

Abstract
Alzheimer’s disease (AD) is a complex neurodegenerative process that involves altered brain immune, neuronal and metabolic functions. Understanding the underlying mechanisms has relied on mouse models that mimic components of AD pathology. We used gel-free, label-free LC/MS/MS to quantify protein and phosphopeptide levels in brains of APPSwDI/NOS2−/− (CVN-AD) mice. CVN-AD mice show a full spectrum of AD-like pathology, including amyloid deposition, hyperphosphorylated and aggregated tau and neuronal loss that worsens with age. Tryptic digests, with or without phosphopeptide enrichment on an automated titanium dioxide LC system, were separated by on-line two-dimensional LC and analyzed on a Waters Synapt G2 HDMS, yielding relative expression for >950 proteins and >1100 phosphopeptides. Among differentially expressed proteins were known markers found in humans with AD, including GFAP and C1Q. Phosphorylation of connexin 43, not previously described in AD, was increased at 42 weeks, consistent with dysregulation of gap junctions and activation of astrocytes. Additional alterations in phosphoproteins suggests dysregulation of mitochondria, synaptic transmission, vesicle trafficking and innate immune pathways. These data validate the CVN-AD mouse model of AD, identify novel disease and age-related changes in the brain during disease progression and demonstrate the utility of integrating unbiased and phosphoproteomics for understanding disease processes in AD.
Keywords: Alzheimer’s disease, phosphorylation, NOS2, proteomics, mass spectrometry, neurodegeneration
INTRODUCTION
Alzheimer’s disease (AD) is characterized by the formation of amyloid plaques consisting of amyloid-β peptide (Aβ) in the brain parenchyma and cerebral vasculature, the formation of neurofibrillary tangles (NFTs) consisting of aggregates of hyperphosphorylated tau within neurons, neuronal loss and cognitive changes 1, 2. Along with these major pathological features there is also the presence of neuroinflammation, which is thought to play a key role in AD pathology 3, 4.
The study and treatment of AD has focused primarily on the Aβ cascade hypothesis, which states that the abnormal production of Aβ peptides and their accumulation into amyloid plaques are the underlying cause of the disease 5, 6. However, the recent failure of clinical trials using anti- Aβ therapies has challenged the validity of the amyloid cascade hypothesis 7, 8 and further underscores the lack of a mechanistic understanding of the onset of disease and those changes that are important to disease progression. Numerous transgenic models including mice expressing mutant or human alleles of genes such as the presenilins 9, 10, tau 11, 12 and amyloid precursor protein (APP) 13, 14 have been developed to mimic the pathological features of AD, in particular the deposition of Aβ. While these models have led to significant advances in our understanding of specific pathologies, most do not reproduce all of the key features of AD and thus are limited in their ability to model the complete disease process 15, 16,17.
Our laboratory has focused on the role of redox balance and chronic inflammation as contributing factors to the Alzheimer’s disease process. In so doing, we have developed the APPSwDI/NOS2−/− mouse (herein termed CVN-AD model) that presents with age-related accumulation of Aβ, cerebral amyloid angiopathy, hyperphosphorylated and aggregated tau and neuronal loss, accompanied by loss of learning and memory behaviors 18, 19. This model expresses the Swedish K670N/M671L and vasculotropic Dutch/Iowa E693Q/D694N mutations in the amyloid precursor protein (APP), under control of the Thy-1 promoter, and has a functional knock out of the NOS2 gene encoding for the inducible form of nitric oxide synthase (iNOS) 19. As a result, the CVN-AD mice produce Aβ peptides under conditions of low NO production. We believe that lack of NOS2/iNOS and the subsequent lowering of brain NO levels alter NO-mediated regulation of the immune response to Aβ production and amyloid deposition. Since human NOS2 gene and iNOS protein expression and activity are significantly restricted during immune challenges compared to murine NOS2 and iNOS 20–22, the lowered NO in the CVN-AD mouse produces a more human-like response during chronic inflammation. Our previous studies suggest, then, that the full spectrum of AD-like pathology can be recreated in mice under these unique immune redox conditions 18, 19, 23, 24. Therefore, an in depth study of this AD model should provide valuable insights into the fundamental events that lead to disease progression in humans with AD.
Mass spectrometry (MS)-based proteomics, including quantitation of differential protein expression and post-translational modification, has been an important tool for the identification of biomarkers of AD and for the investigation of underlying causes of AD-like pathology 25–31. Using proteomics, differentially expressed proteins in post-mortem human AD brain tissue are observed when compared to normal age-matched individuals. Many of these proteins are also found in mouse models of amyloid deposition including glial fibrillary acidic protein (GFAP) 26, 27, 32–34, apolipoprotein E (ApoE) 26, 34, 35, peroxiredoxin 6 26, and complement C1Q 26. Our data further supports a role for these specific proteins in the response to Aβ production and amyloid deposition and identifies additional new proteins and phosphoproteins that may contribute to the disease process in AD. To study these changes, robust, quantitative, label-free proteomics strategies were implemented across a cohort of 15 animals. Four different ages of CVN-AD mice were examined (6, 12, 24 and 42 wks), with emphasis on 42 wks where the full spectrum of AD-like pathology is observed. Enrichment of phosphopeptides was performed utilizing a recently developed automated titania-based enrichment system. LC/MS/MS analysis of both unbiased and enriched samples was performed using an online multidimensional LC separation to improve coverage. Our results demonstrate proteins and phospho-proteins that may contribute to the AD-like pathologies in our unique mouse model of AD.
METHODS
Animal Model
APPSwDI mice were obtained from Davis et al 36 and crossed to NOS2−/− mice yielding APPSwDI/NOS2−/− mice as previously described 18. Three male mice were selected for each age (6, 12, 24 and 42 weeks) taking care to include animals from as many different litters as possible within each age group. C57BL6 mice at 42 wks of age were used as wild-type (WT) comparators. All mice were maintained at Duke University under approved animal protocols. The research in this paper was conducted according to NIH guidelines on animal care. Care was taken to minimize the number of animals used and to diminish their suffering. General pathological features of APPSwDI/NOS2−/− mice include severe cerebral vascular amyloid deposition, tau pathology and neuronal loss associated with a decline in learning and memory as previously described 18, 19.
Sample preparation
Mice (6, 12, 24, 42 weeks) were euthanized with an intra-peritoneal injection of ketamine and perfused intracardially with 25 ml 0.9% saline. Brains were removed and bisected into hemispheres. Right hemispheres were fixed in 4% paraformaldehyde, left hemispheres were snap frozen in liquid nitrogen and stored at −80 °C. Snap frozen hemibrains were pulverized under liquid nitrogen and stored at −80°C. Although AD pathology is readily observed in hippocampus, analysis of this region was unlikely to generate sufficient quantities of protein necessary for robust phosphopeptide enrichment (typically 1 mg) in individual animals. Therefore, whole brain lysates were used. To prepare protein lysates, approximately 40 mg of powdered brain was homogenized by sonication in 200–400 μl of lysis buffer containing 50 mM ammonium bicarbonate, pH 8, 0.25% Rapigest SF (Waters), Complete Protease Inhibitor (Roche) and Phosphatase Inhibitor Cocktails I and III (Sigma-Aldrich). After sonication for up to 5 cycles of 5s followed by 30s on ice, samples were centrifuged at 20,000 × g for 10 min. Supernatant protein concentrations were determined by Bradford assay.
For unbiased proteomic analysis 1050 μg of each supernatant was adjusted to 5 μg/μl with lysis buffer and was reduced by incubation with 10 mM DTT for 10 min at 80°C. Next, 20 mM iodoacetamide was added, and samples were incubated at room temp in the dark for 30 min. Following addition of 1:100 (w/w) Sequencing Grade Modified Trypsin (Promega), samples were incubated with shaking at 37°C overnight. Next, samples were acidified to a final concentration of 1% TFA/2% ACN and incubated at 60°C for 2 h. Samples were centrifuged at 20,000 × g for 5 min and 50 μg of digested protein was transferred to Total Recovery LC Vials (Waters) followed by addition of 50 fmol of MassPrep ADH standard (Waters) per μg of brain protein digests. Sample pH was adjusted by addition of 1:1 vol. of 200 mM ammonium formate, pH 10.
To enrich for phosphopeptides the remaining 1 mg per sample of proteolyzed brain homogenates was dried by SpeedVac. Peptides were reconstituted in 40 μl of enrichment loading buffer (80% ACN, 1% TFA, 1M glycolic acid) containing 30 pmol trypsinized bovine α-casein and were enriched using a newly developed automated process 37. Briefly, phosphopeptides were enriched on a 10 cm × 250 μm capillary column packed with 5 μm Titansphere particles (GL Sciences). A CapLC pump and autosampler (Waters) were used to inject the sample as well as to perform a series of wash steps with 80% ACN, 1% TFA and 1 M glycolic acid, followed by 80% ACN, 1% TFA, and finally elution with 5% aqueous ammonia, 20% ACN. The enrichment was monitored online prior to fraction collection using a Waters 2487 UV detector. Fractions were collected into 30 μl of 30% formic acid using a Honeycomb Fraction Collector (BASi). Enriched, acidified phosphopeptides were dried down in a SpeedVac and reconstituted in 15 μl of 33% aqueous ammonia, 67% 10mM citrate, 200mM ammonium formate with 12.5 fmol/μl of MassPrep ADH standard digest for 2D-LC-MS/MS analysis.
2D-LC-MS/MS
Two-dimensional liquid chromatography in a high-low pH reversed phase/reversed phase configuration was utilized for analysis of peptide and phosphopeptide mixtures in a manner similar to previously described 38, 39. For both sample types, peptides were first trapped at 2 μl/min at 97/3 v/v water/ACN in 20 mM ammonium formate (pH 10) on a 5 μm XBridge BEH130 C18 300 um × 50 mm column (Waters). A step gradient of ACN at 2 μl/min was used to elute peptides from the 1st dimension column. Five steps of 10.8%, 14.0%, 16.7%, 20.4% and 50.0% ACN were utilized for the unbiased analyses, and three steps of 4.7%, 9.4% and 30.0% ACN were used for phosphopeptide analyses. These percentages were optimized for delivery of an approximately equal load to the 2nd dimension column for each fraction. For 2nd dimension separation, the eluent from the 1st dimension was first diluted 10-fold online with 99.8/0.1/0.1 v/v/v water/ACN/formic acid and trapped on a 5 μm Symmetry C18 180 μm × 20 mm trapping column (Waters). For un-enriched peptide digests, 2nd dimension separations were performed on a 1.7 μm Acquity BEH130 C18 75 μm × 150 mm column (Waters) using a linear gradient of 7 to 35% ACN with 0.1% formic acid over 37 min, at a flow rate of 0.5 μl/min and column temperature of 35 °C. Phosphopeptide analysis was performed identically with the exception that the gradient was from 3 to 20% over 30 min and 20 to 30% ACN over the next 6 min. The total analysis time was 6 h each for un-enriched samples and 4 h each for phosphopeptide-enriched samples.
All MS data collection was performed with the 2D nanoAcquity coupled to a Synapt G2 HDMS mass spectrometer (Waters). Three types of data acquisition were performed in this study, including data-independent analysis (MSE), ion mobility assisted data-independent analysis (HDMSE) and data-dependent analysis (DDA). MSE runs used 0.6 s cycle time alternating between low collision energy (6 V) and high collision energy ramp in the trap region (15 to 40 V), while HDMSE runs used 0.6 s cycle time alternating between low collision energy (6 V) and high collision energy ramp in the transfer region (27 to 50 V). For DDA, the instrument performed a 0.6 s MS scan followed by MS/MS acquisition on the top 3 ions with charge greater than 1. MS/MS scans for each ion used an isolation window of approximately 2.3 Da, 0.6 s scan duration with a maximum of 3 s per precursor, and dynamic exclusion of ions within 1.2 Da of the selected precursor m/z for 120 s.
