

Abstract
Transient receptor potential channels are a large superfamily of non-selective and non-voltage gated ion channels that convey signaling information linked to a broad range of sensory inputs. In the cardiovascular system, the canonical (TRPC) family has been particularly found to play a role in vascular and cardiac disease, responding to neurohormonal and mechanical load stimulation. TRPC1, TRPC3, and TRPC6 are often upregulated in models of cardiovascular disease, and their inhibition ameliorates the associated pathophysiology. Studies in gene deletion models, and overexpression models of wild type and dominant negative proteins supports a direct role of these channels, which likely act together as heterotetramers to influence signaling. Recent evidence has further revealed the importance of protein kinase G phosphorylation as a mechanism to suppress TRPC6 channel current and dependent signaling in vascular and cardiac myocytes. This suggests a novel mechanism underlying benefits of drugs such as sildenafil, a phosphodiesterase type 5 inhibitor, nitrates, and atrial natriuretic peptides. This review describes new evidence supporting a pathophysiologic role of these three TRPC channels, and the potential utility of inhibition strategies to treat cardiovascular disease.
Introduction
Cardiovascular disease is a leading cause of morbidity and mortality, accounting for more than a quarter of all deaths worldwide. A common and prominent feature of these disorders is excessive and sustained neurohormonal and mechanical stimulation that induces pathologic remodeling and organ dysfunction. [1;2] Successful therapies such as angiotensin converting enzyme inhibitors, and angiotensin, endothelin, and beta-adrenergic receptor antagonists block such stimulation [1;2]. Yet additional approaches are greatly needed as the prevalence of these diseases continues to rise and exact a huge societal cost. Recent research has begun identifying novel proteins downstream of the surface receptors that might be therapeutic targets. One example that is garnering increased attention are members of transient receptor potential (trp) channels, a superfamily of non-selective and non-voltage gated ion channels that broadly act as cell signaling/sensory transducers [3]. Trp channels were first identified in Drosophila melanogaster photoreceptor cells as a phospholipase C (PLC)-activatable entity that allowed transmembrane calcium flux [4]. A large family of mammalian homologues was discovered that includes 28 genes and six sub-families based on amino acid sequence. The six TRP channel families are TRPC (canonical), TRPV (vanilloid), TRPM (melastatin) TRPP (polycistins), TRPML (mucolipins), and TRPA (ankyrin) [5]. Trp channels are widely expressed in vascular and cardiac tissues, and their upregulation is increasingly thought to contribute to pathophysiology in both systems. Existing channel antagonists are non-selective and unsuitable for clinical pharmacologic use. Small molecule inhibitors remain in development stages, but other studies are revealing that existing drugs that stimulate cyclic GMP dependent kinase (protein kinase G) can suppress members of these channels [6–11]. This review highlights recent insights regarding several TRPC channels that have been particularly noted to play a role in vascular and cardiac disease, and discusses new evidence for their pharmacologic modulation as a mechanism for disease therapy.
The TRPC Channel Sub-family
The canonical transient receptor potential (TRPC) channels are the mammalian homologues that most closely resemble the trp channels found in Drosophila. There are seven TRPC channels that are further categorized into the following subfamilies: TRPC1, TRPC2, TRPC4/5 and TRPC 3/6/7. [12] TRPC 2 is considered a pseudogene in humans, but is a fully functional channel in rats and mice. [3;5] The TRPC3/6/7 subfamily displays the greatest homology covering ~75% of the amino acid sequence [13]. TRPC1, TRPC3 and TRPC6 have been identified to play a role in heart and vessels, and form the focus of this review.
TRPC channels are characterized by six transmembrane domains with the pore-forming region containing conserved leucine, phenylalanine, and tryptophan (LFW) residues between the fifth and sixth transmembrane domains. [13] The cytoplasmic N-terminal sequence contains three to four ankyrin repeats and a coil-coil domain. [5;13] The coil-coil domain may facilitate the formation of tetramers that is necessary for the production of a functional channel. Another coil-coil domain is present in the internal carboxy terminus following the sixth transmembrane domain. This sequence also contains a TRP box motif with the conserved EWKFAR residues and a calmodulin binding domain that overlaps with a conserved sequence that can allow interaction with inositol 1,4,5,-triphosphate receptor (IP3R) collectively known as the calmodulin IP3 receptor binding domain (CIRB). [12;13]
TRPC channels are non-voltage gated cation channels that are activated by various stimuli allowing movement of Na+ and Ca2+ ions across the membrane. Activation may occur after a process known as store operated calcium entry (SOCE) in which depletion of calcium stores in the endoplasmic reticulum leads to channel activation and influx of calcium across the membrane. [14] Another mechanism of channel activation occurs with ligand binding to Gαq/11-protein coupled receptors (GPCRs) or receptor tyrosine kinases (RTKs) that induce phospholipase C activity to convert PIP2 to diacylglycerol (DAG), with IP3 leading to the activation of downstream effector molecules such as protein kinase C. [12] TRPC3, 6, and 7 are also directly activated by DAG binding to the channel. [5] Mechanical stimuli such as stretch, flow, and osmotic pressure can activate TRPC1, 5 and 6. [15;16] After activation, the transmembrane movement of ions occurs with varying selectivity for calcium or sodium; with the Ca2+/Na+ ion permeability ratio ranging from 1 to 9. [16;17]
TRPC expression is regulated by the Ca2+-activated protein phosphatase, calcineurin (Cn) principally via its downstream target nuclear factor of activated T-cell (NFAT) [18]. TRPC activation and inward Ca2+ entry stimulates Cn, which in turn de-phosphorylates NFAT leading to the nuclear translocation of the latter. As the promoter region for TRPC1, 3, and 6, contains NFAT binding sites, this provides a positive-feedback mechanism to further upregulate channel expression and activity [19;20]. TRPC1 upregulation has been also linked to tumor necrosis factor stimulation of an NFκB pathway [21]. Both transcriptional pathways have been linked to maladapative cardiac and vascular remodeling.
