [Google Scholar]
- 16.Soop A, Sunden-Cullberg J, Albert J, Hallstrom L, Treutiger CJ, Sollevi A. Adenosine infusion attenuates soluble RAGE in endotoxin-induced inflammation in human volunteers. Acta Physiol (Oxf) 2009;197:47–53. doi: 10.1111/j.1748-1716.2009.01985.x. [DOI] [PubMed] [Google Scholar]
- 17.Noji T, Takayama M, Mizutani M, et al. KF24345, an adenosine uptake inhibitor, suppresses lipopolysaccharide-induced tumor necrosis factor-alpha production and leukopenia via endogenous adenosine in mice. J Pharmacol Exp Ther. 2002;300:200–5. doi: 10.1124/jpet.300.1.200. [DOI] [PubMed] [Google Scholar]
- 18.Parmely MJ, Zhou WW, Edwards CK, III, Borcherding DR, Silverstein R, Morrison DC. Adenosine and a related carbocyclic nucleoside analogue selectively inhibit tumor necrosis factor-alpha production and protect mice against endotoxin challenge. J Immunol. 1993;151:389–96. [PubMed] [Google Scholar]
- 19.Hasko G, Nemeth ZH, Vizi ES, Salzman AL, Szabo C. An agonist of adenosine A3 receptors decreases interleukin-12 and interferon-gamma production and prevents lethality in endotoxemic mice. Eur J Pharmacol. 1998;358:261–8. doi: 10.1016/s0014-2999(98)00619-0. [DOI] [PubMed] [Google Scholar]
- 20.Sullivan GW, Fang G, Linden J, Scheld WM. A2A adenosine receptor activation improves survival in mouse models of endotoxemia and sepsis. J Infect Dis. 2004;189:1897–904. doi: 10.1086/386311. [DOI] [PubMed] [Google Scholar]
- 21.Bonneau O, Wyss D, Ferretti S, Blaydon C, Stevenson CS, Trifilieff A. Effect of adenosine A2A receptor activation in murine models of respiratory disorders. Am J Physiol Lung Cell Mol Physiol. 2006;290:L1036–43. doi: 10.1152/ajplung.00422.2005. [DOI] [PubMed] [Google Scholar]
- 22.Reutershan J, Cagnina RE, Chang D, Linden J, Ley K. Therapeutic anti-inflammatory effects of myeloid cell adenosine receptor A2A stimulation in lipopolysaccharide-induced lung injury. J Immunol. 2007;179:1254–63. doi: 10.4049/jimmunol.179.2.1254. [DOI] [PubMed] [Google Scholar]
- 23.Ledent C, Vaugeois JM, Schiffmann SN, et al. Aggressiveness, hypoalgesia and high blood pressure in mice lacking the adenosine A2A receptor. Nature. 1997;388:674–8. doi: 10.1038/41771. [DOI] [PubMed] [Google Scholar]
- 24.Erdmann AA, Gao ZG, Jung U, et al. Activation of Th1 and Tc1 cell adenosine A2A receptors directly inhibits IL-2 secretion in vitro and IL-2-driven expansion in vivo. Blood. 2005;105:4707–14. doi: 10.1182/blood-2004-04-1407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Csoka B, Himer L, Selmeczy Z, et al. Adenosine A2A receptor activation inhibits T helper 1 and T helper 2 cell development and effector function. FASEB J. 2008;22:3491–9. doi: 10.1096/fj.08-107458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Naganuma M, Wiznerowicz EB, Lappas CM, Linden J, Worthington MT, Ernst PB. Cutting edge: critical role for A2A adenosine receptors in the T cell-mediated regulation of colitis. J Immunol. 2006;177:2765–9. doi: 10.4049/jimmunol.177.5.2765. [DOI] [PubMed] [Google Scholar]
- 27.Zarek PE, Huang CT, Lutz ER, et al. A2A receptor signaling promotes peripheral tolerance by inducing T-cell anergy and the generation of adaptive regulatory T cells. Blood. 2008;111:251–9. doi: 10.1182/blood-2007-03-081646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Morrison RR, Tan XL, Ledent C, Mustafa SJ, Hofmann PA. Targeted deletionof A2A aden-osine receptors attenuates the protective effects of myocardial postconditioning. Am J Physiol Heart Circ Physiol. 2007;293:H2523–9. doi: 10.1152/ajpheart.00612.2007. [DOI] [PubMed] [Google Scholar]
- 29.Reichelt ME, Willems L, Hack BA, Peart JN, Headrick JP. Cardiac and coronary function in the Langendorff-perfused mouse heart model. Exp Physiol. 2009;94:54–70. doi: 10.1113/expphysiol.2008.043554. [DOI] [PubMed] [Google Scholar]
- 30.Flood A, Headrick JP. Functional characterization of coronary vascular adenosine receptors in the mouse. Br J Pharmacol. 2001;133:1063–72. doi: 10.1038/sj.bjp.0704170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ammann P, Fehr T, Minder EI, Gunter C, Bertel O. Elevation of troponin I in sepsis and septic shock. Intensive Care Med. 2001;27:965–9. doi: 10.1007/s001340100920. [DOI] [PubMed] [Google Scholar]
- 32.Favory R, Neviere R. Significance and interpretation of elevated troponin in septic patients. Crit Care. 2006;10:224. doi: 10.1186/cc4991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wu AH. Increased troponin in patients with sepsis and septic shock: myocardial necrosis or reversible myocardial depression? Intensive Care Med. 2001;27:959–61. doi: 10.1007/s001340100970. [DOI] [PubMed] [Google Scholar]