Quantitative analysis of unenriched samples was preceded by three column conditioning runs using a pooled sample to improve retention time reproducibility during the study. Approximately 3 μg of unenriched peptide digest was injected and data was collected once for each sample (three biological replicates) using data-independent acquisition (MSE) analysis. Samples were analyzed such that the biological replicates for each condition were evenly distributed throughout the run queue, and a pooled quality control (QC) sample was analyzed three times throughout the study to assess quantitative reproducibility. Finally, seven additional analyses of pooled samples by ion-mobility data-independent analysis (HDMSE) or data-dependent acquisition (DDA) were performed and aligned to gain complementary qualitative identifications.
Phosphopeptide analyses were performed in duplicate for each sample using data-dependent analysis (DDA) mode. Two column-conditioning runs of a pooled sample were performed prior to running individual samples. Approximately 25% of each enriched sample was injected. The first analytical replicate of each sample was analyzed prior to analyzing the second analytical replicate of any sample. Samples were randomized in a similar manner to that done for the unbiased samples and a pool of the phosphoprotein-enriched samples was analyzed seven more times interspersed throughout the study to assess quantitative reproducibility.
Data processing
For robust peak detection and label-free alignment across sample injections (125 raw data files for unbiased and 117 for phosphoproteomics), the commercial package Rosetta Elucidator v3.3 (Rosetta Biosoftware) with multidimensional PeakTeller algorithm was utilized, as recently described 40–44. Briefly, accurate-mass and retention time alignment between samples across each fraction was first performed, with a minimum required peak confidence score of 0.5. Aggregate data for each sample was then obtained by summation of peptide intensities across the three or five fractions. Feature intensities for each injection were subjected to robust median scaling (top and bottom 10% excluded) to generate a single intensity measurement for each feature (accurate mass and retention time pair) in each sample. As previously reported 39, the High/Low pH RPLC configuration generated highly unique fractions, with 82% (9,582/11,695) and 97% (1,497/1,546) of peptides eluting in only a single fraction for unbiased and phosphoproteomic samples, respectively.
We utilized both DDA and HD/MSE to generate peptide identifications for the unbiased samples, and DDA alone for phosphopeptides. For DDA acquisition files, .mgf searchable files were produced in Rosetta Elucidator and searches were then submitted to and retrieved from the Mascot v2.2 (Matrix Sciences) search engine in an automated fashion. For MSE data, ProteinLynx Global Server 2.5 (Waters) was used to generate searchable files which were then submitted to the IdentityE search engine (Waters) 45. Results files were then imported back into Elucidator. A total of 1,174,634 and 59,045 search results were obtained for the unbiased and phosphoproteomic samples, respectively. To enable global spectra scoring across results from both search engines used for the un-enriched samples, search results were concurrently validated using the PeptideProphet and ProteinProphet algorithms in Elucidator using independent reverse decoy database validation 46, 47. The PeptideProphet score was set such that the aggregate data set has a 1.1% peptide false discovery rate (FDR) and a <5% protein FDR, which corresponded to a minimum PeptideProphet score of 0.62. Each peptide identified was allowed to be assigned to a single protein entry, and these assignments were made by ProteinProphet according to the rules of parsimony. For the phosphopeptide samples, annotation was applied to the highest scoring MS/MS spectrum for the aligned data, simply using a minimum Mascot score of 11, giving the desired spectral false discovery rate of 1.0% (4.2% at the peptide level). In addition to phosphopeptide identifications, the confidence of phosphorylation site localization was measured using the implementation of the Ascore algorithm within Elucidator. For binary comparisons and downstream analysis relative protein expression between samples was calculated using the sum of the intensities of all peptides assigned to a particular protein, which has been previously shown to produce accurate data 48. P-values for binary comparisons (see Tables 1–2) were calculated after a log2 transformation of the data using a one-way error-weighted ANOVA with Benjamini-Hochberg FDR correction for multiple hypotheses. For the unenriched and phosphopeptide-enriched data, this was performed at the protein level and phosphopeptide level, respectively. Instead of using a set fold-change cutoff, we chose to use a statistical powering analysis to estimate the appropriate effect-size cutoff as a function of protein or phosphopeptide intensity. It has been previously shown that relative variance (%CV) increases as the observed intensity of a peptide or protein decreases, and a powering analysis can be used to make an appropriate effect size measurement for each protein or peptide since a single fold-change cutoff may not be appropriate across a large intensity range 49. This can also be observed in our data, as shown in the Supplemental Data for proteins (Fig. S1A) and for phosphopeptides (Fig. S1B). The best measure of actual data variance at a particular intensity level was calculated using a moving average, which is the dark line shown in Figure S1. We therefore calculated the effect size (as Cohen’s d) for each binary comparison, and determination of significance was made by using an ANOVA p-value <0.05 and a Cohen’s d > 1.7. An effect size of 1.7 indicates that the mean of one group is positioned at the 95th percentile of the other group used in the comparison 50. Effect Size (Cohen’s d) for each statistically significant analyte is given in Table 1 and 2. The variance values and Cohen’s d calculations for all analytes are included in supplemental tables.
Table 1.
Differentially-expressed proteins in 42 week-old CVN-AD versus WT mice.*
| Protein Name | Protein Description | Fold Change | ANOVA P-value | Cohen’s d | # Peptides | AD Association | Ms Model Proteomics | Human AD Proteomics |
|---|---|---|---|---|---|---|---|---|
| C1QB | Complement C1q subcomponent subunit B | 3.9 | 6.15E-04 | 14.25 | 1 | 26, 94, 95 | 26 | - |
| C1QC | Complement C1q subcomponent subunit C | 3.6 | 0.001 | 4.50 | 1 | 26, 94, 95 | 26 | - |
| APOE | Apolipoprotein E | 2.6 | 0.01 | 9.79 | 19 | 26, 34, 35 | 26 | - |
| GFAP | Glial fibrillary acidic protein | 2.4 | 0.039 | 10.30 | 48 | 26–28, 32–34 | 26, 27 | 28 |
| CH047 | Uncharacterized protein C8orf47 homolog | 2.4 | 0.04 | 3.31 | 2 | - | - | - |
| PP2BC | Serine/threonine-protein phosphatase 2B catalytic subunit gamma | 1.9 | 0.019 | 2.93 | 2 | 96 | - | - |
| DYN2 | Dynamin-2 | 1.9 | 1.38E-09 | 5.24 | 5 | 97, 98 | 98 | - |
| MRP6 | Multidrug resistance-associated protein 6 | 1.8 | 0.028 | 3.79 | 2 | - | - | - |
| RAB43 | Ras-related protein Rab-43 | 1.7 | 0.005 | 4.08 | 5 | - | - | - |
| UK114 | Ribonuclease UK114 | 1.7 | 4.34E-04 | 4.58 | 4 | - | - | - |
| PRDX6 | Peroxiredoxin-6 | 1.5 | 0.009 | 5.96 | 24 | 26 | 26 | - |
| LYST | Lysosomal-trafficking regulator | 1.5 | 0.042 | 1.91 | 2 | - | - | - |
| GLOD 4 | Isoform 3 of Glyoxalase domain-containing protein 4 | 1.3 | <1.00E-06 | 2.48 | 10 | - | - | - |
| AMPL | Isoform 2 of Cytosol aminopeptidase | 1.3 | 0.04 | 2.23 | 6 | - | - | - |
| LXN | Latexin | 1.2 | 0.044 | 1.71 | 3 | - | - | - |
| ENPL | Endoplasmin | 1.2 | 1.16E-05 | 1.88 | 18 | 99, 100 | - | - |
| PRDX3 | Thioredoxin-dependent peroxide reductase | 1.2 | 0.014 | 1.83 | 11 | 101 | - | - |
| SIRT2 | NAD-dependent deacetylase sirtuin-2 | −1.2 | 4.12E-06 | 1.79 | 18 | 102, 103 | - | - |
| AINX | Alpha-internexin | −1.2 | <1.00E-06 | 1.85 | 53 | 104 | 104 | - |
| MOG | Myelin-oligodendrocyte glycoprotein | −1.2 | 0.028 | 2.54 | 10 | - | - | - |
| NIN | Ninein | −1.8 | 2.02E-13 | 3.28 | 2 | - | - | - |
Differentially expressed proteins are listed with references citing previously published AD-associations, as well as references of differential expression in AD model and human AD proteomic studies. Determination of significance was made by using an ANOVA p-value <0.05 and a Cohen’s d effect size > 1.7. To compensate for the low number of replicates, an intensity-based error was used in the calculation of effect size (Supplemental Figure 1). This decreases the influence of erroneous high precision in some measurements, enabling the effect size to be determined by using a moving average of variability, which better approximates the variability at a given point. An effect size of 1.7 indicates that the mean of one group is positioned at the 95th percentile of the other group in a binary comparison 50. The use of Cohen’s d along with ANOVA calculations therefore enables a high degree of confidence that low P-values indeed demonstrate significance even for comparisons with lower apparent fold changes.
Table 2.
Differentially-expressed phosphopeptides in 42 week-old CVN-AD versus WT mice.*
| Modified Peptide Sequence (ModLoc Result) | Max ModLoc | Protein Name | Protein Description | Fold Change | ANOVA P-value | Cohen’s d | AD Association | AD Phospho |
|---|---|---|---|---|---|---|---|---|
| RLQS* IGTENTEENRR | 50 | ALDOA | Fructose-bisphosphate aldolase | 5.8 | 1.79E-13 | 4.90 | 60 | - |
| ELDRFS* LDSEDVYSR | 32 | SHANK 2 | SH3 and multiple ankyrin repeat domains protein 2 | 2.7 | 0.039 | 2.44 | 61 | - |
| (S)L(S)* DPKPLSPTAEESAKER | 9 | BSN | Bassoon | 2.2 | 4.66E-05 | 2.32 | - | - |
| TGEPDEEEGTFR(SS)* IR | 7 | PLM | Phospholemman | 1.8 | 7.81E-05 | 2.36 | 62 | - |
| KVAAGHELQPLAIVDQRPSS* R | 39 | CX43 | Connexin 43 | 1.7 | 0.018 | 1.88 | 63, 64 | - |
| FKIAANEET* PER | 1000 | NASP | Nuclear autoantigenic sperm protein | 1.6 | 0.012 | 1.72 | - | - |
| AIS* LEGEPR | 1000 | DLG2 | Disks large homolog 2 | 1.6 | 0.037 | 1.82 | - | - |
| NIVTPRT* PPPSQGK | 31 | MBP | Isoform 4 of Myelin basic protein | −1.5 | 7.81E-05 | 2.23 | 65 | - |
| FS* WGAEGQKPGFGYGGR | 109 | MBP | Isoform 3/4 of Myelin basic protein | −1.5 | 0.009 | 1.80 | 65 | - |
| ASPVTAPSS* GLHAAVR | 32 | K1522 | Uncharacterized protein KIAA1522 | −1.6 | 0.007 | 1.85 | - | - |
| RGS* GKVPWLK | 1000 | MBP | Isoform 4 of Myelin basic protein | −1.7 | 0.01 | 1.79 | 65 | - |
Peptide sequences listed with phosphorylated residues marked with*and deamidated residues underlined. Proteins are listed with references citing published AD-associations and references to the specific phospho modifications in association with AD model and human AD studies. ModLoc scores of ≥20 indicate 99% certainty in identification of modified residue 52. Determination of significance was made by using an ANOVA p-value <0.05 and a Cohen’s d effect size > 1.7. To compensate for the low number of replicates, an intensity-based error was used in the calculation of effect size (Supplemental Figure 1). This decreases the influence of erroneous high precision in some measurements, enabling the effect size to be determined by using a moving average of variability, which better approximates the variability at a given point. An effect size of 1.7 indicates that the mean of one group is positioned at the 95th percentile of the other group in a binary comparison 50. The use of Cohen’s d along with ANOVA calculations therefore enables a high degree of confidence that low P-values indeed demonstrate significance even for comparisons with lower apparent fold changes.