TRPC1
TRPC1 is considered unique in that no other family member shares high sequence homology. Evidence supports several modes of TRPC1 activation including receptor, mechanical, and store-operated stimulation. [14;15;22] Evidence for TRPC1 as store operated calcium channel (SOCC) comes from experiments showing current activation upon exposure to thapsigargin which depletes calcium stored within the endoplasmic and sarcoplasmic reticulum. [3] This is thought to involve TRPC1 interaction with the ER Ca2+ sensing molecule, stromal interacting molecule 1 (STIM1) and ORAI calcium release-activated calcium modulator 1 (Orai1), coupling internal storage levels to TRPC1 calcium entry. [14;23] Recent evidence, however, suggests TRPC channels can also function independently of STIM1 and Orai1. [24] TRPC1 is also post-translationally regulated by protein kinase C (PKC) mediated phosphorylation, which increases channel activity. [25;26]. TRPC1 channels are present in cardiomyocytes, smooth muscle and endothelial cells, and can interact with other family members including TRPC4/5 and TRPC3/7. [14] In expression systems, TRPC1 alone was found in the endoplasmic reticulum (ER); however when expressed with other channels, particularly TRPC4/5, expression was observed at the plasma membrane. [27;28] TRPC1 may only form heterotetramers at the plasma membrane and functional homo-tetramers in the ER. [27] TRPC1 has also been shown to form a heterotetramer with TRPC3 [29;30]. Co-immunoprecipitation experiments show interactions of TRPC1 with TRPC3 in a STIM-1 dependent manner. [23]
A number of studies have suggested an important regulatory role of TRPC1 in the cardiovascular system. It regulates vascular tone in response to endothelin-I-mediated constriction [17] and potentiates smooth muscle depolarization in response to pressure load [15]. TRPC1 also mediates smooth muscle proliferation which may contribute to hypertension and luminal injury response [31]. Pulmonary smooth muscle growth is suppressed by exposure to antisense oligonucleotide for TRPC1 [14;32] and post growth injury involves SOCE and TRPC1 in neointimal smooth muscle cells. [33] TRPC1 is also thought to play a pathologic role in the heart, with increased cardiomyocyte expression observed with cardiac hypertrophy in rat and mouse [34;35]. Recently, the Rosenberg laboratory [35] reported that mice with global TRPC1 gene deletion exhibit less hypertrophy and chamber dysfunction in response to pressure-overload or neurohormonal stimulation than controls (Figure 1A,). This supports an in vivo role of TRPC1 to pathologic heart disease. TRPC1 coupled hypertrophic signaling has been proposed to involve 5HT receptor-cascades as well as the activation of NFAT [36], though whether this is specific or coupled to heterotetramers formed between TRPC1 and other TRPC species is unclear at present.
Figure 1.

A) Effect of gene deletion of TRPC1 on in vivo mouse heart subjected to pressure overload. Panels show baseline and post aortic constriction hearts (top) and myocardial histology (lower) for both control and TRPC1−/− hearts. There was less hypertrophy, chamber enlargement, and fibrosis in hearts lacking TRPC1. B) Mice with myocyte-selective expression of dominant negative TRPC3 display less decline fractional shortening (%FS) and less hypertrophy (LV/body weight, LW/BW) with sustained pressure overload (TAC). Mice with myocyte-targeted overexpression of a dominant negative TRPC6 display less cardiac hypertrophy in response to TAC. There is also less interstitial fibrosis. From Wu et al [48] with permission. C) Mice with cardiac myocyte targeted TRPC3 overexpression develop dilated and depressed heart function after 8 months, with enhanced stimulation of NFAT. From Nakayama et al [46] with permission. D) Myocytes subjected to transverse aortic constriction (TAC) display enhanced store-operated calcium entry, whereas this is blocked in 90% of cells studied from mice E).
TRPC3 and TRPC6
Despite marked sequence homology between TRPC3 and TRPC6, the two isoforms appear to fulfill distinct positions in signaling pathways in cardiovascular physiology and pathology. Both TRPC3 and TRPC6 are activated by different GPCRs or receptor tyrosine kinases that lead to PLC activation. DAG can also directly stimulate both. TRPC6, but not TRPC3, was initially reported to be mechanosensitive to pressure or stretch independent of receptor activation [37]. However, a more recent study performed in vascular cells found mechanical stimuli alone does not activate current, but rather potentiates co-existing receptor–coupled channel activation. [38] TRPC3 has permeability to Ca2+ 1.5-times that of Na+, whereas TRPC6 is more for selective for Ca2+ (6-fold) [39] Both are expressed in cardiomyocytes, smooth muscle, and endothelial cells, and TRPC6 is also in cardiac myofibroblasts. Co-immunoprecipitation assays reveal binding between TRPC3 and TRPC6 and blunting of TRPC3 current by co-expression with dominant negative TRPC6. [28] However, mice lacking TRPC6 were reported to have mRNA upregulation for TRPC3, suggesting a potential negative regulation of one gene by expression of the other [40].
TRPC3
TRPC3 has been suggested to play a pathophysiologic role in both hypertension and cardiac hypertrophy. TRPC3 levels rise in the spontaneous hypertensive rat, pressure-overloaded rat, and in human hypertension [34;41–44]. In the heart, TRPC3 and TRPC6 play a central role in the Ca2+ signaling that leads to the activation of NFAT and subsequent transcription of hypertrophic genes, both in turn regulated by a positive feedback mechanism coupled to NFAT mediated transcription of the channel. [20;42;45;46] Mice with myocyte-targeted overexpression of TRPC3 exhibit spontaneous hypertrophy, dilation, and NFAT activation [46], and worsened apoptosis in ischemia/reperfusion models [47]. In contrast, hearts with myocytes that express a dominant negative TRPC3 exhibit a blunted response to pressure overload or neurohormonal stimulation [48] (Figure 1B). This supports the link between TRPC3 activation and maladaptive hypertrophy and suggests it as a potential therapeutic target. Couplig of TRPC3 channel Ca2+conductance and NFAT activation has been demonstrated both in vivo [48] and in vitro [45] using gain and loss of function models. While the exact sub-cellular localization of TRPC3-modulated Ca2+ involved with this activation has not been defined, evidence supports plasma membrane localization of TRPC3 upon stimulation, with co-localization with the Na+-Ca2+ exchanger [49] and triggering of a reverse mode current via the exchanger. This would be compatible with mechanisms of calcium activation linked to calcineurin/NFAT stimulation [50].