- 34.Bateman RM, Tokunaga C, Kareco T, Dorscheid DR, Walley KR. Myocardial hypoxia-inducible HIF-1alpha, VEGF, and GLUT1 gene expression is associated with microvascular and ICAM-1 heterogeneity during endotoxemia. Am J Physiol Heart Circ Physiol. 2007;293:H448–56. doi: 10.1152/ajpheart.00035.2007. [DOI] [PubMed] [Google Scholar]
- 35.Annane D, Bellissant E, Cavaillon JM. Septic shock. Lancet. 2005;365:63–78. doi: 10.1016/S0140-6736(04)17667-8. [DOI] [PubMed] [Google Scholar]
- 36.Cunnion RE, Schaer GL, Parker MM, Natanson C, Parrillo JE. The coronary circulation in human septic shock. Circulation. 1986;73:637–44. doi: 10.1161/01.cir.73.4.637. [DOI] [PubMed] [Google Scholar]
- 37.Avontuur JA, Bruining HA, Ince C. Nitric oxide causes dysfunction of coronary autoregulation in endotoxemic rats. Cardiovasc Res. 1997;35:368–76. doi: 10.1016/s0008-6363(97)00132-6. [DOI] [PubMed] [Google Scholar]
- 38.Buffington CW, Lechner RB, Martin W. Lack of direct coronary vascular effects of Escherichia coli endotoxin in dogs. Proc Soc Exp Biol Med. 1987;186:218–22. doi: 10.3181/00379727-186-42607. [DOI] [PubMed] [Google Scholar]
- 39.Zatta AJ, Headrick JP. Mediators of coronary reactive hyperaemia in isolated mouse heart. Br J Pharmacol. 2005;144:576–87. doi: 10.1038/sj.bjp.0706099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Nemeth ZH, Csoka B, Wilmanski J, et al. Adenosine A2A receptor inactivation increases survival in polymicrobial sepsis. J Immunol. 2006;176:5616–26. doi: 10.4049/jimmunol.176.9.5616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Chouker A, Thiel M, Lukashev D, et al. Critical role of hypoxia and A2A adenosine receptors in liver tissue-protecting physiological anti-inflammatory pathway. Mol Med. 2008;14:116–23. doi: 10.2119/2007-00075.Chouker. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Csoka B, Nemeth ZH, Virag L, et al. A2A adenosine receptors and C/EBPbeta are crucially required for IL-10 production by macrophages exposed to Escherichia coli. Blood. 2007;110:2685–95. doi: 10.1182/blood-2007-01-065870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Barsig J, Kusters S, Vogt K, Volk HD, Tiegs G, Wendel A. Lipopolysaccharide-induced interleukin-10 in mice: role of endogenous tumor necrosis factor-alpha. Eur J Immunol. 1995;25:2888–93. doi: 10.1002/eji.1830251027. [DOI] [PubMed] [Google Scholar]
- 44.Fijen JW, Zijlstra JG, De Boer P, et al. Suppression of the clinical and cytokine response to endotoxin by RWJ-67657, a p38 mitogen-activated protein-kinase inhibitor, in healthy human volunteers. Clin Exp Immunol. 2001;124:16–20. doi: 10.1046/j.1365-2249.2001.01485.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ulich TR, Guo KZ, Irwin B, Remick DG, Davatelis GN. Endotoxin-induced cytokine gene expression in vivo. II. Regulation of tumor necrosis factor and interleukin-1 alpha/beta expression and suppression. Am J Pathol. 1990;137:1173–85. [PMC free article] [PubMed] [Google Scholar]
- 46.Wang J, Wicker LS, Santamaria P. IL-2 and its high-affinity receptor: genetic control of immunoregulation and autoimmunity. Semin Immunol. 2009;21:363–71. doi: 10.1016/j.smim.2009.04.004. [DOI] [PubMed] [Google Scholar]
- 47.Zhang H, Conrad DM, Butler JJ, Zhao C, Blay J, Hoskin DW. Adenosine acts through A2 receptors to inhibit IL-2-induced tyrosine phosphorylation of STAT5 in T lymphocytes: role of cyclic adenosine 3′, 5′-monophosphate and phosphatases. J Immunol. 2004;173:932–44. doi: 10.4049/jimmunol.173.2.932. [DOI] [PubMed] [Google Scholar]
- 48.Ezeamuzie CI, Philips E. Adenosine A3 receptors on human eosinophils mediate inhibition of degranulation and superoxide anion release. Br J Pharmacol. 1999;127:188–94. doi: 10.1038/sj.bjp.0702476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ridker PM, Glynn RJ, Hennekens CH. C-reactive protein adds to the predictive value of total and HDL cholesterol in determining risk of first myocardial infarction. Circulation. 1998;97:2007–11. doi: 10.1161/01.cir.97.20.2007. [DOI] [PubMed] [Google Scholar]
- 50.Griselli M, Herbert J, Hutchinson WL, et al. C-reactive protein and complement are important mediators of tissue damage in acute myocardial infarction. J Exp Med. 1999;190:1733–40. doi: 10.1084/jem.190.12.1733. [DOI] [PMC free article] [PubMed] [Google Scholar]