Both DDA and MSE data were searched against the Swissprot database v57.1 with mus musculus taxonomy along with the sequences for ADH1_YEAST, CASA1_BOVIN and CASA2_BOVIN, plus reverse decoy database (48,026 total entries, downloaded 04/21/11). Mascot searches used 10 ppm precursor and 0.04 Da product ion tolerances, and PLGS 2.5 was searched with auto tolerance resulting in final tolerances of ~5 ppm and ~14 ppm for precursor and product ions, respectively. Both search engines utilized fixed carbamidomethyl (C) and variable oxidized (M) and deamidation (NQ) modifications. MS/MS spectra from phosphopeptide-enriched samples were also searched using variable pST and pY modifications. Enzyme specificity was set to tryptic and a maximum of 2 and 3 missed cleavages were allowed for the unbiased and phosphopeptide searches, respectively. The full set of quantitative data at the peptide-level for each sample injection, including annotation of peptide and protein scores are available in Supplemental Data.
To check for common contaminants the samples acquired using DDA methods were also searched against a concatenated database comprised of the SP_Mouse DB (UniProt) used for the primary analysis and the cRAP DB (common Repository of Adventitious Proteins) v2102.01.1from the Global Proteome Machine (http://www.thegpm.org/crap/). The same search tolerances and allowed modifications used here were identical to that for the principal analysis. No contaminants were identified in the unenriched (<3% FDR at protein level, <1% FDR at the peptide level) or in the phosphopeptide-enriched samples (3% FDR at the protein level, <1% FDR at the peptide level). In addition, data was searched for variable lysine and prolyl hydroxylation. These supplemental data, in the form of a Scaffold file, are available for download at at https://discovery.genome.duke.edu/express/resources/2699/2699_open_mouse_cRAP_073113.sf3 and https://discovery.genome.duke.edu/express/resources/2699/2699_open_mouse_cRAP_pSTY_073113.sf3 and https://discovery.genome.duke.edu/express/resources/2699/2699_open_mouse_cRAP_073113_oK_oP.sf3
Quantitative Data QC
Several steps were taken in order to ensure and check for quantitative reproducibility. Three pooled QC samples were interspersed throughout LC-MS analysis of the un-enriched samples and six pooled phosphopeptide QC samples were interspersed throughout analysis of the enriched samples. All pools and individual samples, both enriched and unenriched, were spiked with digested yeast alcohol dehydrogenase prior to LC-MS analysis (see above). The median CVs of the peptides identified to ADH were 14.3% and 21.8% in the unenriched and enriched data sets, respectively. To monitor the phosphopeptide enrichment, digested alpha-casein was spiked in prior to enrichment and the median CV of the enriched phosphopeptides range was 27%. Binned CV histograms of peptides and phosphopeptides across the pooled QC samples run throughout each sample set are shown in Supplemental Figure S2. Additionally, principle component analysis was performed to identify potential outliers (Supplemental Figure S3).
Immunohistochemistry
Paraformaldehyde fixed hemibrains were incubated for 24 h in 10%, 20% and 30% sucrose sequentially for cryoprotection. Frozen sagittal sections were collected at a thickness of 25 microns on a microtome. Sections were equally spaced at 600 μm apart. Sections were stained free-floating using standard techniques with either AT8 antibody (1:500, Thermo Scientific), or anti-GFAP antibody (1:3000, Invitrogen). Secondary antibodies, ABC kit, and DAB kit were from Vector Laboratories.
Western blot analysis
Forty micrograms of protein supernatants from brain lysates were separated on 4–20% denaturing SDS-PAGE gels (Criterion, BioRad) at a constant voltage of 200 V followed by transfer to PVDF membranes at 4°C overnight at 50 V. Membranes were blocked in TBS, 0.1% Tween-20 (TBST), with 5% non-fat milk and incubated overnight at 4°C in either anti-Cx43 (1:1000, Chemicon), anti-Phos (S368) Cx43 (1:1000, Cell Signaling), anti-pS202/pT205 Tau (AT8) (1:500, Thermo Scientific), anti-pT212/pS214 Tau (AT100) (1:1000, Thermo Scientific), anti-pT231/pS235 Tau (AT180) (1:500, Thermo Scientific), anti-Tau (1:1000, Upstate Biotech), or anti-actin (1:2000, Santa Cruz) in TBST, 1% milk. Membranes were washed in TBST with 1% milk and incubated 1 h in either anti-goat HRP conjugated or anti-rabbit HRP conjugated antibodies (1:2000, Santa Cruz). Membranes were washed and developed using Western Lightning Plus ECL reagent (Perkin Elmer). Digital images were captured using a FluorChem Q imaging system (Cell Biosciences) and quantitative analysis was performed using Alpha View software (Cell Biosciences). Individual densitometry values were adjusted using actin. In the case of phospho-Cx43 analysis, these values were also normalized to Cx43 values for each sample. By normalizing values to 42 week WT samples, which were run on all gels from a single preparation, densitometric comparisons between gels was made possible. Phospho-Tau densitometric data were normalized to total tau.
RESULTS
Development of the integrated analysis of brain sample protein expression and phosphorylation
Since both expression level changes and alterations in phosphorylation can be phenotypic of AD or AD-like pathology, we developed a method to quantitate changes in both levels and in phosphorylation of proteins in individual brain digests from CVN-AD versus wild-type mice. Principal comparisons were made between 42 wk old CVN-AD mice and age matched WT mice. Proteins that were judged to be significantly different at this age were then re-examined in 6, 12 or 24 wk old CVN-AD mice for patterns of change.
Fig. 1 shows a simplified view of the work- flow plan we developed to quantitate both global protein expression and phosphoproteins in the same brain digest. For analysis of changes in global protein expression, a small portion (3 μg per sample) of the digested brain lysate (see methods for details) was separated using five-fraction, two-dimensional liquid chromatography and analyzed in-line with a Waters Synapt G2 HDMS mass spectrometer. For quantitative analysis, each sample was analyzed singly by data-independent acquisition (MSE) with a QC pool also analyzed throughout the study. Representative chromatograms from each fraction of one of the QC pool injections are shown in Figure 1B, highlighting the orthogonal nature of the approach. In addition, pools were analyzed by data-dependent acquisition (DDA) and by ion mobility-assisted MSE (HD-MSE) to increase qualitative identifications. Using Rosetta Elucidator, a total of 25 analyses were aligned independently and the relative abundance of co-aligned features was determined by measuring area-under-the-curve. At 1% peptide FDR, the analysis resulted in the quantitation of 11,695 peptides and 1,814 proteins across the 15 samples, with 955 proteins being confidently identified by two or more peptides (see Supplemental Tables).
Figure 1.

Proteomic workflow. A, Powdered brain lysates from five treatment groups were trypsinized and each was split into two fractions. Un-enriched peptides were analyzed by 5 fraction RP-RP 2D-LC. Phosphopeptides were enriched using an automated capillary TiO2 column, and were analyzed by 3 fraction RP-RP 2D-LC followed by label-free quantitation. B, Representative base peak intensity chromatograms from each fraction for the 5-fraction analysis of the un-enriched samples. C, Representative base peak intensity chromatograms from each fraction for the 3-fraction analysis of the phosphopeptide samples.
Phosphopeptides were enriched from a second aliquot of the tryptic digest sample in an automated fashion using a titania-based enrichment platform. In contrast to our previous label-free phosphoproteomic studies 51, which utilized a single dimension LC separation and a Thermo Orbitrap instrument for analysis of phosphopeptide-enriched samples, for this study, we optimized a three-fraction 2D-LC separation interfaced to a Waters Synapt G2 based on the same high/low pH separation utilized for the un-enriched samples. Using rat brain tissue for method development, a ~30% increase in the number of phosphopeptide identifications was observed (data not shown). Each of the 15 TiO2-enriched mouse brain digests was analyzed in duplicate for more robust quantitation at the peptide level. Representative chromatograms of each fraction from a pooled sample run throughout the study (Fig. 1C) show excellent use of the separation space with the optimized first dimension fraction cuts and second dimension gradient. In addition, the fractions were highly unique, with 97% of phosphopeptides identified in only one of the three fractions (Table S4). Data was processed in Elucidator essentially as described for the un-enriched samples (see above and Methods) except that confidence of site localization was measured using the implementation of the Ascore algorithm within Elucidator 52. At a 1% spectral FDR, we quantified 1,170 phosphopeptides belonging to 560 proteins across the 15 samples. Importantly, using the label-free alignment strategy, these peptides were quantified in virtually all samples. We observed 80% specificity for phosphopeptides based on area under the curve (AUC) measurements of phosphopeptides versus AUC measurements of all identified peptides.
To gauge the increase in proteome coverage by this novel integrated approach, we compared the overlap between the un-enriched and phosphopeptide-enriched proteomes (Fig. 2A). 274 of the 955 quantified proteins (those identified with 2+ peptides) had a corresponding quantified phosphopeptide. For these 274 proteins, changes in phosphopeptide expression can be compared to changes in overall protein expression to elucidate whether the phosphorylation change is likely due to a change in modification stoichiometry or a change in overall protein abundance 53. We also used a top3 protein quantitation that uses the average intensity of the three peptides that yield the highest MS signal for each protein for the estimation of relative protein abundance (Fig. 2B) 54, 55. This is based on the theory that the average of the three most efficiently ionized peptides directly correlates with the input amount of the corresponding protein. A similar plot was made for the phosphopeptide abundance in the enriched samples using only the AUC intensity of each peptide (Fig. 2C). Both plots include all quantified species with those significantly dysregulated at 42 weeks plotted in black. These analyses clearly show that the proteins and phosphopeptides that were significantly changed were distributed across the entire quantifiable dynamic range (Fig. 2B–C).
Figure 2.

Summary of quantified proteomes. A, Venn diagram comparing the overlap between proteins quantified with 2+ peptides in the un-enriched sample versus identified proteins in the phosphoproteome B-C, The intensity of quantified proteins (B) and phosphopeptides (C) plotted against ranked abundance. Proteins/phosphopeptides that were significantly different between CVN-AD and WT mice brain lysates at 42 wks of age were determined by using an ANOVA p-value <0.05 and a Cohen’s d effect size > 1.7 as described in the Methods and are denoted with black circles.
Differential protein expression in CVN-AD mice vs. WT
We first examined relative protein expression in un-enriched samples from aged CVN-AD mice, where an advanced AD-like pathology is clearly evident. We found that 15 proteins with at least 2+ peptides (p<0.05; d>1.7) were increased compared to WT mice at 42 wks of age (Table 1). Two additional proteins quantified by a single peptide were also included in Table 1 (see below). In contrast, only four proteins were significantly down-regulated in CVN-AD versus WT mice and identified by at least 2+ peptides. The Cohen’s d test (see methods) was used to provide increased validation for the significance of the protein changes in CVN-AD mice compared to WT mice.
Age-related changes in protein expression
In order to probe for possible trends in age-associated protein expression that might provide insight into the development of an AD-like phenotype, we compared the levels of proteins known to be significantly different in CVN-AD mice at 42 wks of age (see Table 1) with the levels of the same proteins in CVN-AD mice at 6,12 or 24 wks of age. Examples of these expression patterns are shown in Fig. 3. Protein levels of Multidrug resistance associated protein 6 (MRP6), Ras related protein RAB43 and UK114 ribonuclease in CVN-AD mice increased over the 42 wks to a level significantly greater than WT mice at 42 wks of age (Fig. 3A, E, F, respectively). In contrast, glyoxalase domain containing protein-4 (GLOD-4) levels were initially high at 6 wks and remained elevated throughout the times studied (Fig. 3B). Similarly, patterns were found for proteins that decreased in CVN-AD mice at 42 wks compared to WT mice. For example,, ninein (NIN) levels decreased from 6 wks to 42 wks, while α-Internexin (AINX) protein levels were initially lower or remained low (Fig. 3C, D, respectively).
Figure 3.

Age-related protein intensity patterns in CVN-AD mice. A subset of proteins that were determined to be significantly different between CVN-AD and WT mice at 42wks of age (identified in Table 1) were further assessed to determine patterns of expression in CVN-AD mice at earlier time points (6,12, 24wks). Patterns of change are shown for A, MRP6; B, GLOD 4; C, NIN; D, AINX; E, RAB43; F, UK114. Data are mean protein intensity (Supplemental Table S3) ± S.E.M., *p<0.05 compared to 6wks (one-way ANOVA). P-values indicated above horizontal bars correspond to 42wk CVN-AD vs 42wk WT comparison (also found in Table 1).