In addition to expression modulation, TRPC3 activity is regulated by post-translational change such as glycosylation, interaction with calcium/calmodulin, and phosphorylation of serine and threonine residues. Glycosylation at one site between transmembrane domains in the channel regulates constitutive expression levels. [51] Channel activity can be inhibited by Ca/CaM binding to the CIRB domain [52], and by phosphorylation by PKC at serine 712, and phosphorylation at T11 and S263 by PKG. [25]. The potential therapeutic impact of these latter modifications remains unknown. The importance of TRPC3 stimulation to both cardiac hypertrophy and vascular disease, poses the hypothesis that it may prove a valuable therapeutic target for various forms of heart and vascular disease. However, selective inhibitors that can be used in vivo remain lacking as discussed in more detail below.
TRPC6
TRPC6 is thought to participate in the pathophysiology of systemic and pulmonary vascular disease, and cardiac hypertrophy and dysfunction. In lung, the channel is central to hypoxic vasoconstriction, an important physiologic mechanism to direct blood from poorly ventilated areas in the lung to areas that are well-ventilated. [53] TRPC6 also plays a key role in the smooth muscle constrictor response to α-agonists [40]. In heart, TRPC6 may serve as a stretch activated channel providing mechanoelectrical feedback [15;54], and provide a mechanism for Na+ entry, contributing to membrane depolarization and opening of voltage-gated calcium channels. Coupling between TRPC6 and the Na+/Ca2+ exchanger has been proposed which could lead to increased reverse mode transport of Ca2+ to modulate excitation-contraction coupling [55]. TRPC6 expression is markedly upregulated in hypertrophied myocardium [9;20;45], and overexpression of the gene itself results in heart failure [20;45], whereas cardiomyocyte-targeted overexpression of a dominant negative TRPC6 suppresses maladapative remodeling to pressure overload (Fig 1B). [48]
Though all three TRPC species (1,3, and 6) all have NFAT sequences in their promoter, the presence of a positive feedback loop coupling channel activation and expression via this pathway has been directly confirmed to date only for TRPC6 [20]. In this study, expression of a luciferase-reporter coupled to the TRPC6 promoter that either lacked or had mutated NFAT binding sites suppressed upregulated gene expression in response to calcineurin or endothelin.
TRPC6 channel activity can also be post-translationally modulated by several mechanisms. TRPC6 has two N-glycosylation sites in the first two extracellular loops (N473 and N561) of the channel, and the prevention of glycosylation at one site by gene modification increased basal activity in expressed channels. [33;51]. TRPC6 has several known phosphorylation sites that can positively or negatively regulate activity. Phosphorylation by Fyn, a non-receptor tyrosine kinase, increases TRPC6 activity. [12] Channel activity is also positively modulated by cytoplasmic Ca2+ and Ca2+/calmodulin binding to the carboxy terminus [56;57]. PKG phosphorylates TRPC6 at T70 and S322 (homologous to T11 and S263 in TRPC3), both in the cytoplasmic N-terminus of the channel. Recent studies have focused on PKG modulation, as this appears to have quite potent suppressive effects on channel conductance and associated NFAT signaling [8–11]. This has posed a potential therapeutic approach using existing agents that enhance PKG activity, as discussed in the next section.
TRPC inhibition as a therapeutic target
Recent findings supporting a role for TRPC1, 3, and 6 in cardiovascular disease suggests they might themselves be a pharmacologic target. Unfortunately, all current available channel inhibitors are nonselective and often times alter other modes of calcium handling. For example, SKF96365, an inhibitor of IP3R, has been used experimentally to hamper the current of TRPC channels and SOCE yet its action is not specific for any TRPC subtype. Stretch activated channel inhibitors like gadolinium chloride and the tarantula toxin GsMTx-4 are useful to inhibit TRPC1 and TRPC6 channel currents likely due to the function of the channels as potential mechanosensors. [54] However, this action is also non-specific for a given channel, and may influence other mechanisms. Bistrifluoromethyl pyrazoles (BTPs) acts more specifically to inhibit only TRPC channels without affecting K+ channels or voltage-gated channels. [58] One bistrifluoromethyl pyrazole derivative (BTP2) has been used in experimental systems to inhibit multiple forms of TRPC current. Another pyrazole compound (Pyr3) may be more selective for TRPC3 current in an expression system and diminish hypertrophic signaling in pressure-overloaded hearts in vivo [59]. To date, clinical pharmaceuticals that inhibit TRPC species remain to be developed. Another avenue is developing TRPC isoform-selective channel activators which would allow more careful examination of TRPC channel function in various model systems. Flufenamic acid may provide stimulation of TRPC6 in experimental systems. [60] It could further prove useful as it can inhibit other members of the TRPC3/6/7 subset and so provide an ability to activate TRPC6 subunits present in heterotetramers.
Suppressing TRPC channel activity and expression by PKG activation
As noted, several very recent studies have revealed the potency of activators of cGMP/PKG signaling as a means to blunt TRPC3 and particularly TRPC6 channel activity. This has been achieved both by activating cGMP synthesis via natriuretic peptide or nitric oxide signaling pathways, or by inhibiting cGMP hydrolysis with phosphodiesterase type-V inhibitors such as sildenafil.
Nitric-oxide/cGMP stimulation has been shown to suppress TRPC1–3 heterotetramers, as well as TRPC6. The former appears a relevant mechanism for PKG modulation of vascular tone [6] as a mechanism of vasorelaxation. NO-dependent PKG phosphorylation of TRPC6 at mouse T69 (T70 in human gene) inhibited current in immortalized aortic cells, and was attentutated in cells expression TRPC6 with a T69A mutation [10].
