

Abstract
The addicted phenotype is characterized as a long-lasting, chronically relapsing disorder that persists following long periods of abstinence, suggesting that the underlying molecular changes are stable and endure for long periods even in the absence of drug. Here, we investigated Transforming Growth Factor-Beta Type I receptor (TGF-β R1) expression in the nucleus accumbens (NAc) following periods of withdrawal from cocaine self-administration (SA) and a sensitizing regimen of non-contingent cocaine. Rats were exposed to either (i) repeated systemic injections (cocaine or saline), or (ii) self-administration (cocaine or saline) and underwent a period of forced abstinence (either 1 or 7 days of drug cessation). Withdrawal from cocaine self-administration resulted in an increase in TGF-β R1 protein expression in the NAc compared to saline controls. This increase was specific for volitional cocaine intake as no change in expression was observed following a sensitizing regimen of experimenter-administered cocaine. These findings implicate TGF-β signaling as a novel potential therapeutic target for treating drug addiction.
Introduction
Drug addiction is a chronic disorder represented by persistent drug-seeking and reoccurring episodes of relapse [1]. Psychomotor stimulant abuse and addiction leads to large economic and societal burdens, yet to date, there is no effective intervention. While recent reports have shed a great deal of insight into the neurobiology of addiction, a more complete understanding of how drug abuse leads to long-term behavioral, cellular, and morphological plasticity is desperately needed in order to establish a treatment for this disabling disease [1]–[6]. The neuroadaptions that are initiated following drug exposure and that remain stable over periods of drug abstinence are of particular interest, as these changes occur in the absence of the drug itself, and may confer a neurobiological mechanism that leads to long-term behavioral changes such as craving and relapse [7].
Time-dependent adaptations in synaptic connectivity, glutamatergic and dopaminergic receptor expression and signaling, and neurotrophic levels have been reported following cessation of cocaine treatment [8]–[17]. Transforming Growth Factor Beta (TGF-β) is a signaling cascade that may be a prospective facilitator of these long-term changes in drug-induced plasticity. TGF-β signaling cascades are widely distributed throughout the central nervous system and have a variety of cellular functions in the adult organism, including apoptosis, cellular homeostasis and tissue repair [18]. The binding of TGF-β to the TGF-β Type I Receptor (TGF-β R1) initiates signal propagation through two mechanisms: a canonical mechanism via SMAD proteins, and a non-canonical SMAD-independent mechanism via activation of extracellular signal-related kinases (ERKs), and signaling cascades associated with actin dynamics such as GTPases [18].
TGF-β R1 and downstream signaling cascades have been implicated in numerous psychiatric disorders, including diseases that are largely comorbid with addiction, such as depression and anxiety [19]–[22]. Moreover, TGF-β has been shown to have a role in mediating adult neurogenesis, a neural mechanism shown to be involved in mediating drug-taking and relapse [23]–[25]. The involvement of TGF-β signaling in mediating neural plasticity marks this pathway as a potential regulator of cellular changes in response to drug taking. To this end, we have investigated changes in TGF-β signaling using two models of drug exposure over varying periods of drug abstinence.
Methods
Subjects
Sprague Dawley rats weighing between 300–400 g at the time of testing were used in the experiments. All rats were undisturbed for two days upon arrival to the colony room to allow for habituation, and housed on a 12 hr light-dark cycle with ad libitum access to food and water. Rats were housed two per cage for the experimenter-administered cocaine experiments. For the self-administration (SA) experiments, rats were singly housed following surgery and for the duration of the experiment in order to protect the catheter/harness assembly. Testing took place seven days/week during the rats’ dark phase of the light-dark cycle. This study was conducted in accordance with the guidelines set up by the Institutional Animal Care and Use Committee of the State University of New York at Buffalo.
Locomotor Apparatus
Locomotor activity was recorded by an infrared motion-sensor system (AccuScan Instruments) fitted outside plastic cages (40×40×30 cm) containing a thin layer of corncob bedding that were cleaned between each test session. The Fusion activity-monitoring system software monitors infrared beam breaks at a frequency of 0.01 sec. The interruption of any beam not interrupted during the previous sample was interpreted as an activity score.