To further understand the relationship of protein expression patterns to disease, we immunostained fixed sections of CVN-AD mice brain at 6, 12, 24, and 42 wks of age for GFAP (Fig. 4). Increased immunoreactivity for GFAP is a common indicator of AD-like disease progression 56, 57, As shown in Fig. 4A–H, intense GFAP immunostaining indicative of reactive astrocytes was observed in the CA1 and CA2 regions of the hippocampus, in the subiculum, and in the molecular layer of the dentate gyrus beginning at 12 weeks of age, but was not observed in WT control mice of the corresponding age. Immunopositive staining was also observed in the cortex and was consistent with activated astrocytes surrounding Aβ deposits (arrows Fig. 4). Reactive gliosis increased with age and by 42 weeks was observed throughout a large area of the brain. Thus, these data correlate with the relative protein expression of GFAP in the CVN-AD brains as observed using LC/MS/MS (Fig. 4I).
Figure 4.

Age-related increase in GFAP expression in CVN-AD mice. Representative immunopositive staining for GFAP in mouse brain fixed sections from CVN-AD and from wild-type mice at 6, 12, 24 and 42 weeks of age. GFAP immunoreactivity is evident in the cortex (C, arrows), CA1, CA2, and dentate gyrus (DG) at 12 weeks in CVN-AD mice and progresses into the subiculum (S), CA3, and throughout the cortex with age. Scale bars = 500um. I, Data represent the corresponding mean protein intensity found in brain lysates from CVN-AD mice (Supplemental Table S3) ± S.E.M for GFAP at 6, 12, 24, and 42 wks. *p<0.05 compared to 6wks (one-way ANOVA). P-values indicated above horizontal bars correspond to 42wk CVN-AD vs 42wk WT comparison (also found in Table 1).
Analysis of C1Q proteins
It is increasingly appreciated that “single-hit” proteins may be considered significant based on additional, supplementary criteria 58. Among quantified proteins with only a single peptide, two biologically important proteins, C1QB and C1QC, were markedly upregulated at 42 weeks in CVN-AD mice (Table 1). To justify inclusion of these proteins as significant, we considered the following criteria: a) these two peptides are the most commonly identified to the respective proteins in the PeptideAtlas database (www.peptideatlas.org) and thus the most likely to be identified if the protein is low abundance; b) the C1QB peptide was identified by both Mascot and PLGS search engines in the same 2DLC fraction; c) the C1QC peptide was identified in three independent biological samples (in the same 2DLC fraction); and d) the two peptides to these closely related proteins have virtually identical expression patterns across the samples (Fig. 5A). Using these peptide identification criteria, we determined that complement C1QB and C1QC were significantly changed in CVN-AD mice compared to WT mice, with both subunits of the C1Q complex induced >3.6-fold compared to WT at 42wks. Comparing the expression of C1Q peptides to that of Peroxiredoxin-6 (Prdx6), a protein identified with 24 peptides, we can see a similar increase over time (Fig. 5B), and statistically significant difference between expression in CVN-AD and WT at 42 weeks. Expression patterns of other known markers of AD, including APOE, GFAP, and GRN are also highly consistent (Table S1 and S2).
Figure 5.
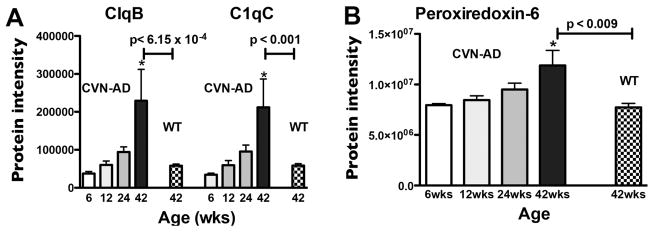
Comparison of single-hit age-related C1Q protein intensity changes with Prdx6 age-related protein changes in CVN-AD mice. Prdx6 was chosen to compare against these single-hit proteins due to its high number (24) of identified peptides. Protein intensity values of C1QB and C1QC (A) and Prdx6 (B) were plotted for mice at 6, 12, 24 and 42 wks of age in CVN-AD mice and at 42wks of age in WT mice. Mean protein intensity (Supplemental Table S3) ± S.E.M. (n = 3) significantly increased in CVN-AD mice with age, *p<0.05 compared to 6wks (one-way ANOVA). P-values indicated above horizontal bars correspond to 42wk CVN-AD vs 42wk WT comparison (Table 1).
Analysis of Phosphopeptides
We identified 7 phosphopeptides that were significantly increased by greater than 1.5-fold, and 4 that were decreased by 1.5-fold or more in CVN-AD versus wild-type mice at 42 weeks (Table 2). Potential associations of the specific phosphopeptide with AD (if known) are also shown and include Aldoa 60, Shank2 61, phospholemman 62, Cx43 63, 64, and MBP 65.
Of the phosphoproteins in Table 2 we chose to examine connexin 43 (Cx43) in more detail. Cx43 is an integral membrane protein that forms hemichannels consisting of six Cx subunits. Adjacent cells align these hemichannels to form intercellular gap junctions that promote communication between astrocytes and facilitate neuronal-glial interactions. Phosphorylation of the Cx43 C-terminus has been previously correlated with multiple cellular functions including gap junction formation and disassembly, regulation of hemichannel permeability, membrane targeting of Cx subunits, alterations in intercellular communication, and astrocyte activation 66,67. We quantified a total of three Cx43 phosphopeptides corresponding to two unique phosphorylation sites, pSer365 and pSer373. The non-phosphorylated forms of these peptides were also found in the un-enriched sample set. All three phosphopeptides were altered with age in the CVN-AD mice and were significantly up-regulated versus WT mice at 42 weeks (Fig. 6A). On the other hand, the intensity values of the non-phosphorylated forms of these peptides in the un-enriched data set did not change at 6, 12, 24, or 42 wk in CVN-AD mice nor in 42wk WT mice (Fig. 6B). To validate our findings of increased Cx43 phosphorylation we measured phosphorylation at Ser368 using a commercially available antibody against pSer368 and Western blot. This site lies between the two phospho sites identified in our study and was not specifically identified among the enriched peptides. The inability to find the Ser368 site in this study is likely due to the trypsin digest which would generate a 4-mer peptide that is too small to be confidently identified in such a complex sample. However, our western blot shows that pSer368 was significantly increased at 24 and 42 weeks in brain lysates of CVN-AD mice versus aged-matched wild-type brain lysates while total levels of Cx43 were unchanged across all treatment groups (Fig. 6C–E). Collectively, these data demonstrate that Cx43 phosphorylation is altered in CVN-AD mice and is consistent with studies demonstrating altered gap junction permeability in Alzheimer’s disease 63, 64.
Figure 6.

Phosphorylation of connexin 43 in CVN-AD versus wild-type mice. A, Average intensities of three different CX43 phosphopeptides, corresponding to two unique phospho-sites, pSer365 and pSer373, are shown for CVN-AD mice at 6, 12, 24 and 42wks of age and for WT mice at 42wk of age. Data are mean intensities ± S.E.M. (n=3 per group), **p<0.01 for CVN-AD 42 weeks versus CVN-AD at 6 weeks (one-way ANOVA); * P< 0.05 for CVN-AD mice 42 weeks versus wild-type (pSer365 peptides only; Rosetta Elucidator error model) B, The corresponding intensities of un-enriched Cx43 peptides (containing non-phosphorylated Ser365 and Ser373) are shown for the same brain lysates as in A. Data are mean ± S.E.M. (n=3 per group). C,D, Cx43 phosphorylation at Ser368 and total Cx43 levels were measured by western blot using whole brain lysates at 6, 12, 24 and 42 wks from either CVN-AD (C) or WT (D) mice. Images are representative of triplicate assays for each mouse strain. E, Data represent the average fold change of pSer368 levels in CVN-AD mice with respect to WT mice. Fold change was determined from the ratios of Cx43-Ser368/total CX43 levels for CVN-AD mice compared to Cx43-pSer368/total CX43 levels for WT mice. Phosphorylation of Ser368 at 24 and 42 weeks was significantly higher in CVN-AD. Data are mean ± S.E.M. (n=3 per group), *p<0.05, CVN-AD versus wild-type (student’s t-test).
Tau phosphorylation
The formation of neurofibrillary tangles (NFT) consisting of hyperphosphorylated and aggregated tau protein is a major hallmark of Alzheimer’s disease. Our previously published studies have shown the formation of hyperphosphorylated tau in aged CVN-AD mice using well-described antibodies (AT8, AT180) to detect specific phosphorylated epitopes 18. We confirmed the presence and distribution of pathologically phosphorylated tau in 6 to 42 week-old CVN-AD mice by immunohistochemistry (Fig. 7) using the AT8 antibody, which is specific for the double phospho-epitope pS202/pT205. Immunostaining was observed in the subiculum and hippocampus of CVN-AD versus wild-type mice beginning at 12 weeks of age and increased with age. A significant increase in phosphorylation of two additional disease associated epitopes, (AT100 (pT212/pS214) and AT180 (pT231/pS235)) was also observed by western blot in CVN-AD mice at 42 weeks compared to WT mice (Fig. 8). The phosphoproteomic analysis identified 24 tau phosphopeptides, many of which are known to be associated with tau pathology, over the entire time course. However, we were unable to detect a statistically significant increase in total or phosphorylated tau at any site with age, nor was there a difference observed when compared to wild-type mice (Supplemental Fig. 4A–B). Since immunohistochemistry clearly shows increased phospho-tau, the lack of detection in our phosphopeptide analysis appears discordant. To better understand this discrepancy we examined the abundance of phospho-tau peptides and compared this to the abundance of other phosphopeptides in our data set. Phospho-tau is estimated to be the 4th most abundant phosphoprotein in mouse brain, accounting for almost 4% of the total phosphopeptide signal, but was estimated to be only the 136th most abundant protein quantified in the un-enriched dataset (Supplemental Fig. 4C–D). Collectively, these data show that tau is highly phosphorylated in mouse brain irrespective of disease-state and suggests that the hyperphosphorylation recognized by the AT8, AT100, and AT180 antibodies represents small and localized increases in phosphotau relative to the total.
Figure 7.

Immunohistochemical analysis of phospho Tau in CVN-AD versus wild-type mice. The phosphorylation of tau was compared between CVN-AD and wild-type mice at 6, 12, 24 and 42 weeks of age using immunohistochemistry on brain sections. AT8 immunoreactivity, corresponding to double phosphorylation at pS202/pT205, is evident in the CA1 and subiculum (S) at 12 weeks in CVN and increases in severity with age. At 42 weeks immunoreactivity is evident in the dentate gyrus (DG). Subiculum scale bars = 100um. Hippocampus scale bars = 500um.
Figure 8.

Western blot analysis of Tau in CVN-AD versus wild-type mice. A. Tau phosphorylation at pT231/pS235 and pT212/pS214 was measured by western blotting using AT180 and AT100 antibodies, respectively, along with total tau and actin in 42 week WT and CVN animals. B. Densitometric analysis of AT180 and AT100 immunoreactivity from whole brain lysates of 42 week CVN-AD mice versus 42 week old WT mice. Data values are normalized to total tau. AT180 and AT100 phospho tau epitopes were significantly higher in CVN-AD. Data are mean ± S.E.M. (n=3 per group), *p<0.05 CVN-AD versus wild-type (student’s t-test).