Several studies have used sildenafil and shown it too suppresses TRPC1 and TRPC6 current and dependent signaling. In vessels, sildenafil suppressed TRPC1 activation coupled to NFAT stimulation in pulmonary vessels [61], which may be a mechanism for its benefits in pulmonary hypertension. In the heart, we first reported that sildenafil blocks upregulation of TRPC6 gene and protein expression in hearts subjected to pressure overload [9](Fig 2A). The blockade is dependent upon PKG activation, and involves the positive feedback look coupled to NFAT in the promoter. Expression of wild-type TRPC6 in HEK cells demonstrated marked suppression of current in cells exposed to a cGMP analog, but this was blocked in cells expressing mutant channels with either T70 or S322 mutated to alanine or glutamine (Fig 2A and B). Thus both threonine and serine sites appear to regulate current. Further evidence for the potency of this interaction was demonstrated by the impact of PKG activation on TRPC6-dependent NFAT stimulation. As shown in Fig 3d, cells transfected with the angiotensin II receptor (AT1) and wild-type TRPC6 display enhanced NFAT activation with angiotensin stimulation. This is partially suppressed if the TRPC6 channel expresses a phosphomimetic form at either one of the identified sites, and is fully blocked if both are mutated (i.e. T70E, S322E). Sildenafil’s efficacy to suppress myocyte hypertrophy has been independently reported [11] in a study that further showed this was primarily a direct effect on the channel. They further showed that the hypertrophic response to mechanical stretch or neurohormonal stimulation were comparably suppressed by expression of a dominant negative TRPC6, or blockade of TRPC channels with BTP2 or sildenafil treatment.
Figure 2.
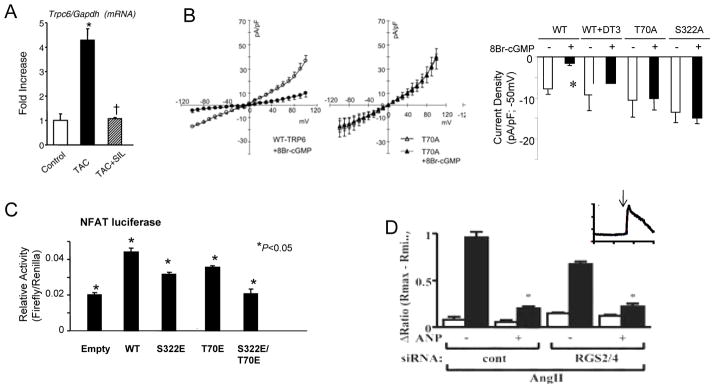
A) ( Left) Myocardial TRPC6 gene expression is upregulated in hearts subjected to TAC and this is markedly suppressed if animals are co-treated with sildenafil (SIL), a PDE5 inhibitor. B) (Left) TRPC6 current in Trpc6 transfected HEK-293 cells at rest (open circles) and after exposure to 8-Br-cGMP (activator of PKG; closed circles). The latter markedly suppressed wild-type (WT) current, but not in cells transfected with phospho-silenced mutant of TRPC6 T70A. (Right) Summary data for electrophysiology studies (max current) shows dependence of cGMP-medicated suppression on PKG activation (DT3 is PKG inhibitor), and lack of response in cells expressing TRPC6 phospho-silenced mutants at both T70 and S322. C) Angiotensin II (ATII) stimuates NFAT promoter activity in HEK-293 cells (all co-transfected with the AT1R receptor). This is blunted if channels containing phospho-mimetic mutations are expressed, and fully blocked if both mutations are present. Modified C) Myocytes with gene suppression of RGS2/4 combined have less AngII stimulated TRPC current compared with controls, but this current is still markedly further suppressed by ANP supporting the role of direct phosphorylation by PKG. Inset is an example of calcium influx stimulated by TRPC6 activation. Panels A–C, modified from from Koitabashi et al. [9], Panel D modified from Kinoshida [8] with permission.
Figure 3.

Schematic of PKG regulation of TRPC current and signaling. Stretch, and Gaq/11 coupled activation (the latter via direct DAG stimulation, or indirectly via IP3 depletion of intracellular stores) activates the TRPC channel (TRPC1, 3, 6); many likely combining different isoforms into a heterotetramer. The resulting cation entry stimulates Ca2+/calmodulin (CaM) activation of calcineurin (Cn). Cn dephosphorylates NFAT, sending it to the nucleus where it can modify gene transcription. TRPC1, 3, 6 are sensitive to NFAT and are upregulated in this process. PKG can be activated by a nitric oxide synthase (NOS) pathway or natriuretic peptide (NP) pathway, likely within different intracellular pools. PKG in turn phosphorylates and activates RGS2/4 protein that can suppress Gaq/11 and thus TRPC activation, but also directly phosphorylates TRPC6 (and TRPC3) to inhibit channel conductance. This in turn blocks the positive NFAT feedback loop.
Lastly, atrial natriuretic peptide (ANP) via stimulation of particulate guanylyl cyclase also has antihypertrophic effects that are coupled to TRPC6 inhibition [11;62]. As true in the prior NO and sildenafil modulation studies, the investigators showed suppression of TRPC6 mediated calcium entry was blocked by ANP (Fig. 3d) and this was blocked if cells expression a TRPC6 with T69A. Regulator of G-coupled signaling 2 and 4 have been linked to sildenafil and ANP-mediated inhibition of Gq-coupled signaling. [63;64] Since this reduces DAG levels, it could by itself be a mechanism for reduced TRPC6 activation. However, the authors found that cells with RGS2/4 reduced by gene silencing still demonstrated reduction of a TRPC channel current by ANP (Fig. 3e).
Figure 4 displays a summary schematic regarding TRPC 1, 3, and 6 modulation, and the new role of PKG phosphorylation of TRPC6 as a negative modulator of this signaling.