Self-administration Test Chambers
Sixteen standard experimental test chambers (MED Associates, Inc.) equipped with two snout-poke holes located on one wall of the test chamber monitored with infrared detectors were used for SA experiments. Two stimulus lights were mounted above the snout-poke holes, and a houselight was mounted in the middle of the back wall of the test chamber. All test chambers were housed in sound attenuating chambers, which mitigate all external light sources and sounds, including sounds from the syringe infusion pumps. Test chambers were computer controlled through a MED Associates interface with MED-PC with a temporal resolution of 0.01 sec.
Drug
(−)-Cocaine hydrochloride (gifted by NIDA) was dissolved in sterile 0.9% saline. For the experimenter-administered cocaine experiment, a constant injection volume of 1.0 ml/kg was used. Saline or 10.0 mg/kg cocaine was injected intraperitoneally (i.p.) immediately prior to the start of each session in the home cage or in locomotor chamber. For SA experiments, cocaine solutions (4.5 mg/ml) made on a weekly basis were delivered via a syringe pump. Pump durations were adjusted according to body weight on a daily basis in order to deliver the correct dose of drug (1.0 mg/kg/infusion cocaine).
Jugular Catheterization and Patency Test
Rats were implanted with chronic indwelling jugular catheters and allowed 7 days to recover following surgery as previously described [26], [27]. Catheters were flushed daily with 0.2 ml solution of enrofloxacin (4 mg/ml) mixed in a heparinized saline solution (50 IU/ml in 0.9% sterile saline) to preserve catheter patency. At the end of behavioral testing, each animal received an intravenous (IV) infusion of ketamine hydrochloride (0.5 mg/kg in 0.05 ml saline) and the behavioral response was observed to verify catheter patency. Loss of muscle tone and righting reflexes served as behavioral indicators of patency. Only rats with patent catheters were used in data analysis.
Self-administration
One week after jugular catheter surgery, the rats were assigned to self-administer either 1.0 mg/kg/inf cocaine or saline. Rats were tested for SA over 10 test sessions, during which responses to the active snout-poke resulted in IV injections of cocaine (or saline) according to a Fixed Ratio 1 (FR1) schedule of reinforcement followed by a 30 sec time-out period. Infusions were accompanied by a 5 sec illumination of the stimulus light above the active snout-poke hole and the houselight was extinguished for the duration of the time-out period. Snout-poke responses to the inactive alternative resulted in no programmed consequences. Session durations were terminated after either a 2-hr duration or 20 infusions had been earned (cumulative dose 20 mg/kg), whichever occurred first. Following testing, catheters were flushed and rats were returned to the colony room. The criterion for acquisition of cocaine SA was an average of 10 infusions per day during the 10-session cocaine test phase.
Withdrawal
Following SA, rats were counterbalanced according to SA performance and assigned to one of two withdrawal time points (1 or 7 days). In the 1-day withdrawal group (cocaine, n = 7; saline, n = 6), brains were harvested 24 hrs after the last day of SA testing. Rats were sacrificed by rapid decapitation, brains were removed and sliced into 1 mm thick sections using a brain matrix (Braintree Scientific), and 2 mm diameter tissue punches from the nucleus accumbens (NAc) were collected and rapidly frozen on dry ice. In the 7-day withdrawal group (cocaine, n = 8; saline, n = 7), rats were returned to their home cages in the colony room and left undisturbed for one week, following which brains were removed and NAc tissue punches were collected in an identical manner.
Locomotor Response to Experimenter-administered Cocaine
In order to control for any basal differences in motor responses, rats were tested for locomotor response to novelty (1 hr duration using the Accuscan Monitoring system). Rats were then counterbalanced according to the locomotor scores (data not shown) and were assigned to receive seven daily i.p. injections of either 10 mg/kg cocaine or saline. Injections occurred in the test room on days 1, 3, 5 & 7 and animals were placed in the locomotor chambers for 1 hr. Injections on days 2, 4 & 6 occurred in the home cage [28], [29]. Following the last day of injections, rats were returned to the colony room and remained undisturbed in their home cages. In the 1-day withdrawal group (cocaine, n = 7; saline, n = 8), brains were removed and NAc tissue punches collected 24 hrs after the last injection using the same procedures as described for the SA experiments. In the 7-day withdrawal group (cocaine, n = 10; saline, n = 10), rats were left undisturbed in the colony room for one week, following which brains were removed and NAc tissue punches were collected in an identical manner.