DISCUSSION
In this study we have shown that label-free quantitative proteomics can be easily implemented for accurate quantitation of brain tissue proteomes and (phospho)proteomes. These techniques, coupled with the CVN-AD mouse model that mimics the full spectrum of human AD-like pathology, have collectively allowed us to examine proteins and phosphoproteins that may be involved in AD. Using this approach we have identified a subset of proteins that when compared to non-diseased wild type mice, are significantly changed as a result of the disease process. A number of the up-regulated proteins have been previously found to be induced in humans with AD and in other mouse AD models, including: glial fibrillary acidic protein (GFAP) 26, 27, 32–34, apolipoprotein E (ApoE) 26, 34, 35, and complement C1Q74, 75. Each of these 3 proteins are believed to be an early component of the response to amyloid and are produced by astrocytes and/or microglia. For example, C1Q initiates complement-mediated pro-inflammatory events, that may be involved in immune-mediated neuronal damage in AD 68, 69. However despite its known involvement in immune toxicity, the neuronal expression of C1Q, its participation in clearance of apoptotic cells, and its ability to induce neurotrophic factors strongly indicate a neuroprotective function for C1Q 70–72.
Peroxiredoxin 6 (Prdx6) is an additional protein found in our study that has been previously associated with AD 26. Oxidative stress has been tightly linked to neurodegenerative diseases including AD and increased expression of anti-oxidant genes and proteins are invariantly observed at some stage of the disease process 73–75. In this study five members of the peroxiredoxin family were identified in the unbiased proteome (Prdx1, 2, 3, 5 and 6), with Prdx4 as a notable exception likely due to its inherently low expression levels in mouse brain 76. Of these family members we detected significantly elevated intensities for Prdx6 and Prdx3 but no change in the expression level of Prdx1, Prdx2, and Prdx5. Prdx6 induction has been found in human AD brain, particularly in plaque associated, GFAP positive astrocytes 77 and is up-regulated in brains of APPSwLon mice 26. Although increased Prdx2 has also been found in AD hippocampus 78, we did not observe a significant change in levels of this specific protein in our study. This may be due, at least in part, to regionalization of Prdx2 expression in brain making detection in whole brain lysates difficult. Compared to the other Prdx isoforms, which are of the 2-Cys peroxiredoxin class, Prdx6 is a bifunctional 1-Cys peroxiredoxin that exhibits both peroxidase and Ca2+-independent phospholipase A2 activities 79. Prdx6 is a stress-responsive protein, and is induced in numerous disease models, although neither the precise regulatory mechanism(s), nor the relevant substrates of Prdx6, are known in AD. Although little is known with regards to its specific induction versus other isoforms, cumulative data suggests that Prdx6 has a protective, rather than deleterious role, in AD 77.
Several significantly up regulated and down-regulated proteins were also found in the proteome but their functions are unknown and/or have not yet been associated with AD. For example, UK114 is a ribonuclease involved in immune regulation of chemokines 80 while latexin, which is also increased in the CVN-AD mice is a negative regulator of stem cells and is associated with aging 81. A 1.8-fold decrease in level was found for the protein, ninein, in CVN-AD versus WT mice at 42 weeks. Ninein expression is increased when neuronal dendrites are formed thus, decreased levels may result in disruption of plasticity. Loss of ninein is also associated with depletion of progenitors in the ventricular zone (VZ) 82, 83. Alpha-internexin, a second protein that is likely to impact neuronal plasticity, was also significantly decreased 84, 85.
An additional strength of our analysis is the ability to achieve high throughput and robust quantitation of phosphopeptides on the same brain samples using an automated phosphopeptide enrichment1. For example, CVN-AD mice demonstrate increased phosphorylation of bassoon, and to a lesser extent of complexin 2 (see Supplemental Table S4). Because of the involvement of these proteins in synaptic transmission 86, 87 these phosphorylation changes suggest that synaptic active zone organization and the regulation of excitatory synaptic activity are altered in our mouse model. Altered regulation of synaptic activity is further indicated by the modifications to Shank2, and DLG2, proteins of the post-synaptic density. The phosphorylation of phospholemman (FXYD1) and the decreased expression of FXYD7 (Supplemental Table S3) both point to an alteration of ion transporter activity, which is regulated by FXYD family members 88 and is critical to proper neuronal excitation. Nonetheless, a major challenge of this (and other) phosphoproteomic analyses is that the functional relevance of only a small fraction of annotated phosphorylation sites have been characterized. Thus, although phosphorylation was dysregulated on a number of AD-associated proteins in our study, the implications of many of these site-specific modifications are for the most part not readily interpretable.
Astrocyte proteins were also found to have abnormal phosphorylation patterns that could affect normal functioning. For example, gap junction protein connexin 43 (Cx43) affects glutamate uptake, a key process involved in excitotoxicity 67. Alterations to Cx43 phosphorylation have been reported to correlate with the activation of astrocytes in AD and in the response to traumatic brain injury (TBI) 89. Phosphorylation at specific sites regulates membrane targeting, gap junction assembly and permeability 66. In our study we identified two sites of increased phosphorylation on Cx43, namely, Ser365, and Ser368. Increased phosphorylation at Ser368 has been functionally linked to a decrease in gap junction permeability and is often correlated to regions of TBI and decreased glutamate clearance 90. It is also interesting to note that phosphorylation at Ser365 may in fact prevent phosphorylation of Ser368 66, suggesting that these specific changes in Cx43 phosphorylation may be mutually exclusive and occur in a regionally distinct manner.
Phosphorylation of the tau protein at specific sites is clearly an important pathological finding that is relevant to AD 91, 92. Previously, we have reported that the AT8 phospho-epitope (pS202/pT205) was phosphorylated in CVN-AD mice and was localized to affected neurons 18. In this study we performed a longitudinal immunohistochemical analysis of brain sections in the CVN-AD mouse and show that disease-associated AT8 positive tau is markedly increased beginning at 12 weeks and further increases with age. We also detected increased phosphorylation by western blot analysis of the AT180 (pT231/pS235), and AT100 (pT212/pS214) disease-associated phospho epitopes. However, while our phosphoproteomic analysis clearly shows that tau is abundantly phosphorylated in whole brain, representing greater than 4% of the total phosphopeptide intensity, global levels of tau phosphorylation do not appear significantly altered in whole brain homogenates of CVN-AD versus wild-type mice assayed by this method. Two potential explanations are possible. First, while use of whole tissue is not an uncommon approach, regional specificity of the tau pathology may limit the ability to detect changes in whole brain lysates. Additionally, the sensitivity of unlabeled MS/MS quantitation for singular proteins in complex mixtures is generally less than the sensitivity obtained through more specific detection methods, such as immunodetection 93. As indicted by immunohistochemistry, the phosphorylation of disease-associated tau in AD and CVN-AD mice is regionally constrained. Thus, although localized increases in specific phospho-tau epitopes are detectable by specific antibodies, these changes are masked by high levels of global tau phosphorylation when assayed using whole brain MS/MS analysis. These data re-enforce the requirement for the combination of localized (regional, subcellular and PTM-specific) analyses with phosphoproteomic analyses of disease models.
Overall, our analysis identified alterations in protein expression associated with immune reactivity, lipid metabolism and white matter changes, cytoskeletal integrity, synaptic regulation, axonal transport, vesicle trafficking, signal transduction, and redox balance. Many of the identified proteins in Table 1 are known to be found in humans with AD through either biochemical analyses or direct proteomic quantification. These data show early involvement of specific proteins and phospho-proteins such as C1qb/c, GFAP and Phospho-Cx43 as well as proteins and phospho-proteins not commonly associated with AD such as RAB43, MRP6 or ninein. Insight into the role of these proteins in the disease process may be useful to further understanding AD pathology and mechanisms behind the disease. In addition, the extensive overlap of proteins and protein phospho-modifications observed in CVN-AD mice with proteins and phospho-proteins known to be altered in humans with AD provides significant validation of this mouse model of AD. Finally, these data show that gel and label-free methodologies, combined with automated phosphopeptide enrichment, are well-suited to statistically-robust investigations of longitudinal protein expression and post-translational modifications in disease processes.
Supplementary Material
Acknowledgments
This work was supported by National Institutes of Health grants: AG031845; AG031124
ABBREVIATIONS
- AD
Alzheimer’s disease
- APP
amyloid precursor protein
- APPSwDI
amyloid precursor protein (K670N/M671L, E693Q, D694N)
- NOS2
nitric oxide synthase 2
- WT
wild-type
- CVN-AD
(APPSwDI/NOS2−/−)
- Ser
serine
- NFT
neurofibrillary tangle
- Aβ
amyloid beta
- CSF
cerebral spinal fluid
- PIB
Pittsburgh compound B
- PET
positron emission tomography
- iNOS
inducible nitric oxide synthase
- NO
nitric oxide
- GFAP
glial fibrillary acidic protein
- ApoE
apolipoprotein E
- ADH
alcohol dehydrogenase
- DAB
3,3′-diaminobenzidine
- Cx43
connexin 43
- HRP
horseraddish peroxidase
- ECL
enhanced chemiluminescence
- DDA
data dependent analysis
- QC
quality control
- RPLC
reverse phase liquid chromatography
- FDR
false discovery rate
- CASA
casein A
- TiO2
titanium dioxide
- Prdx6
peroxiredoxin 6
- Prdx1
peroxiredoxin 1
- Prdx2
peroxiredoxin 2
- Prdx3
peroxiredoxin 3
- Prdx5
peroxiredoxin 5
- PLGS
ProteinLynx Global Server
- MW
molecular weight
- PHF
paired helical filaments
- MAP
microtubule associated protein
- GSK3β
glycogen synthase kinase 3 beta
- CDK
cyclin dependent kinase
- PKC
protein kinase C
- Sir2
silent information regulation 2
- HDAC
histone deacetylase
- SIRT2
sirtuin 2
- TBI
traumatic brain injury
- PTEN
phosphatase and tensin homolog
- PI3k
phosphatidylinositol-3-OH kinase
- Akt
protein kinase B
- PKB
protein kinase B
- PKA
protein kinase A
- PKN
protein kinase N
- CaMII
Ca2+/calmodulin protein kinase II
- C1qB
complement C1q subcomponent B
- C1qC
complement C1q subcomponent C
- CHO47
uncharacterized protein C8orf47 homolog
- PP2BC
serine/threonine-protein phosphatase 2B catalytic subunit gamma
- Dyn2
dynamin 2
- Mrp6
multidrug resistance-associated protein 6
- Rab43
ras-related protein Rab-43
- UK114
ribonuclease UK114
- Lyst
lysosomal-trafficking regulator
- NIN
ninein
- A4
amyloid precursor protein
- AldoA
fructose-bisphosphate aldolase
- Shank2
SH3 and multiple ankyrin repeat domains protein 2
- Bsn
bassoon
- NDRG2
N-myc down-regulated family member 2
- Plm
phospholemman
- Rims1
regulating synaptic membrane exocytosis protein 1
- NASP
nuclear autoantigenic sperm protein
- DLG2
Disks large homolog 2
- Spir1
Spire 1 homolog
- GAP43
neuromodulin
- MBP
myelin basic protein
- K1522
uncharacterized protein KIAA 1522
- NHERF1
Na+/H+ exchange regulatory cofactor 1
- DG
dentate gyrus
Footnotes
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.
Supporting information includes: an Excel file, “Hoos_JPR_Supporting_Information.xlxs”, which contains: Supplemental Table S1 – Peptide expression data from un-enriched dataset containing peptide sequences, precursor mass and charge, product ion mass, fixed and variable modifications, peptide identity scores, protein accession numbers, Supplemental Table S2 – Peptide best flier values used in determination of protein expression data. Supplemental Table S3 – Protein expression data from un-enriched dataset including number of distinct peptides per protein, quantitative expression values, CV, ANOVA and Cohen’s d calculations for age group comparisons. Supplemental Table S4 – Peptide expression data from phosphopeptide-enriched dataset containing peptide sequences, precursor mass and charge, product ion mass, fixed and variable modifications, Mascot ion scores, protein accession numbers, quantitative expression values and site localization scores (Ascore). Supplemental Table S5 – Spectrum output from Scaffold file containing un-enriched dataset. Includes protein sequence coverage, accession numbers, indication of shared peptides and number of unique peptides per protein. Supplemental Table S6 – Spectrum output from Scaffold file containing phosphopeptide-enriched dataset. Includes protein sequence coverage, accession numbers, indication of shared peptides and number of unique peptides per protein. In addition, “Hoos_JPR_Supporting_Information.pdf,” which contains: Supplemental Figure S1 – Moving average used in Cohen’s d calculation for significance.. Supplemental Figure S2 – Principle component analysis to identify potential outliers. Supplemental Figure S3 – Binned CV histograms of peptides and phosphopeptides across the pooled QC samples run throughout each sample set. Supplemental Figure S4- Phosphorylation of Tau in CVN-AD versus wild-type mice. A, Intensities of all tau phosphopeptides in the enriched samples from the proteomic data. B, Intensities of all tau peptides from the un-enriched proteomic data. C, The five most abundant phosphoproteins in terms of the percent of total phosphopeptide signal. D, The five most abundant proteins in terms of intensity, along with the top five phosphoproteins. Data in C and D are mean ± S.E.M. (n=18 analyses, un-enriched; n=39 analyses, phospho-enriched).