TRPCs as Therapeutic Targets
As the role of TRPC channels in cardiac myocytes and vascular tissues continues to be revealed, data increasingly points to these as potentially useful targets for disease therapy. Most of our understanding of specific isoform behavior and regulation comes from expression in heterologous systems, and even there, whether one is studying homodimers or heterodimers is unclear. Lack of selective inhibitors has limited progress, as has the difficulty in precisely defining a specific TRPC channel current from other cation currents – particularly in excitable tissue like myocytes. As virtually all existing pharmacologic reagents have been broad in their inhibitory effects, and embryonic genetic deletion models may induce counter-changes in other isoforms, we do not know whether a highly specific drug would be equally effective. The new data showing potent PKG-mediated modulation of TRPC6 is intriguing, as it may have implications for the use of cGMP-enhancing strategies in diseases where TRPC6 upregulation is observed. Based on current data, potential cardiovascular targets include pulmonary and systemic vascular hypertension, cardiac hypertrophy, and maladaptive stress remodeling with heart failure. In addition to these diseases, gain of function or expression mutations have been described for TRPC6 leading to familial glomerulosclerosis [65], and pulmonary hypertension. TRPC hyperactivation has also been suggested to play a role in skeletal muscle diseases such as muscular dystrophy [66;67] and recently in hyperactive airways in asthma [68]. One intriguing feature of TRPC3/6 antagonists is that their effects in normal heart and vessels may be less marked, as their basal expression levels are low. The induced expression in disease in selected organs might help confer targeting selectivity from inhibitors. Further evolution of more selective, orally bioavailable, and longer-acting TRPC inhibitors will hopefully open up this area of novel therapeutics.
References
- 1.Hill JA, Olson EN. Cardiac plasticity. N Engl J Med. 2008;358:1370–1380. doi: 10.1056/NEJMra072139. [DOI] [PubMed] [Google Scholar]
- 2.Mudd JO, Kass DA. Tackling heart failure in the twenty-first century. Nature. 2008;451:919–928. doi: 10.1038/nature06798. [DOI] [PubMed] [Google Scholar]
- 3.Montell C. The TRP superfamily of cation channels. Sci STKE. 2005;2005:re3. doi: 10.1126/stke.2722005re3. [DOI] [PubMed] [Google Scholar]
- 4.Hardie RC, Minke B. Phosphoinositide-mediated phototransduction in Drosophila photoreceptors: the role of Ca2+ and trp. Cell Calcium. 1995;18:256–274. doi: 10.1016/0143-4160(95)90023-3. [DOI] [PubMed] [Google Scholar]
- 5.Clapham DE. TRP channels as cellular sensors. Nature. 2003;426:517–524. doi: 10.1038/nature02196. [DOI] [PubMed] [Google Scholar]
- 6.Chen J, Crossland RF, Noorani MM, Marrelli SP. Inhibition of TRPC1/TRPC3 by PKG contributes to NO-mediated vasorelaxation. Am J Physiol Heart Circ Physiol. 2009;297:H417–H424. doi: 10.1152/ajpheart.01130.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kwan HY, Huang Y, Yao X. Regulation of canonical transient receptor potential isoform 3 (TRPC3) channel by protein kinase G. Proc Natl Acad Sci U S A. 2004;101:2625–2630. doi: 10.1073/pnas.0304471101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kinoshita H, Kuwahara K, Nishida M, Jiang Z, Rong X, Kiyonaka S, Kuwabara Y, Kurose H, Inoue R, Mori Y, et al. Inhibition of TRPC6 Channel Activity Contributes to the Antihypertrophic Effects of Natriuretic Peptides-Guanylyl Cyclase-A Signaling in the Heart. Circ Res. 2010 doi: 10.1161/CIRCRESAHA.109.208314. [DOI] [PubMed] [Google Scholar]
- 9.Koitabashi N, Aiba T, Hesketh GG, Rowell J, Zhang M, Takimoto E, Tomaselli GF, Kass DA. Cyclic GMP/PKG-dependent inhibition of TRPC6 channel activity and expression negatively regulates cardiomyocyte NFAT activation Novel mechanism of cardiac stress modulation by PDE5 inhibition. J Mol Cell Cardiol. 2009;48:713–724. doi: 10.1016/j.yjmcc.2009.11.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Takahashi S, Lin H, Geshi N, Mori Y, Kawarabayashi Y, Takami N, Mori MX, Honda A, Inoue R. Nitric oxide-cGMP-protein kinase G pathway negatively regulates vascular transient receptor potential channel TRPC6. J Physiol. 2008;586:4209–4223. doi: 10.1113/jphysiol.2008.156083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Nishida M, Watanabe K, Sato Y, Nakaya M, Kitajima N, Ide T, Inoue R, Kurose H. Phosphorylation of TRPC6 channels at Thr69 is required for anti-hypertrophic effects of phosphodiesterase 5 inhibition. J Biol Chem. 2010;285:13244–13253. doi: 10.1074/jbc.M109.074104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ramsey IS, Delling M, Clapham DE. An introduction to TRP channels. Annu Rev Physiol. 2006;68:619–647. doi: 10.1146/annurev.physiol.68.040204.100431. [DOI] [PubMed] [Google Scholar]
- 13.Vazquez G, Wedel BJ, Aziz O, Trebak M, Putney JW., Jr The mammalian TRPC cation channels. Biochim Biophys Acta. 2004;1742:21–36. doi: 10.1016/j.bbamcr.2004.08.015. [DOI] [PubMed] [Google Scholar]