Western Blot Quantification of TGF-β R1
Protein expression levels of TGF-β R1 were analyzed by Western blotting as previously described [30], [31]. Briefly, frozen NAc tissue punches from each rat were homogenized in 30 μl of homogenization buffer containing 320 mM sucrose, 5 mM HEPES buffer, 1% SDS, phosphatase inhibitor cocktails I and II (Sigma), and protease inhibitors (Roche). Protein concentrations were determined, and a total of 30 μg of protein was loaded onto 10% Tris-SDS polyacrylamide gels for electrophoresis fractionation. Proteins were transferred to nitrocellulose membranes, blocked with 5% non-fat milk, and incubated overnight at 4°C with primary antibodies (anti-rabbit TGF-β Receptor I, Cell Signaling, 1∶500; anti-mouse β-actin, Cell Signaling, 1∶10,000) in Odyssey blocking buffer. After thorough washing with 0.1% Tween-20 in phosphate-buffered saline, membranes were incubated with IRDye secondary antibodies (1∶5000; Li-Cor) dissolved in Odyssey blocking buffer for 1 hr at room temperature. The blots were imaged with the Odyssey Infrared Imaging system (Li-Cor) and quantified by densitometry using NIH Image J. The amount of protein blotted onto each lane was normalized to levels of β-actin.
Data Analysis
Locomotor activity during the experimenter-administered cocaine experiment, and performance during SA were analyzed using a two-factor within-subject Analyses of Variance (ANOVAs) with the between-session variable as drug group (cocaine/saline) and the within-subject variable as time (day of injections or sessions, respectively). The primary dependent measure of locomotor activity used for statistical analysis was the total number of beam breaks, and the primary dependent measure for SA acquisition was the number of infusions. Western blot data was compared using two-tailed Student’s t-tests. Statistical significance was set at p<0.05 using SPSS statistical software. All data are represented as the mean ± SEM.
Results
Analysis of the results for the SA experiments showed that there was a main effect of drug (cocaine/saline) [F(1,250) = 617.6; p<0.0001] and a significant interaction between cocaine and saline SA across the 10 days of testing [F(9,250) = 9.297; p<0.001]. Follow-up post hoc (Tukey’s) tests revealed that rats responding for infusions of cocaine had a significantly greater number of infusions than animals responding for saline on sessions 2–10, indicating that rats acquired cocaine self-administration ( Figure 1 ).
Figure 1. Self-administration of cocaine or saline.
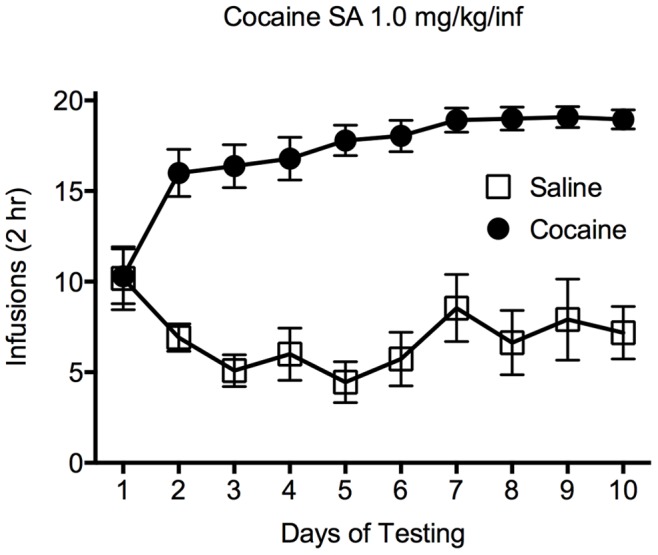
This plot shows the number of infusions earned across ten days of self-administration of cocaine (1.0 mg/kg/inf) or saline. Closed circles represent animals self-administering cocaine, open squares indicate animals receiving infusions of saline. Data are expressed as the average number of infusions (± SEM) over the ten days of cocaine/saline self-administration; *p<0.05.