Supplemental data, in the form of a Scaffold file, are available for download at at https://discovery.genome.duke.edu/express/resources/2699/2699_open_mouse_cRAP_073113.sf3 and https://discovery.genome.duke.edu/express/resources/2699/2699_open_mouse_cRAP_pSTY_073113.sf3 and https://discovery.genome.duke.edu/express/resources/2699/2699_open_mouse_cRAP_073113_oK_oP.sf3
Supporting material is available free of charge via the Internet at http://pubs.acs.org. Scaffold files containing MS/MS spectra of identified peptides and phosphopeptides are available for download at https://discovery.genome.duke.edu/express/resources/2699/MSMS_Supplement.zip and can be viewed using the Scaffold Free Viewer (Proteome Software) available for download at http://www.proteomesoftware.com.
References
- 1.Nagy Z, Esiri MM, Joachim C, Jobst KA, Morris JH, King EM, Hindley NJ, McDonald B, Litchfield S, Barnetson L, Smith AD. Comparison of pathological diagnostic criteria for Alzheimer disease. Alzheimer Dis Assoc Disord. 1998;12(3):182–9. doi: 10.1097/00002093-199809000-00010. [DOI] [PubMed] [Google Scholar]
- 2.Selkoe DJ. Alzheimer’s disease: genes, proteins, and therapy. Physiol Rev. 2001;81(2):741–66. doi: 10.1152/physrev.2001.81.2.741. [DOI] [PubMed] [Google Scholar]
- 3.Rogers J. The inflammatory response in Alzheimer’s disease. J Periodontol. 2008;79(8 Suppl):1535–43. doi: 10.1902/jop.2008.080171. [DOI] [PubMed] [Google Scholar]
- 4.Colton CA, Chernyshev ON, Gilbert DL, Vitek MP. Microglial contribution to oxidative stress in Alzheimer’s disease. Ann N Y Acad Sci. 2000;899:292–307. doi: 10.1111/j.1749-6632.2000.tb06195.x. [DOI] [PubMed] [Google Scholar]
- 5.Selkoe DJ. The genetics and molecular pathology of Alzheimer’s disease: roles of amyloid and the presenilins. Neurol Clin. 2000;18(4):903–22. doi: 10.1016/s0733-8619(05)70232-2. [DOI] [PubMed] [Google Scholar]
- 6.Gilbert BJ. The role of amyloid beta in the pathogenesis of Alzheimer’s disease. J Clin Pathol. 2013;66(5):362–6. doi: 10.1136/jclinpath-2013-201515. [DOI] [PubMed] [Google Scholar]
- 7.Mullane K, Williams M. Alzheimer’s therapeutics: continued clinical failures question the validity of the amyloid hypothesis-but what lies beyond? Biochem Pharmacol. 2013;85(3):289–305. doi: 10.1016/j.bcp.2012.11.014. [DOI] [PubMed] [Google Scholar]
- 8.Castellani RJ, Perry G. Pathogenesis and disease-modifying therapy in Alzheimer’s disease: the flat line of progress. Arch Med Res. 2012;43(8):694–8. doi: 10.1016/j.arcmed.2012.09.009. [DOI] [PubMed] [Google Scholar]
- 9.Guo Q, Fu W, Sopher BL, Miller MW, Ware CB, Martin GM, Mattson MP. Increased vulnerability of hippocampal neurons to excitotoxic necrosis in presenilin-1 mutant knock-in mice. Nat Med. 1999;5(1):101–6. doi: 10.1038/4789. [DOI] [PubMed] [Google Scholar]
- 10.Wang R, Dineley KT, Sweatt JD, Zheng H. Presenilin 1 familial Alzheimer’s disease mutation leads to defective associative learning and impaired adult neurogenesis. Neuroscience. 2004;126(2):305–12. doi: 10.1016/j.neuroscience.2004.03.048. [DOI] [PubMed] [Google Scholar]
- 11.Lewis J, McGowan E, Rockwood J, Melrose H, Nacharaju P, Van Slegtenhorst M, Gwinn-Hardy K, Paul Murphy M, Baker M, Yu X, Duff K, Hardy J, Corral A, Lin WL, Yen SH, Dickson DW, Davies P, Hutton M. Neurofibrillary tangles, amyotrophy and progressive motor disturbance in mice expressing mutant (P301L) tau protein. Nat Genet. 2000;25(4):402–5. doi: 10.1038/78078. [DOI] [PubMed] [Google Scholar]
- 12.Andorfer C, Kress Y, Espinoza M, de Silva R, Tucker KL, Barde YA, Duff K, Davies P. Hyperphosphorylation and aggregation of tau in mice expressing normal human tau isoforms. J Neurochem. 2003;86(3):582–90. doi: 10.1046/j.1471-4159.2003.01879.x. [DOI] [PubMed] [Google Scholar]
- 13.Hsiao K, Chapman P, Nilsen S, Eckman C, Harigaya Y, Younkin S, Yang F, Cole G. Correlative memory deficits, Abeta elevation, and amyloid plaques in transgenic mice. Science. 1996;274(5284):99–102. doi: 10.1126/science.274.5284.99. [DOI] [PubMed] [Google Scholar]
- 14.Games D, Adams D, Alessandrini R, Barbour R, Berthelette P, Blackwell C, Carr T, Clemens J, Donaldson T, Gillespie F, et al. Alzheimer-type neuropathology in transgenic mice overexpressing V717F beta-amyloid precursor protein. Nature. 1995;373(6514):523–7. doi: 10.1038/373523a0. [DOI] [PubMed] [Google Scholar]
- 15.Schwab C, Hosokawa M, McGeer PL. Transgenic mice overexpressing amyloid beta protein are an incomplete model of Alzheimer disease. Exp Neurol. 2004;188(1):52–64. doi: 10.1016/j.expneurol.2004.03.016. [DOI] [PubMed] [Google Scholar]
- 16.Radde R, Duma C, Goedert M, Jucker M. The value of incomplete mouse models of Alzheimer’s disease. Eur J Nucl Med Mol Imaging. 2008;35 (Suppl 1):S70–4. doi: 10.1007/s00259-007-0704-y. [DOI] [PubMed] [Google Scholar]
- 17.Oddo S, Caccamo A, Shepherd JD, Murphy MP, Golde TE, Kayed R, Metherate R, Mattson MP, Akbari Y, LaFerla FM. Triple-transgenic model of Alzheimer’s disease with plaques and tangles: intracellular Abeta and synaptic dysfunction. Neuron. 2003;39(3):409–21. doi: 10.1016/s0896-6273(03)00434-3. [DOI] [PubMed] [Google Scholar]
- 18.Wilcock DM, Lewis MR, Van Nostrand WE, Davis J, Previti ML, Gharkholonarehe N, Vitek MP, Colton CA. Progression of amyloid pathology to Alzheimer’s disease pathology in an amyloid precursor protein transgenic mouse model by removal of nitric oxide synthase 2. J Neurosci. 2008;28(7):1537–45. doi: 10.1523/JNEUROSCI.5066-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Colton CA, Wilcock DM, Wink DA, Davis J, Van Nostrand WE, Vitek MP. The effects of NOS2 gene deletion on mice expressing mutated human AbetaPP. J Alzheimers Dis. 2008;15(4):571–87. doi: 10.3233/jad-2008-15405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Geller DA, Billiar TR. Molecular biology of nitric oxide synthases. Cancer Metastasis Rev. 1998;17(1):7–23. doi: 10.1023/a:1005940202801. [DOI] [PubMed] [Google Scholar]
- 21.Weinberg JB, Misukonis MA, Shami PJ, Mason SN, Sauls DL, Dittman WA, Wood ER, Smith GK, McDonald B, Bachus KE, et al. Human mononuclear phagocyte inducible nitric oxide synthase (iNOS): analysis of iNOS mRNA, iNOS protein, biopterin, and nitric oxide production by blood monocytes and peritoneal macrophages. Blood. 1995;86(3):1184–95. [PubMed] [Google Scholar]
- 22.Mestas J, Hughes CC. Of mice and not men: differences between mouse and human immunology. J Immunol. 2004;172(5):2731–8. doi: 10.4049/jimmunol.172.5.2731. [DOI] [PubMed] [Google Scholar]
- 23.Colton CA, Vitek MP, Wink DA, Xu Q, Cantillana V, Previti ML, Van Nostrand WE, Weinberg JB, Dawson H. NO synthase 2 (NOS2) deletion promotes multiple pathologies in a mouse model of Alzheimer’s disease. Proc Natl Acad Sci U S A. 2006;103(34):12867–72. doi: 10.1073/pnas.0601075103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ridnour LA, Dhanapal S, Hoos M, Wilson J, Lee J, Cheng RY, Brueggemann EE, Hines HB, Wilcock DM, Vitek MP, Wink DA, Colton CA. Nitric oxide-mediated regulation of beta-amyloid clearance via alterations of MMP-9/TIMP-1. J Neurochem. 2012;123(5):736–49. doi: 10.1111/jnc.12028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sowell RA, Owen JB, Butterfield DA. Proteomics in animal models of Alzheimer’s and Parkinson’s diseases. Ageing Res Rev. 2009;8(1):1–17. doi: 10.1016/j.arr.2008.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sizova D, Charbaut E, Delalande F, Poirier F, High AA, Parker F, Van Dorsselaer A, Duchesne M, Diu-Hercend A. Proteomic analysis of brain tissue from an Alzheimer’s disease mouse model by two-dimensional difference gel electrophoresis. Neurobiol Aging. 2007;28(3):357–70. doi: 10.1016/j.neurobiolaging.2006.01.011. [DOI] [PubMed] [Google Scholar]
- 27.Shin SJ, Lee SE, Boo JH, Kim M, Yoon YD, Kim SI, Mook-Jung I. Profiling proteins related to amyloid deposited brain of Tg2576 mice. Proteomics. 2004;4(11):3359–68. doi: 10.1002/pmic.200400961. [DOI] [PubMed] [Google Scholar]
- 28.Shiozaki A, Tsuji T, Kohno R, Kawamata J, Uemura K, Teraoka H, Shimohama S. Proteome analysis of brain proteins in Alzheimer’s disease: subproteomics following sequentially extracted protein preparation. J Alzheimers Dis. 2004;6(3):257–68. doi: 10.3233/jad-2004-6306. [DOI] [PubMed] [Google Scholar]
- 29.Di Domenico F, Sultana R, Barone E, Perluigi M, Cini C, Mancuso C, Cai J, Pierce WM, Butterfield DA. Quantitative proteomics analysis of phosphorylated proteins in the hippocampus of Alzheimer’s disease subjects. J Proteomics. 2011;74(7):1091–103. doi: 10.1016/j.jprot.2011.03.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Robinson RA, Lange MB, Sultana R, Galvan V, Fombonne J, Gorostiza O, Zhang J, Warrier G, Cai J, Pierce WM, Bredesen DE, Butterfield DA. Differential expression and redox proteomics analyses of an Alzheimer disease transgenic mouse model: effects of the amyloid-beta peptide of amyloid precursor protein(Xi) Neuroscience. 2011;177:207–22. doi: 10.1016/j.neuroscience.2011.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Rudrabhatla P, Jaffe H, Pant HC. Direct evidence of phosphorylated neuronal intermediate filament proteins in neurofibrillary tangles (NFTs): phosphoproteomics of Alzheimer’s NFTs. FASEB J. 2011;25(11):3896–905. doi: 10.1096/fj.11-181297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ross GW, O’Callaghan JP, Sharp DS, Petrovitch H, Miller DB, Abbott RD, Nelson J, Launer LJ, Foley DJ, Burchfiel CM, Hardman J, White LR. Quantification of regional glial fibrillary acidic protein levels in Alzheimer’s disease. Acta Neurol Scand. 2003;107(5):318–23. doi: 10.1034/j.1600-0404.2003.02098.x. [DOI] [PubMed] [Google Scholar]
- 33.Noppe M, Crols R, Andries D, Lowenthal A. Determination in human cerebrospinal fluid of glial fibrillary acidic protein, S-100 and myelin basic protein as indices of non-specific or specific central nervous tissue pathology. Clin Chim Acta. 1986;155(2):143–50. doi: 10.1016/0009-8981(86)90275-5. [DOI] [PubMed] [Google Scholar]