- 14.Ambudkar IS, Ong HL, Liu X, Bandyopadhyay BC, Cheng KT. TRPC1: the link between functionally distinct store-operated calcium channels. Cell Calcium. 2007;42:213–223. doi: 10.1016/j.ceca.2007.01.013. [DOI] [PubMed] [Google Scholar]
- 15.Inoue R, Jian Z, Kawarabayashi Y. Mechanosensitive TRP channels in cardiovascular pathophysiology. Pharmacol Ther. 2009;123:371–385. doi: 10.1016/j.pharmthera.2009.05.009. [DOI] [PubMed] [Google Scholar]
- 16.Sharif-Naeini R, Folgering JH, Bichet D, Duprat F, Delmas P, Patel A, Honore E. Sensing pressure in the cardiovascular system: Gq-coupled mechanoreceptors and TRP channels. J Mol Cell Cardiol. 2010;48:83–89. doi: 10.1016/j.yjmcc.2009.03.020. [DOI] [PubMed] [Google Scholar]
- 17.Abramowitz J, Birnbaumer L. Physiology and pathophysiology of canonical transient receptor potential channels. FASEB J. 2009;23:297–328. doi: 10.1096/fj.08-119495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Oh-hora M. Calcium signaling in the development and function of T-lineage cells. Immunol Rev. 2009;231:210–224. doi: 10.1111/j.1600-065X.2009.00819.x. [DOI] [PubMed] [Google Scholar]
- 19.Pigozzi D, Ducret T, Tajeddine N, Gala JL, Tombal B, Gailly P. Calcium store contents control the expression of TRPC1, TRPC3 and TRPV6 proteins in LNCaP prostate cancer cell line. Cell Calcium. 2006;39:401–415. doi: 10.1016/j.ceca.2006.01.003. [DOI] [PubMed] [Google Scholar]
- 20.Kuwahara K, Wang Y, McAnally J, Richardson JA, Bassel-Duby R, Hill JA, Olson EN. TRPC6 fulfills a calcineurin signaling circuit during pathologic cardiac remodeling. J Clin Invest. 2006;116:3114–3126. doi: 10.1172/JCI27702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Paria BC, Malik AB, Kwiatek AM, Rahman A, May MJ, Ghosh S, Tiruppathi C. Tumor necrosis factor-alpha induces nuclear factor-kappaB-dependent TRPC1 expression in endothelial cells. J Biol Chem. 2003;278:37195–37203. doi: 10.1074/jbc.M304287200. [DOI] [PubMed] [Google Scholar]
- 22.Di A, Malik AB. TRP channels and the control of vascular function. Curr Opin Pharmacol. 2010;10:127–132. doi: 10.1016/j.coph.2009.11.010. [DOI] [PubMed] [Google Scholar]
- 23.Yuan JP, Zeng W, Huang GN, Worley PF, Muallem S. STIM1 heteromultimerizes TRPC channels to determine their function as store-operated channels. Nat Cell Biol. 2007;9:636–645. doi: 10.1038/ncb1590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.DeHaven WI, Jones BF, Petranka JG, Smyth JT, Tomita T, Bird GS, Putney JW., Jr TRPC channels function independently of STIM1 and Orai1. J Physiol. 2009;587:2275–2298. doi: 10.1113/jphysiol.2009.170431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Yao X. TRPC, cGMP-dependent protein kinases and cytosolic Ca2+ Handb Exp Pharmacol. 2007:527–540. doi: 10.1007/978-3-540-34891-7_31. [DOI] [PubMed] [Google Scholar]
- 26.Ahmmed GU, Mehta D, Vogel S, Holinstat M, Paria BC, Tiruppathi C, Malik AB. Protein kinase Calpha phosphorylates the TRPC1 channel and regulates store-operated Ca2+ entry in endothelial cells. J Biol Chem. 2004;279:20941–20949. doi: 10.1074/jbc.M313975200. [DOI] [PubMed] [Google Scholar]
- 27.Alfonso S, Benito O, Alicia S, Angelica Z, Patricia G, Diana K, Vaca L. Regulation of the cellular localization and function of human transient receptor potential channel 1 by other members of the TRPC family. Cell Calcium. 2008;43:375–387. doi: 10.1016/j.ceca.2007.07.004. [DOI] [PubMed] [Google Scholar]
- 28.Hofmann T, Schaefer M, Schultz G, Gudermann T. Subunit composition of mammalian transient receptor potential channels in living cells. Proc Natl Acad Sci U S A. 2002;99:7461–7466. doi: 10.1073/pnas.102596199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Strubing C, Krapivinsky G, Krapivinsky L, Clapham DE. Formation of novel TRPC channels by complex subunit interactions in embryonic brain. J Biol Chem. 2003;278:39014–39019. doi: 10.1074/jbc.M306705200. [DOI] [PubMed] [Google Scholar]
- 30.Lintschinger B, Balzer-Geldsetzer M, Baskaran T, Graier WF, Romanin C, Zhu MX, Groschner K. Coassembly of Trp1 and Trp3 proteins generates diacylglycerol- and Ca2+-sensitive cation channels. J Biol Chem. 2000;275:27799–27805. doi: 10.1074/jbc.M002705200. [DOI] [PubMed] [Google Scholar]
- 31.Ng LC, McCormack MD, Airey JA, Singer CA, Keller PS, Shen XM, Hume JR. TRPC1 and STIM1 mediate capacitative Ca2+ entry in mouse pulmonary arterial smooth muscle cells. J Physiol. 2009;587:2429–2442. doi: 10.1113/jphysiol.2009.172254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sweeney M, Yu Y, Platoshyn O, Zhang S, McDaniel SS, Yuan JX. Inhibition of endogenous TRP1 decreases capacitative Ca2+ entry and attenuates pulmonary artery smooth muscle cell proliferation. Am J Physiol Lung Cell Mol Physiol. 2002;283:L144–L155. doi: 10.1152/ajplung.00412.2001. [DOI] [PubMed] [Google Scholar]
- 33.Dietrich A, Kalwa H, Fuchs B, Grimminger F, Weissmann N, Gudermann T. In vivo TRPC functions in the cardiopulmonary vasculature. Cell Calcium. 2007;42:233–244. doi: 10.1016/j.ceca.2007.02.009. [DOI] [PubMed] [Google Scholar]