To examine if TGF-β receptor expression is regulated following cocaine SA, protein levels of TGF-β R1 from whole cell lysates of NAc tissue punches from rats with a history of cocaine and following a period of 1 or 7 days of withdrawal were analyzed. TGF-β R1 expression was unchanged following 1 day of withdrawal from cocaine SA compared to the saline group ( Figure 2 ). In comparison, there was a significant increase in TGF-β R1 expression in the NAc of animals with a history of cocaine SA following a 7-day withdrawal period compared to saline controls [t(13) = 3.269; p<0.001], indicating that TGF-β R1 expression is up-regulated following a period of drug cessation in a time-dependent manner ( Figure 2 ).
Figure 2. TGF-β R1 expression following active cocaine exposure.

Relative TGF-β R1 protein expression in the NAC of rats following 1 or 7 days of withdrawal from cocaine (1.0 mg/kg/inf; 10 days) or saline self-administration; *p<0.05 compared to saline.
We next asked if this increase in TGF-β R1 expression occurred following all cocaine regimens, and thus simply a result of drug exposure. To this end, we used a regimen of cocaine known to induce behavioral sensitization, which thought to have, at least in part, common neural substrates that underlie addiction [32]. As shown in Figure 3 , animals receiving repeated systemic injections of cocaine exhibited an increase in locomotor activity over time, suggesting the development of behavioral sensitization. There was a significant interaction between group (cocaine/saline) and day [F(4,120) = 7.718; p<0.01], and a main effect of drug group (cocaine/saline) [F(1,30) = 19.01; p<0.001]. Post-hoc tests showed that rats in the cocaine group had significantly greater locomotor activity on days 2–4 of injections compared to day 1 and compared to saline (all p’s <0.05), whereas no differences in locomotor activity were observed across time in animals injected with saline. Furthermore, post-hoc tests showed that animals injected with cocaine exhibited significantly more locomotor activity compared to rats receiving injections of saline at Days 2–4 of testing.
Figure 3. Locomotor response to a sensitizing regimen of cocaine.
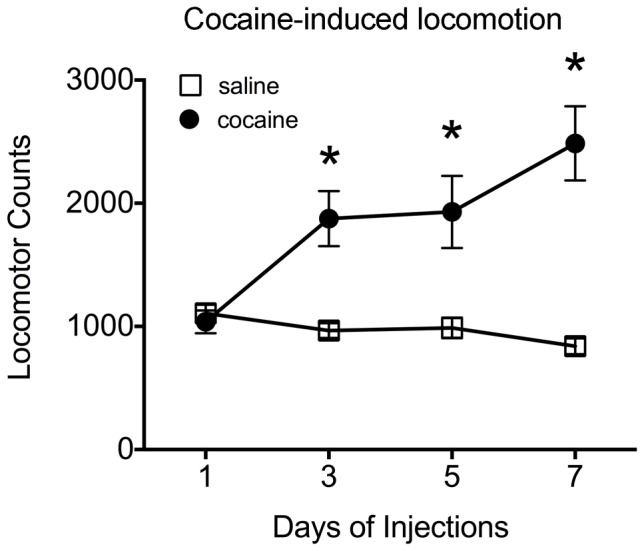
This plot shows locomotor activity in response to repeated injections of cocaine (10 mg/kg, i.p.) or saline. Closed circles represent animals receiving injections of cocaine; open squares indicate animals receiving injections of saline. Data are shown as group averages (± SEM); *p<0.05 compared to saline.
Levels of TGF-β R1 expression were examined following experimenter-administered cocaine, and surprisingly, we found a different expression pattern to that observed following SA. Twenty-four hours following experimenter administered cocaine, we found no change in TGF-β receptor protein expression compared to saline treated [t(18) = 0.9140, p>0.05], which remained unchanged after a 7-day withdrawal period [t(13) = 0.2850, p>0.05] ( Figure 4 ). Taken together, these data indicate TGF-β R1 signaling is regulated only following withdrawal from cocaine SA, but not experimenter-administered cocaine.