- 34.Diedrich JF, Minnigan H, Carp RI, Whitaker JN, Race R, Frey W, 2nd, Haase AT. Neuropathological changes in scrapie Alzheimer’s disease are associated with increased expression of apolipoprotein E and cathepsin D in astrocytes. J Virol. 1991;65(9):4759–68. doi: 10.1128/jvi.65.9.4759-4768.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Barger SW, DeWall KM, Liu L, Mrak RE, Griffin WS. Relationships between expression of apolipoprotein E and beta-amyloid precursor protein are altered in proximity to Alzheimer beta-amyloid plaques: potential explanations from cell culture studies. J Neuropathol Exp Neurol. 2008;67(8):773–83. doi: 10.1097/NEN.0b013e318180ec47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Davis J, Xu F, Deane R, Romanov G, Previti ML, Zeigler K, Zlokovic BV, Van Nostrand WE. Early-onset and robust cerebral microvascular accumulation of amyloid beta-protein in transgenic mice expressing low levels of a vasculotropic Dutch/Iowa mutant form of amyloid beta-protein precursor. J Biol Chem. 2004;279(19):20296–306. doi: 10.1074/jbc.M312946200. [DOI] [PubMed] [Google Scholar]
- 37.Richardson BM, Soderblom EJ, Thompson JW, Moseley MA. Automated, Reproducible, Titania-Based Phosphopeptide Enrichment Strategy for Label-Free Quantitative Phosphoproteomics. J Biomol Tech. 2013 doi: 10.7171/jbt.13-2401-002. Published online 2012 December 22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Gilar M, Olivova P, Daly AE, Gebler JC. Orthogonality of separation in two-dimensional liquid chromatography. Anal Chem. 2005;77(19):6426–34. doi: 10.1021/ac050923i. [DOI] [PubMed] [Google Scholar]
- 39.Dowell JA, Frost DC, Zhang J, Li L. Comparison of two-dimensional fractionation techniques for shotgun proteomics. Anal Chem. 2008;80(17):6715–23. doi: 10.1021/ac8007994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Paweletz CP, Wiener MC, Bondarenko AY, Yates NA, Song Q, Liaw A, Lee AY, Hunt BT, Henle ES, Meng F, Sleph HF, Holahan M, Sankaranarayanan S, Simon AJ, Settlage RE, Sachs JR, Shearman M, Sachs AB, Cook JJ, Hendrickson RC. Application of an end-to-end biomarker discovery platform to identify target engagement markers in cerebrospinal fluid by high resolution differential mass spectrometry. J Proteome Res. 2010;9(3):1392–401. doi: 10.1021/pr900925d. [DOI] [PubMed] [Google Scholar]
- 41.Meng F, Wiener MC, Sachs JR, Burns C, Verma P, Paweletz CP, Mazur MT, Deyanova EG, Yates NA, Hendrickson RC. Quantitative analysis of complex peptide mixtures using FTMS and differential mass spectrometry. J Am Soc Mass Spectrom. 2007;18(2):226–33. doi: 10.1016/j.jasms.2006.09.014. [DOI] [PubMed] [Google Scholar]
- 42.Saka HA, Thompson JW, Chen YS, Kumar Y, Dubois LG, Moseley MA, Valdivia RH. Quantitative proteomics reveals metabolic and pathogenic properties of Chlamydia trachomatis developmental forms. Molecular Microbiology. 2011;82(5):1185–1203. doi: 10.1111/j.1365-2958.2011.07877.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Soderblom EJ, Philipp M, Thompson JW, Caron MG, Moseley MA. Quantitative label-free phosphoproteomics strategy for multifaceted experimental designs. Analytical Chemistry. 2011;83(10):3758–64. doi: 10.1021/ac200213b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Reidel B, Thompson JW, Farsiu S, Moseley MA, Skiba NP, Arshavsky VY. Proteomic profiling of a layered tissue reveals unique glycolytic specializations of photoreceptor cells. Molecular & Cellular Proteomics. 2011;10(3):M110 002469. doi: 10.1074/mcp.M110.002469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Geromanos SJ, Vissers JP, Silva JC, Dorschel CA, Li GZ, Gorenstein MV, Bateman RH, Langridge JI. The detection, correlation, and comparison of peptide precursor and product ions from data independent LC-MS with data dependant LC-MS/MS. Proteomics. 2009;9(6):1683–95. doi: 10.1002/pmic.200800562. [DOI] [PubMed] [Google Scholar]
- 46.Keller A, Nesvizhskii AI, Kolker E, Aebersold R. Empirical statistical model to estimate the accuracy of peptide identifications made by MS/MS and database search. Anal Chem. 2002;74(20):5383–92. doi: 10.1021/ac025747h. [DOI] [PubMed] [Google Scholar]
- 47.Nesvizhskii AI, Keller A, Kolker E, Aebersold R. A statistical model for identifying proteins by tandem mass spectrometry. Anal Chem. 2003;75(17):4646–58. doi: 10.1021/ac0341261. [DOI] [PubMed] [Google Scholar]
- 48.Reidel B, Thompson JW, Farsiu S, Moseley MA, Skiba NP, Arshavsky VY. Proteomic profiling of a layered tissue reveals unique glycolytic specializations of photoreceptor cells. Mol Cell Proteomics. 2011;10(3):M110 002469. doi: 10.1074/mcp.M110.002469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Levin Y. The role of statistical power analysis in quantitative proteomics. Proteomics. 2011;11(12):2565–7. doi: 10.1002/pmic.201100033. [DOI] [PubMed] [Google Scholar]
- 50.Cohen J. Statistical power analysis for the behavioral sciences. 2. L. Erlbaum Associates; Hillsdale, N.J: 1988. p. xxi.p. 567. [Google Scholar]
- 51.Soderblom EJ, Philipp M, Thompson JW, Caron MG, Moseley MA. Quantitative label-free phosphoproteomics strategy for multifaceted experimental designs. Anal Chem. 2011;83(10):3758–64. doi: 10.1021/ac200213b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Beausoleil SA, Villen J, Gerber SA, Rush J, Gygi SP. A probability-based approach for high-throughput protein phosphorylation analysis and site localization. Nat Biotechnol. 2006;24(10):1285–92. doi: 10.1038/nbt1240. [DOI] [PubMed] [Google Scholar]
- 53.Wu R, Dephoure N, Haas W, Huttlin Edward L, Zhai B, Sowa Mathew E, Gygi Steven P. Correct interpretation of comprehensive phosphorylation dynamics requires normalization by protein expression changes. Mol Cell Proteomics. 2011;10(8):M111 009654. doi: 10.1074/mcp.M111.009654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Silva JC, Gorenstein MV, Li G-z, Vissers JPC, Geromanos SJ. Absolute quantification of proteins by LCMS. A virtue of parallel MS acquisition. Mol Cell Proteomics. 2006;5(1):144–156. doi: 10.1074/mcp.M500230-MCP200. [DOI] [PubMed] [Google Scholar]
- 55.Grossmann J, Roschitzki B, Panse C, Fortes C, Barkow-Oesterreicher S, Rutishauser D, Schlapbach R. Implementation and evaluation of relative and absolute quantification in shotgun proteomics with label-free methods. J Proteomics. 2010;73(9):1740–6. doi: 10.1016/j.jprot.2010.05.011. [DOI] [PubMed] [Google Scholar]
- 56.Nagele RG, Wegiel J, Venkataraman V, Imaki H, Wang KC. Contribution of glial cells to the development of amyloid plaques in Alzheimer’s disease. Neurobiol Aging. 2004;25(5):663–74. doi: 10.1016/j.neurobiolaging.2004.01.007. [DOI] [PubMed] [Google Scholar]
- 57.Rodriguez JJ, Olabarria M, Chvatal A, Verkhratsky A. Astroglia in dementia and Alzheimer’s disease. Cell Death Differ. 2009;16(3):378–85. doi: 10.1038/cdd.2008.172. [DOI] [PubMed] [Google Scholar]
- 58.Shteynberg D, Deutsch EW, Lam H, Eng JK, Sun Z, Tasman N, Mendoza L, Moritz RL, Aebersold R, Nesvizhskii AI. iProphet: multi-level integrative analysis of shotgun proteomic data improves peptide and protein identification rates and error estimates. Mol Cell Proteomics. 2011;10(12):M111 007690. doi: 10.1074/mcp.M111.007690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Carroll MV, Sim RB. Complement in health and disease. Adv Drug Deliv Rev. 2011;63(12):965–75. doi: 10.1016/j.addr.2011.06.005. [DOI] [PubMed] [Google Scholar]
- 60.Gawinecka J, Dieks J, Asif AR, Carimalo J, Heinemann U, Streich JH, Dihazi H, Schulz-Schaeffer W, Zerr I. Codon 129 polymorphism specific cerebrospinal fluid proteome pattern in sporadic Creutzfeldt-Jakob disease and the implication of glycolytic enzymes in prion-induced pathology. J Proteome Res. 2010;9(11):5646–57. doi: 10.1021/pr1004604. [DOI] [PubMed] [Google Scholar]
- 61.Gong Y, Lippa CF, Zhu J, Lin Q, Rosso AL. Disruption of glutamate receptors at Shank-postsynaptic platform in Alzheimer’s disease. Brain Res. 2009;1292:191–8. doi: 10.1016/j.brainres.2009.07.056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Jahn H, Wittke S, Zurbig P, Raedler TJ, Arlt S, Kellmann M, Mullen W, Eichenlaub M, Mischak H, Wiedemann K. Peptide fingerprinting of Alzheimer’s disease in cerebrospinal fluid: identification and prospective evaluation of new synaptic biomarkers. PLoS One. 2011;6(10):e26540. doi: 10.1371/journal.pone.0026540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Nagy JI, Li W, Hertzberg EL, Marotta CA. Elevated connexin43 immunoreactivity at sites of amyloid plaques in Alzheimer’s disease. Brain Res. 1996;717(1–2):173–8. doi: 10.1016/0006-8993(95)01526-4. [DOI] [PubMed] [Google Scholar]
- 64.Mei X, Ezan P, Giaume C, Koulakoff A. Astroglial connexin immunoreactivity is specifically altered at beta-amyloid plaques in beta-amyloid precursor protein/presenilin1 mice. Neuroscience. 2010;171(1):92–105. doi: 10.1016/j.neuroscience.2010.08.001. [DOI] [PubMed] [Google Scholar]
- 65.David DC, Ittner LM, Gehrig P, Nergenau D, Shepherd C, Halliday G, Gotz J. Beta-amyloid treatment of two complementary P301L tau-expressing Alzheimer’s disease models reveals similar deregulated cellular processes. Proteomics. 2006;6(24):6566–77. doi: 10.1002/pmic.200600634. [DOI] [PubMed] [Google Scholar]
- 66.Solan JL, Marquez-Rosado L, Sorgen PL, Thornton PJ, Gafken PR, Lampe PD. Phosphorylation at S365 is a gatekeeper event that changes the structure of Cx43 and prevents down-regulation by PKC. J Cell Biol. 2007;179(6):1301–9. doi: 10.1083/jcb.200707060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Pannasch U, Vargova L, Reingruber J, Ezan P, Holcman D, Giaume C, Sykova E, Rouach N. Astroglial networks scale synaptic activity and plasticity. Proc Natl Acad Sci U S A. 2011;108(20):8467–72. doi: 10.1073/pnas.1016650108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.McGeer PL, McGeer EG. The possible role of complement activation in Alzheimer disease. Trends Mol Med. 2002;8(11):519–23. doi: 10.1016/s1471-4914(02)02422-x. [DOI] [PubMed] [Google Scholar]