- 34.Ohba T, Watanabe H, Murakami M, Takahashi Y, Iino K, Kuromitsu S, Mori Y, Ono K, Iijima T, Ito H. Upregulation of TRPC1 in the development of cardiac hypertrophy. J Mol Cell Cardiol. 2007;42:498–507. doi: 10.1016/j.yjmcc.2006.10.020. [DOI] [PubMed] [Google Scholar]
- 35.Seth M, Zhang ZS, Mao L, Graham V, Burch J, Stiber J, Tsiokas L, Winn M, Abramowitz J, Rockman HA, et al. TRPC1 channels are critical for hypertrophic signaling in the heart. Circ Res. 2009;105:1023–1030. doi: 10.1161/CIRCRESAHA.109.206581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Vindis C, D’Angelo R, Mucher E, Negre-Salvayre A, Parini A, Mialet-Perez J. Essential role of TRPC1 channels in cardiomyoblasts hypertrophy mediated by 5-HT2A serotonin receptors. Biochem Biophys Res Commun. 2010;391:979–983. doi: 10.1016/j.bbrc.2009.12.001. [DOI] [PubMed] [Google Scholar]
- 37.Spassova MA, Hewavitharana T, Xu W, Soboloff J, Gill DL. A common mechanism underlies stretch activation and receptor activation of TRPC6 channels. Proc Natl Acad Sci U S A. 2006;103:16586–16591. doi: 10.1073/pnas.0606894103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Inoue R, Jensen LJ, Jian Z, Shi J, Hai L, Lurie AI, Henriksen FH, Salomonsson M, Morita H, Kawarabayashi Y, et al. Synergistic activation of vascular TRPC6 channel by receptor and mechanical stimulation via phospholipase C/diacylglycerol and phospholipase A2/omega-hydroxylase/20-HETE pathways. Circ Res. 2009;104:1399–1409. doi: 10.1161/CIRCRESAHA.108.193227. [DOI] [PubMed] [Google Scholar]
- 39.Dietrich A, Gudermann T. TRPC6 Handb Exp Pharmacol. 2007:125–141. doi: 10.1007/978-3-540-34891-7_7. [DOI] [PubMed] [Google Scholar]
- 40.Dietrich A, Mederos YS, Gollasch M, Gross V, Storch U, Dubrovska G, Obst M, Yildirim E, Salanova B, Kalwa H, et al. Increased vascular smooth muscle contractility in TRPC6−/− mice. Mol Cell Biol. 2005;25:6980–6989. doi: 10.1128/MCB.25.16.6980-6989.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Liu D, Yang D, He H, Chen X, Cao T, Feng X, Ma L, Luo Z, Wang L, Yan Z, et al. Increased transient receptor potential canonical type 3 channels in vasculature from hypertensive rats. Hypertension. 2009;53:70–76. doi: 10.1161/HYPERTENSIONAHA.108.116947. [DOI] [PubMed] [Google Scholar]
- 42.Bush EW, Hood DB, Papst PJ, Chapo JA, Minobe W, Bristow MR, Olson EN, McKinsey TA. Canonical transient receptor potential channels promote cardiomyocyte hypertrophy through activation of calcineurin signaling. J Biol Chem. 2006;281:33487–33496. doi: 10.1074/jbc.M605536200. [DOI] [PubMed] [Google Scholar]
- 43.Thilo F, Loddenkemper C, Berg E, Zidek W, Tepel M. Increased TRPC3 expression in vascular endothelium of patients with malignant hypertension. Mod Pathol. 2009;22:426–430. doi: 10.1038/modpathol.2008.200. [DOI] [PubMed] [Google Scholar]
- 44.Bush EW, Hood DB, Papst PJ, Chapo JA, Minobe W, Bristow MR, Olson EN, McKinsey TA. Canonical transient receptor potential channels promote cardiomyocyte hypertrophy through activation of calcineurin signaling. J Biol Chem. 2006;281:33487–33496. doi: 10.1074/jbc.M605536200. [DOI] [PubMed] [Google Scholar]
- 45.Onohara N, Nishida M, Inoue R, Kobayashi H, Sumimoto H, Sato Y, Mori Y, Nagao T, Kurose H. TRPC3 and TRPC6 are essential for angiotensin II-induced cardiac hypertrophy. EMBO J. 2006;25:5305–5316. doi: 10.1038/sj.emboj.7601417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Nakayama H, Wilkin BJ, Bodi I, Molkentin JD. Calcineurin-dependent cardiomyopathy is activated by TRPC in the adult mouse heart. FASEB J. 2006;20:1660–1670. doi: 10.1096/fj.05-5560com. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Shan D, Marchase RB, Chatham JC. Overexpression of TRPC3 increases apoptosis but not necrosis in response to ischemia-reperfusion in adult mouse cardiomyocytes. Am J Physiol Cell Physiol. 2008;294:C833–C841. doi: 10.1152/ajpcell.00313.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Wu X, Eder P, Chang B, Molkentin JD. TRPC channels are necessary mediators of pathologic cardiac hypertrophy. Proc Natl Acad Sci U S A. 2010;107:7000–7005. doi: 10.1073/pnas.1001825107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Eder P, Probst D, Rosker C, Poteser M, Wolinski H, Kohlwein SD, Romanin C, Groschner K. Phospholipase C-dependent control of cardiac calcium homeostasis involves a TRPC3-NCX1 signaling complex. Cardiovasc Res. 2007;73:111–119. doi: 10.1016/j.cardiores.2006.10.016. [DOI] [PubMed] [Google Scholar]
- 50.Rinne A, Banach K, Blatter LA. Regulation of nuclear factor of activated T cells (NFAT) in vascular endothelial cells. J Mol Cell Cardiol. 2009;47:400–410. doi: 10.1016/j.yjmcc.2009.06.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Dietrich A, Schnitzler M, Emmel J, Kalwa H, Hofmann T, Gudermann T. N-linked protein glycosylation is a major determinant for basal TRPC3 and TRPC6 channel activity. J Biol Chem. 2003;278:47842–47852. doi: 10.1074/jbc.M302983200. [DOI] [PubMed] [Google Scholar]