Figure 4. TGF-β R1 expression following experimenter-administered cocaine.

Relative TGF-β R1 protein expression in the NAc of rats following 1 or 7 days of withdrawal from a sensitizing regimen of cocaine (10 mg/kg; i.p, 7 days) or saline; *p<0.05 compared to saline.
Discussion
The results of this study identify the TGF-β receptor as a previously unknown molecular adaption following periods of cocaine cessation. The time-dependent regulation of TGF-β R1 occurred following active but not passive cocaine exposure, as the increase in protein expression was observed only after withdrawal from cocaine SA, and not after withdrawal from a sensitizing regimen of cocaine.
It is intriguing to speculate about the involvement of the TGF-β receptor-signaling cascade following a history of cocaine self-administration. TGF-β signaling pathways may potentially regulate actin cycling directly to alter structural changes in the NAc. Repeated exposure to psychomotor stimulants, such as cocaine, results in morphological changes to NAc medium spiny neurons (MSNs) including increases in dendritic spine density [30], [33]–[35]. One mechanism that directly links TGF-β signaling to cocaine-induced morphological plasticity is through a documented ability of TGF-β to alter the actin cytoarchitecture through Rho GTPase signaling, which has previously been linked to cocaine-induced structural plasticity [30], [36]–[39]. Moreover, drug-induced structural plasticity of dendritic spine morphology exists along a continuum that appears to be a function of time from cessation of drug exposure, method of intake, drug paradigm and re-exposure to cues previously associated with drug availability [2], [40]–[44]. Further studies are needed to identify the exact role of TGF-β receptor signaling in mediating cocaine-induced structural changes, and how such alterations may impact relapse behaviors.
Our findings demonstrate that TGF-β receptor expression is increased only in the NAc of animals that self-administer cocaine and not following experimenter-administered drug. There are numerous examples of neurobiological changes that occur differentially following active versus passive drug exposure [45]–[51] and these changes may underlie distinct behavioral changes such as drug sensitivity [52], [53]. It is important to note that several procedural differences between the SA and experimenter-administered cocaine protocols, such as dosing regimens and pharmacokinetics, may have a role in the selective increase in TGF-β R1 expression following SA and future studies are needed to examine the contributions of these factors.
It is worth noting that our findings differ from those of Maze et al. [54], in which the authors report an increase in TGF-β R1 following experimenter-administered cocaine. However, there are significant procedural differences between the Maze et al. study and the current experiments that may account for such paradoxical results: (i) Maze et al. tested mice, rather than rats as used in the current experiments; (ii) mRNA were measured in the Maze study, whereas we report protein expression.
While future studies are needed to develop a more complete temporal profile of TGF-β receptor expression following cocaine SA and withdrawal (i.e., prolonged periods of forced abstinence), our results demonstrate that the TGF-β Type I receptor is a potential target for intervention towards an effective pharmacotherapy in treating addiction. Further studies will determine how cocaine mediates down-stream signaling of TGF-β and how such cascades result in long-term cellular, morphological, and behavioral plasticity.
Acknowledgments
We would like to thank Karen Dietz for her comments on this manuscript. The cocaine tested in these experiments was gifted by NIDA. The authors on this manuscript reported no biomedical financial interests or potential conflicts of interest.