- 69.Fonseca MI, Zhou J, Botto M, Tenner AJ. Absence of C1q leads to less neuropathology in transgenic mouse models of Alzheimer’s disease. J Neurosci. 2004;24(29):6457–65. doi: 10.1523/JNEUROSCI.0901-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Benoit ME, Tenner AJ. Complement protein C1q-mediated neuroprotection is correlated with regulation of neuronal gene and microRNA expression. J Neurosci. 2011;31(9):3459–69. doi: 10.1523/JNEUROSCI.3932-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Fraser DA, Pisalyaput K, Tenner AJ. C1q enhances microglial clearance of apoptotic neurons and neuronal blebs, and modulates subsequent inflammatory cytokine production. J Neurochem. 2010;112(3):733–43. doi: 10.1111/j.1471-4159.2009.06494.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Pisalyaput K, Tenner AJ. Complement component C1q inhibits beta-amyloid- and serum amyloid P-induced neurotoxicity via caspase- and calpain-independent mechanisms. J Neurochem. 2008;104(3):696–707. doi: 10.1111/j.1471-4159.2007.05012.x. [DOI] [PubMed] [Google Scholar]
- 73.Honda K, Casadesus G, Petersen RB, Perry G, Smith MA. Oxidative stress and redox-active iron in Alzheimer’s disease. Ann N Y Acad Sci. 2004;1012:179–82. doi: 10.1196/annals.1306.015. [DOI] [PubMed] [Google Scholar]
- 74.Sultana R, Boyd-Kimball D, Poon HF, Cai J, Pierce WM, Klein JB, Merchant M, Markesbery WR, Butterfield DA. Redox proteomics identification of oxidized proteins in Alzheimer’s disease hippocampus and cerebellum: an approach to understand pathological and biochemical alterations in AD. Neurobiol Aging. 2006;27(11):1564–76. doi: 10.1016/j.neurobiolaging.2005.09.021. [DOI] [PubMed] [Google Scholar]
- 75.Belkacemi A, Ramassamy C. Time sequence of oxidative stress in the brain from transgenic mouse models of Alzheimer’s disease related to the amyloid-beta cascade. Free Radic Biol Med. 2012;52(3):593–600. doi: 10.1016/j.freeradbiomed.2011.11.020. [DOI] [PubMed] [Google Scholar]
- 76.Huttlin EL, Jedrychowski MP, Elias JE, Goswami T, Rad R, Beausoleil SA, Villen J, Haas W, Sowa ME, Gygi SP. A tissue-specific atlas of mouse protein phosphorylation and expression. Cell. 2010;143(7):1174–89. doi: 10.1016/j.cell.2010.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Power JH, Asad S, Chataway TK, Chegini F, Manavis J, Temlett JA, Jensen PH, Blumbergs PC, Gai WP. Peroxiredoxin 6 in human brain: molecular forms, cellular distribution and association with Alzheimer’s disease pathology. Acta Neuropathol. 2008;115(6):611–22. doi: 10.1007/s00401-008-0373-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Sultana R, Boyd-Kimball D, Cai J, Pierce WM, Klein JB, Merchant M, Butterfield DA. Proteomics analysis of the Alzheimer’s disease hippocampal proteome. J Alzheimers Dis. 2007;11(2):153–64. doi: 10.3233/jad-2007-11203. [DOI] [PubMed] [Google Scholar]
- 79.Chen JW, Dodia C, Feinstein SI, Jain MK, Fisher AB. 1-Cys peroxiredoxin, a bifunctional enzyme with glutathione peroxidase and phospholipase A2 activities. J Biol Chem. 2000;275(37):28421–7. doi: 10.1074/jbc.M005073200. [DOI] [PubMed] [Google Scholar]
- 80.Dhawan L, Liu B, Pytlak A, Kulshrestha S, Blaxall BC, Taubman MB. Y-box binding protein 1 and RNase UK114 mediate monocyte chemoattractant protein 1 mRNA stability in vascular smooth muscle cells. Mol Cell Biol. 2012;32(18):3768–75. doi: 10.1128/MCB.00846-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Liang Y, Van Zant G. Aging stem cells, latexin, and longevity. Exp Cell Res. 2008;314(9):1962–72. doi: 10.1016/j.yexcr.2008.01.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Ohama Y, Hayashi K. Relocalization of a microtubule-anchoring protein, ninein, from the centrosome to dendrites during differentiation of mouse neurons. Histochem Cell Biol. 2009;132(5):515–24. doi: 10.1007/s00418-009-0631-z. [DOI] [PubMed] [Google Scholar]
- 83.Wang X, Tsai JW, Imai JH, Lian WN, Vallee RB, Shi SH. Asymmetric centrosome inheritance maintains neural progenitors in the neocortex. Nature. 2009;461(7266):947–55. doi: 10.1038/nature08435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Liem RK, Messing A. Dysfunctions of neuronal and glial intermediate filaments in disease. J Clin Invest. 2009;119(7):1814–24. doi: 10.1172/JCI38003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Lariviere RC, Julien JP. Functions of intermediate filaments in neuronal development and disease. J Neurobiol. 2004;58(1):131–48. doi: 10.1002/neu.10270. [DOI] [PubMed] [Google Scholar]
- 86.Schoch S, Gundelfinger ED. Molecular organization of the presynaptic active zone. Cell Tissue Res. 2006;326(2):379–91. doi: 10.1007/s00441-006-0244-y. [DOI] [PubMed] [Google Scholar]
- 87.Sudhof TC. Calcium control of neurotransmitter release. Cold Spring Harb Perspect Biol. 2012;4(1):a011353. doi: 10.1101/cshperspect.a011353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Garty H, Karlish SJ. Role of FXYD proteins in ion transport. Annu Rev Physiol. 2006;68:431–59. doi: 10.1146/annurev.physiol.68.040104.131852. [DOI] [PubMed] [Google Scholar]
- 89.Ohsumi A, Nawashiro H, Otani N, Ooigawa H, Toyooka T, Shima K. Temporal and spatial profile of phosphorylated connexin43 after traumatic brain injury in rats. J Neurotrauma. 2010;27(7):1255–63. doi: 10.1089/neu.2009.1234. [DOI] [PubMed] [Google Scholar]
- 90.Wang Y, Song JH, Denisova JV, Park WM, Fontes JD, Belousov AB. Neuronal gap junction coupling is regulated by glutamate and plays critical role in cell death during neuronal injury. J Neurosci. 2012;32(2):713–25. doi: 10.1523/JNEUROSCI.3872-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Iqbal K, Liu F, Gong CX, Grundke-Iqbal I. Tau in Alzheimer disease and related tauopathies. Curr Alzheimer Res. 2010;7(8):656–64. doi: 10.2174/156720510793611592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Trojanowski JQ, Lee VM. Phosphorylation of neuronal cytoskeletal proteins in Alzheimer’s disease and Lewy body dementias. Ann N Y Acad Sci. 1994;747:92–109. doi: 10.1111/j.1749-6632.1994.tb44403.x. [DOI] [PubMed] [Google Scholar]
- 93.Kito K, Ito T. Mass spectrometry-based approaches toward absolute quantitative proteomics. Curr Genomics. 2008;9(4):263–74. doi: 10.2174/138920208784533647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Tenner AJ, Fonseca MI. The double-edged flower: roles of complement protein C1q in neurodegenerative diseases. Adv Exp Med Biol. 2006;586:153–76. doi: 10.1007/0-387-34134-X_11. [DOI] [PubMed] [Google Scholar]
- 95.Fan R, DeFilippis K, Van Nostrand WE. Induction of complement proteins in a mouse model for cerebral microvascular A beta deposition. J Neuroinflammation. 2007;4:22. doi: 10.1186/1742-2094-4-22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Liu F, Grundke-Iqbal I, Iqbal K, Gong CX. Contributions of protein phosphatases PP1, PP2A, PP2B and PP5 to the regulation of tau phosphorylation. Eur J Neurosci. 2005;22(8):1942–50. doi: 10.1111/j.1460-9568.2005.04391.x. [DOI] [PubMed] [Google Scholar]
- 97.Aidaralieva NJ, Kamino K, Kimura R, Yamamoto M, Morihara T, Kazui H, Hashimoto R, Tanaka T, Kudo T, Kida T, Okuda J, Uema T, Yamagata H, Miki T, Akatsu H, Kosaka K, Takeda M. Dynamin 2 gene is a novel susceptibility gene for late-onset Alzheimer disease in non-APOE-epsilon4 carriers. J Hum Genet. 2008;53(4):296–302. doi: 10.1007/s10038-008-0251-9. [DOI] [PubMed] [Google Scholar]
- 98.Takano M, Maekura K, Otani M, Sano K, Nakamura-Hirota T, Tokuyama S, Min KS, Tomiyama T, Mori H, Matsuyama S. Proteomic analysis of the brain tissues from a transgenic mouse model of amyloid beta oligomers. Neurochem Int. 2012;61(3):347–55. doi: 10.1016/j.neuint.2012.05.018. [DOI] [PubMed] [Google Scholar]
- 99.Katayama T, Imaizumi K, Sato N, Miyoshi K, Kudo T, Hitomi J, Morihara T, Yoneda T, Gomi F, Mori Y, Nakano Y, Takeda J, Tsuda T, Itoyama Y, Murayama O, Takashima A, St George-Hyslop P, Takeda M, Tohyama M. Presenilin-1 mutations downregulate the signalling pathway of the unfolded-protein response. Nat Cell Biol. 1999;1(8):479–85. doi: 10.1038/70265. [DOI] [PubMed] [Google Scholar]
- 100.Viana RJ, Nunes AF, Rodrigues CM. Endoplasmic reticulum enrollment in Alzheimer’s disease. Mol Neurobiol. 2012;46(2):522–34. doi: 10.1007/s12035-012-8301-x. [DOI] [PubMed] [Google Scholar]
- 101.Kim SH, Fountoulakis M, Cairns N, Lubec G. Protein levels of human peroxiredoxin subtypes in brains of patients with Alzheimer’s disease and Down syndrome. J Neural Transm Suppl. 2001;(61):223–35. doi: 10.1007/978-3-7091-6262-0_18. [DOI] [PubMed] [Google Scholar]
- 102.Polito L, Kehoe PG, Davin A, Benussi L, Ghidoni R, Binetti G, Quadri P, Lucca U, Tettamanti M, Clerici F, Bagnoli S, Galimberti D, Nacmias B, Sorbi S, Guaita A, Scarpini E, Mariani C, Forloni G, Albani D. The SIRT2 polymorphism rs10410544 and risk of Alzheimer’s disease in two Caucasian case-control cohorts. Alzheimers Dement. 2012 doi: 10.1016/j.jalz.2012.02.003. [DOI] [PubMed] [Google Scholar]
- 103.Green KN, Steffan JS, Martinez-Coria H, Sun X, Schreiber SS, Thompson LM, LaFerla FM. Nicotinamide restores cognition in Alzheimer’s disease transgenic mice via a mechanism involving sirtuin inhibition and selective reduction of Thr231-phosphotau. J Neurosci. 2008;28(45):11500–10. doi: 10.1523/JNEUROSCI.3203-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Tilleman K, Stevens I, Spittaels K, Haute CV, Clerens S, Van Den Bergh G, Geerts H, Van Leuven F, Vandesande F, Moens L. Differential expression of brain proteins in glycogen synthase kinase-3 transgenic mice: a proteomics point of view. Proteomics. 2002;2(1):94–104. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.