- 52.Zhang Z, Tang J, Tikunova S, Johnson JD, Chen Z, Qin N, Dietrich A, Stefani E, Birnbaumer L, Zhu MX. Activation of Trp3 by inositol 1,4,5-trisphosphate receptors through displacement of inhibitory calmodulin from a common binding domain. Proc Natl Acad Sci U S A. 2001;98:3168–3173. doi: 10.1073/pnas.051632698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Weissmann N, Dietrich A, Fuchs B, Kalwa H, Ay M, Dumitrascu R, Olschewski A, Storch U, Schnitzler M, Ghofrani HA, et al. Classical transient receptor potential channel 6 (TRPC6) is essential for hypoxic pulmonary vasoconstriction and alveolar gas exchange. Proc Natl Acad Sci U S A. 2006;103:19093–19098. doi: 10.1073/pnas.0606728103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Sharif-Naeini R, Folgering JH, Bichet D, Duprat F, Delmas P, Patel A, Honore E. Sensing pressure in the cardiovascular system: Gq-coupled mechanoreceptors and TRP channels. J Mol Cell Cardiol. 2009 doi: 10.1016/j.yjmcc.2009.03.020. [DOI] [PubMed] [Google Scholar]
- 55.Poburko D, Liao CH, Lemos VS, Lin E, Maruyama Y, Cole WC, van BC. Transient receptor potential channel 6-mediated, localized cytosolic [Na+] transients drive Na+/Ca2+ exchanger-mediated Ca2+ entry in purinergically stimulated aorta smooth muscle cells. Circ Res. 2007;101:1030–1038. doi: 10.1161/CIRCRESAHA.107.155531. [DOI] [PubMed] [Google Scholar]
- 56.Boulay G. Ca(2+)-calmodulin regulates receptor-operated Ca(2+) entry activity of TRPC6 in HEK-293 cells. Cell Calcium. 2002;32:201–207. doi: 10.1016/s0143416002001550. [DOI] [PubMed] [Google Scholar]
- 57.Kwon Y, Hofmann T, Montell C. Integration of phosphoinositide- and calmodulin-mediated regulation of TRPC6. Mol Cell. 2007;25:491–503. doi: 10.1016/j.molcel.2007.01.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Soboloff J, Spassova M, Hewavitharana T, He LP, Luncsford P, Xu W, Venkatachalam K, van RD, Patterson RL, Gill DL. TRPC channels: integrators of multiple cellular signals. Handb Exp Pharmacol. 2007:575–591. doi: 10.1007/978-3-540-34891-7_34. [DOI] [PubMed] [Google Scholar]
- 59.Kiyonaka S, Kato K, Nishida M, Mio K, Numaga T, Sawaguchi Y, Yoshida T, Wakamori M, Mori E, Numata T, et al. Selective and direct inhibition of TRPC3 channels underlies biological activities of a pyrazole compound. Proc Natl Acad Sci U S A. 2009;106:5400–5405. doi: 10.1073/pnas.0808793106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Foster RR, Zadeh MA, Welsh GI, Satchell SC, Ye Y, Mathieson PW, Bates DO, Saleem MA. Flufenamic acid is a tool for investigating TRPC6-mediated calcium signalling in human conditionally immortalised podocytes and HEK293 cells. Cell Calcium. 2009;45:384–390. doi: 10.1016/j.ceca.2009.01.003. [DOI] [PubMed] [Google Scholar]
- 61.Wang C, Li JF, Zhao L, Liu J, Wan J, Wang YX, Wang J, Wang C. Inhibition of SOC/Ca2+/NFAT pathway is involved in the anti-proliferative effect of sildenafil on pulmonary artery smooth muscle cells. Respir Res. 2009;10:123. doi: 10.1186/1465-9921-10-123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Kinoshita H, Kuwahara K, Nishida M, Jiang Z, Rong X, Kiyonaka S, Kuwabara Y, Kurose H, Inoue R, Mori Y, et al. Inhibition of TRPC6 Channel Activity Contributes to the Antihypertrophic Effects of Natriuretic Peptides-Guanylyl Cyclase-A Signaling in the Heart. Circ Res. 2010 doi: 10.1161/CIRCRESAHA.109.208314. [DOI] [PubMed] [Google Scholar]
- 63.Takimoto E, Koitabashi N, Hsu S, Ketner EA, Nagayama T, Bedja D, Gabrielson K, Blanton R, Siderovski DP, Mendelsohn ME, et al. RGS2 mediates cardiac compensation to pressure-overload and anti-hypertrophic effects of PDE5 inhibition. J Clin Invest. 2009;119:408–420. doi: 10.1172/JCI35620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Tokudome T, Kishimoto I, Horio T, Arai Y, Schwenke DO, Hino J, Okano I, Kawano Y, Kohno M, Miyazato M, et al. Regulator of G-protein signaling subtype 4 mediates antihypertrophic effect of locally secreted natriuretic peptides in the heart. Circulation. 2008;117:2329–2339. doi: 10.1161/CIRCULATIONAHA.107.732990. [DOI] [PubMed] [Google Scholar]
- 65.Winn MP, Conlon PJ, Lynn KL, Farrington MK, Creazzo T, Hawkins AF, Daskalakis N, Kwan SY, Ebersviller S, Burchette JL, et al. A mutation in the TRPC6 cation channel causes familial focal segmental glomerulosclerosis. Science. 2005;308:1801–1804. doi: 10.1126/science.1106215. [DOI] [PubMed] [Google Scholar]
- 66.Lansman JB, Franco-Obregon A. Mechanosensitive ion channels in skeletal muscle: a link in the membrane pathology of muscular dystrophy. Clin Exp Pharmacol Physiol. 2006;33:649–656. doi: 10.1111/j.1440-1681.2006.04393.x. [DOI] [PubMed] [Google Scholar]
- 67.Williams IA, Allen DG. Intracellular calcium handling in ventricular myocytes from mdx mice. Am J Physiol Heart Circ Physiol. 2007;292:H846–H855. doi: 10.1152/ajpheart.00688.2006. [DOI] [PubMed] [Google Scholar]
- 68.Colsoul B, Nilius B, Vennekens R. On the putative role of transient receptor potential cation channels in asthma. Clin Exp Allergy. 2009;39:1456–1466. doi: 10.1111/j.1365-2222.2009.03315.x. [DOI] [PubMed] [Google Scholar]