Funding Statement
This work was supported by the State University of New York at Buffalo and a grant from NIAAA T32-AA007583-11 (A.M.G). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1. Hyman SE, Malenka RC, Nestler EJ (2006) Neural mechanisms of addiction: the role of reward-related learning and memory. Annu Rev Neurosci 29: 565–598. [DOI] [PubMed] [Google Scholar]
- 2. Robison AJ, Nestler EJ (2011) Transcriptional and epigenetic mechanisms of addiction. Nat Rev Neurosci 12: 623–637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Nestler EJ (2001) Molecular basis of long-term plasticity underlying addiction. Nat Rev Neurosci 2: 119–128. [DOI] [PubMed] [Google Scholar]
- 4. Nestler EJ (2004) Molecular mechanisms of drug addiction. Neuropharmacology 47 Suppl 1 24–32. [DOI] [PubMed] [Google Scholar]
- 5. Kalivas PW (2009) The glutamate homeostasis hypothesis of addiction. Nat Rev Neurosci 10: 561–572. [DOI] [PubMed] [Google Scholar]
- 6. Luscher C, Malenka RC (2011) Drug-evoked synaptic plasticity in addiction: from molecular changes to circuit remodeling. Neuron 69: 650–663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Pierce RC, Kalivas PW (1997) A circuitry model of the expression of behavioral sensitization to amphetamine-like psychostimulants. Brain Res Brain Res Rev 25: 192–216. [DOI] [PubMed] [Google Scholar]
- 8. Loweth JA, Tseng KY, Wolf ME (2013) Using metabotropic glutamate receptors to modulate cocaine's synaptic and behavioral effects: mGluR1 finds a niche. Curr Opin Neurobiol 23: 500–506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Li X, DeJoseph MR, Urban JH, Bahi A, Dreyer JL, et al. (2013) Different roles of BDNF in nucleus accumbens core versus shell during the incubation of cue-induced cocaine craving and its long-term maintenance. J Neurosci 33: 1130–1142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Loweth JA, Tseng KY, Wolf ME (2013) Adaptations in AMPA receptor transmission in the nucleus accumbens contributing to incubation of cocaine craving. Neuropharmacology. [DOI] [PMC free article] [PubMed]
- 11. Conrad KL, Ford K, Marinelli M, Wolf ME (2010) Dopamine receptor expression and distribution dynamically change in the rat nucleus accumbens after withdrawal from cocaine self-administration. Neuroscience 169: 182–194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Kourrich S, Chapman CA (2003) NMDA receptor-dependent long-term synaptic depression in the entorhinal cortex in vitro. J Neurophysiol 89: 2112–2119. [DOI] [PubMed] [Google Scholar]
- 13. Grimm JW, Lu L, Hayashi T, Hope BT, Su TP, et al. (2003) Time-dependent increases in brain-derived neurotrophic factor protein levels within the mesolimbic dopamine system after withdrawal from cocaine: implications for incubation of cocaine craving. J Neurosci 23: 742–747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Otaka M, Ishikawa M, Lee BR, Liu L, Neumann PA, et al. (2013) Exposure to cocaine regulates inhibitory synaptic transmission in the nucleus accumbens. J Neurosci 33: 6753–6758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Boudreau AC, Reimers JM, Milovanovic M, Wolf ME (2007) Cell surface AMPA receptors in the rat nucleus accumbens increase during cocaine withdrawal but internalize after cocaine challenge in association with altered activation of mitogen-activated protein kinases. J Neurosci 27: 10621–10635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Boudreau AC, Wolf ME (2005) Behavioral sensitization to cocaine is associated with increased AMPA receptor surface expression in the nucleus accumbens. J Neurosci 25: 9144–9151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Guan X, Wang L, Chen CL, Guan Y, Li S (2010) Roles of two subtypes of corticotrophin-releasing factor receptor in the corticostriatal long-term potentiation under cocaine withdrawal condition. J Neurochem 115: 795–803. [DOI] [PubMed] [Google Scholar]
- 18. Kunwar AJ, Rickmann M, Backofen B, Browski SM, Rosenbusch J, et al. (2011) Lack of the endosomal SNAREs vti1a and vti1b led to significant impairments in neuronal development. Proc Natl Acad Sci U S A 108: 2575–2580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Ageta H, Murayama A, Migishima R, Kida S, Tsuchida K, et al. (2008) Activin in the brain modulates anxiety-related behavior and adult neurogenesis. PLoS One 3: e1869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Dow AL, Russell DS, Duman RS (2005) Regulation of activin mRNA and Smad2 phosphorylation by antidepressant treatment in the rat brain: effects in behavioral models. J Neurosci 25: 4908–4916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Schuckit MA (2006) Comorbidity between substance use disorders and psychiatric conditions. Addiction 101 Suppl 1 76–88. [DOI] [PubMed] [Google Scholar]
- 22. Zilberman ML, Tavares H, Blume SB, el-Guebaly N (2003) Substance use disorders: sex differences and psychiatric comorbidities. Can J Psychiatry 48: 5–13. [DOI] [PubMed] [Google Scholar]
- 23. Wachs FP, Winner B, Couillard-Despres S, Schiller T, Aigner R, et al. (2006) Transforming growth factor-beta1 is a negative modulator of adult neurogenesis. J Neuropathol Exp Neurol 65: 358–370. [DOI] [PubMed] [Google Scholar]
- 24.Deschaux O, Vendruscolo LF, Schlosburg JE, Diaz-Aguilar L, Yuan CJ, et al. (2012) Hippocampal neurogenesis protects against cocaine-primed relapse. Addict Biol. [DOI] [PMC free article] [PubMed]
- 25. Noonan MA, Bulin SE, Fuller DC, Eisch AJ (2010) Reduction of adult hippocampal neurogenesis confers vulnerability in an animal model of cocaine addiction. J Neurosci 30: 304–315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Chandra R, Lenz JD, Gancarz AM, Chaudhury D, Schroeder GL, et al. (2013) Optogenetic inhibition of D1R containing nucleus accumbens neurons alters cocaine-mediated regulation of Tiam1. Front Mol Neurosci 6: 13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Gancarz AM, Kausch MA, Lloyd DR, Richards JB (2012) Between-session progressive ratio performance in rats responding for cocaine and water reinforcers. Psychopharmacology (Berl) 222: 215–223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Dietz DM, Tapocik J, Gaval-Cruz M, Kabbaj M (2005) Dopamine transporter, but not tyrosine hydroxylase, may be implicated in determining individual differences in behavioral sensitization to amphetamine. Physiol Behav 86: 347–355. [DOI] [PubMed] [Google Scholar]
- 29. Hooks MS, Jones GH, Smith AD, Neill DB, Justice JB Jr (1991) Individual differences in locomotor activity and sensitization. Pharmacol Biochem Behav 38: 467–470. [DOI] [PubMed] [Google Scholar]
- 30. Dietz DM, Sun H, Lobo MK, Cahill ME, Chadwick B, et al. (2012) Rac1 is essential in cocaine-induced structural plasticity of nucleus accumbens neurons. Nat Neurosci 15: 891–896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Damez-Werno D, LaPlant Q, Sun H, Scobie KN, Dietz DM, et al. (2012) Drug experience epigenetically primes Fosb gene inducibility in rat nucleus accumbens. J Neurosci 32: 10267–10272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Steketee JD, Kalivas PW (2011) Drug wanting: behavioral sensitization and relapse to drug-seeking behavior. Pharmacol Rev 63: 348–365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Russo SJ, Dietz DM, Dumitriu D, Morrison JH, Malenka RC, et al. (2010) The addicted synapse: mechanisms of synaptic and structural plasticity in nucleus accumbens. Trends Neurosci 33: 267–276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Dietz DM, Dietz KC, Nestler EJ, Russo SJ (2009) Molecular mechanisms of psychostimulant-induced structural plasticity. Pharmacopsychiatry 42 Suppl 1 S69–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Robinson TE, Kolb B (2004) Structural plasticity associated with exposure to drugs of abuse. Neuropharmacology 47 Suppl 1 33–46. [DOI] [PubMed] [Google Scholar]
- 36. Edlund S, Landstrom M, Heldin CH, Aspenstrom P (2002) Transforming growth factor-beta-induced mobilization of actin cytoskeleton requires signaling by small GTPases Cdc42 and RhoA. Mol Biol Cell 13: 902–914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Vardouli L, Moustakas A, Stournaras C (2005) LIM-kinase 2 and cofilin phosphorylation mediate actin cytoskeleton reorganization induced by transforming growth factor-beta. J Biol Chem 280: 11448–11457. [DOI] [PubMed] [Google Scholar]
- 38. Kim WY, Shin SR, Kim S, Jeon S, Kim JH (2009) Cocaine regulates ezrin-radixin-moesin proteins and RhoA signaling in the nucleus accumbens. Neuroscience 163: 501–505. [DOI] [PubMed] [Google Scholar]
- 39. Wang X, Cahill ME, Werner CT, Christoffel DJ, Golden SA, et al. (2013) Kalirin-7 mediates cocaine-induced AMPA receptor and spine plasticity, enabling incentive sensitization. J Neurosci 33: 11012–11022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Gipson CD, Kupchik YM, Shen H, Reissner KJ, Thomas CA, et al. (2013) Relapse induced by cues predicting cocaine depends on rapid, transient synaptic potentiation. Neuron 77: 867–872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Golden SA, Russo SJ (2012) Mechanisms of psychostimulant-induced structural plasticity. Cold Spring Harb Perspect Med 2. [DOI] [PMC free article] [PubMed]
- 42. Ferrario CR, Gorny G, Crombag HS, Li Y, Kolb B, et al. (2005) Neural and behavioral plasticity associated with the transition from controlled to escalated cocaine use. Biol Psychiatry 58: 751–759. [DOI] [PubMed] [Google Scholar]
- 43. Li Y, Acerbo MJ, Robinson TE (2004) The induction of behavioural sensitization is associated with cocaine-induced structural plasticity in the core (but not shell) of the nucleus accumbens. Eur J Neurosci 20: 1647–1654. [DOI] [PubMed] [Google Scholar]
- 44. Shen HW, Toda S, Moussawi K, Bouknight A, Zahm DS, et al. (2009) Altered dendritic spine plasticity in cocaine-withdrawn rats. J Neurosci 29: 2876–2884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Chen BT, Bowers MS, Martin M, Hopf FW, Guillory AM, et al. (2008) Cocaine but not natural reward self-administration nor passive cocaine infusion produces persistent LTP in the VTA. Neuron 59: 288–297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. McCutcheon JE, Wang X, Tseng KY, Wolf ME, Marinelli M (2011) Calcium-permeable AMPA receptors are present in nucleus accumbens synapses after prolonged withdrawal from cocaine self-administration but not experimenter-administered cocaine. J Neurosci 31: 5737–5743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. McFarland K, Lapish CC, Kalivas PW (2003) Prefrontal glutamate release into the core of the nucleus accumbens mediates cocaine-induced reinstatement of drug-seeking behavior. J Neurosci 23: 3531–3537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Robinson TE, Gorny G, Savage VR, Kolb B (2002) Widespread but regionally specific effects of experimenter- versus self-administered morphine on dendritic spines in the nucleus accumbens, hippocampus, and neocortex of adult rats. Synapse 46: 271–279. [DOI] [PubMed] [Google Scholar]
- 49. Mark GP, Hajnal A, Kinney AE, Keys AS (1999) Self-administration of cocaine increases the release of acetylcholine to a greater extent than response-independent cocaine in the nucleus accumbens of rats. Psychopharmacology (Berl) 143: 47–53. [DOI] [PubMed] [Google Scholar]
- 50. Dworkin SI, Smith JE (1986) Behavioral contingencies involved in drug-induced neurotransmitter turnover changes. NIDA Res Monogr 74: 90–106. [PubMed] [Google Scholar]
- 51.Caffino L, Cassina C, Giannotti G, Orru A, Moro F, et al. (2013) Short-term abstinence from cocaine self-administration, but not passive cocaine infusion, elevates alphaCaMKII autophosphorylation in the rat nucleus accumbens and medial prefrontal cortex. Int J Neuropsychopharmacol: 1–7. [DOI] [PubMed]
- 52. Ator NA, Griffiths RR (1993) Differential sensitivity to midazolam discriminative-stimulus effects following self-administered versus response-independent midazolam. Psychopharmacology (Berl) 110: 1–4. [DOI] [PubMed] [Google Scholar]
- 53. Dworkin SI, Mirkis S, Smith JE (1995) Response-dependent versus response-independent presentation of cocaine: differences in the lethal effects of the drug. Psychopharmacology (Berl) 117: 262–266. [DOI] [PubMed] [Google Scholar]
- 54. Maze I, Covington HE 3rd, Dietz DM, LaPlant Q, Renthal W, et al. (2010) Essential role of the histone methyltransferase G9a in cocaine-induced plasticity. Science 327: 213–216. [DOI] [PMC free article] [PubMed] [Google Scholar]