

Abstract
Emerging evidence indicates that airway epithelial barrier function is compromised in asthma, a disease characterized by Th2-skewed immune response against inhaled allergens, but the mechanisms involved are not well understood. The purpose of this study was to investigate the effects of Th2-type cytokines on airway epithelial barrier function. 16HBE14o- human bronchial epithelial cells monolayers were grown on collagen coated Transwell inserts. The basolateral or apical surfaces of airway epithelia were exposed to human interleukin-4 (IL-4), IL-13, IL-25, IL-33, thymic stromal lymphopoietin (TSLP) alone or in combination at various concentrations and time points. We analyzed epithelial apical junctional complex (AJC) function by measuring transepithelial electrical resistance (TEER) and permeability to FITC-conjugated dextran over time. We analyzed AJC structure using immunofluorescence with antibodies directed against key junctional components including occludin, ZO-1, β-catenin and E-cadherin. Transepithelial resistance was significantly decreased after both basolateral and apical exposure to IL-4. Permeability to 3 kDa dextran was also increased in IL-4-exposed cells. Similar results were obtained with IL-13, but none of the innate type 2 cytokines examined (TSLP, IL-25 or IL-33) significantly affected barrier function. IL-4 and IL-13-induced barrier dysfunction was accompanied by reduced expression of membrane AJC components but not by induction of claudin- 2. Enhanced permeability caused by IL-4 was not affected by wortmannin, an inhibitor of PI3 kinase signaling, but was attenuated by a broad spectrum inhibitor of janus associated kinases. Our study indicates that IL-4 and IL-13 have disruptive effect on airway epithelial barrier function. Th2-cytokine induced epithelial barrier dysfunction may contribute to airway inflammation in allergic asthma.
Keywords: IL-13, IL-25, IL-33, IL-4, Janus associated kinase (JAK), TSLP, Th2 cytokines, adherens junction, airway epithelial cell, allergy, apical junctional complex, asthma, barrier dysfunction, tight junction
Introduction
Airway epithelial cell (AEC) barrier dysfunction is increasingly associated with allergic inflammation and asthma,1-3 but the mechanisms involved remain poorly understood. The AEC barrier consists of surface layer mucus as well as apical junctional complexes (AJC) that assemble between neighboring cells and include tight junctions and adherens junctions. Apical tight junctions are composed of three types of transmembrane proteins: (1) members of claudin family, (2) tight junction-associated MARVEL family members (i.e., occludin, tricellulin and MARVELD3) and (3) immunoglobulin-like proteins such as junctional adhesion molecule and coxsackie adenovirus receptor.4,5 E-cadherin and members of the nectin family represent the major transmembrane proteins of epithelial adherens junctions.6,7 Several intracellular membrane proteins cluster and stabilize adhesive components of the AJC including the zonula occludens (ZO) proteins and catenin family that link transmembrane AJC components with actin-binding proteins and the cellular cytoskeleton via mechanisms under active study.8
Recent clinical and experimental studies uncovered functional and structural abnormalities of the AEC barrier in the inflamed airway. For example, an analysis of primary AEC obtained from asthmatic subjects, revealed diminished epithelial barrier function that was associated with decreased ZO-1 expression.2 Interestingly, reduced ZO-1 expression and perturbed barrier function persisted in asthmatic AEC after passage in vitro indicating that it was a stable phenotype, although the specific molecular mechanisms involved were not identified. Using model airway epithelia, we recently reported that the synthetic double-stranded RNA polyI:C caused potent disruption of AEC apical junctional complexes in a protein-kinase D-dependent manner.3 This was associated with peri-junctional actin remodeling and likely involves AJC endocytosis (unpublished data).3 Inflammatory cytokines are known to disrupt barrier function and AJC’s in intestinal epithelial cells. For example, both Th1 (IFN-γ), Th2 (IL-13) and innate immune (TNF-α) cytokines can disrupt the structure and function of intestinal epithelial AJC’s via distinct mechanisms.8-12 Although airway and intestinal epithelial barrier structures are similar, we currently have limited understanding of the regulation of AEC barrier structure and function during inflammation, and it remains to be seen whether principles established in the GI tract translate directly to the lung.
We studied the effects of Th2-type cytokines on AEC barrier function and structure using model epithelia grown in vitro. We focused on CD4+ Th2 derived cytokines (IL-4 and IL-13), as well as Th2-type cytokines derived from innate immune cells (IL-25, IL-33 and TSLP), because these cytokines are fundamental to asthma and allergic airway inflammation.13,14
Results and Discussion
We examined the effects of Th2-type cytokines alone or in combination on 16HBE14o- human bronchial epithelial cells. When grown on semi-permeable membranes 16HBE14o- cells formed monolayers of well-differentiated columnar cells with high transepithelial electrical resistance (TEER) typically greater than 550 Ohms × cm2. Further differentiation at air-liquid interface did not further enhance barrier integrity (data not shown), thus cells were maintained under submerged conditions. We first studied the canonical Th2 cytokine IL-4 applied either to the apical or basal surfaces for different time points. Figure 1A shows that at 6 h after IL-4 exposure there was a slight increase in TEER (9−12%) but this followed by progressive and sustained reduction in TEER up to 72 h (Fig. 1A). The effects of IL-4 were slightly more pronounced following basolateral compared with apical exposure (Fig. 1A). In subsequent experiments, we applied IL-4 (and other Th2-type cytokines) to both the apical and basolateral compartments and focused on barrier disruption at later time points. We found that the disruptive effects of IL-4 were dose-dependent, beginning around 5 ng/ml and increasing up to 50 ng/ml (Fig. 1B). The innate type 2 cytokines IL-25, IL-33 and TSLP have been recently implicated with initiation of Th2-type immune responses by acting on multiple cell types including dendritic cells and innate lymphoid cells.14,15 These cytokines are thought to act at mucosal surfaces, but their effects on permeability of AEC barrier have not been well-studied. Interestingly, none of the innate type 2 cytokines alone or in combination significantly disrupted barrier function as determined by TEER measurements (Fig. 1C).
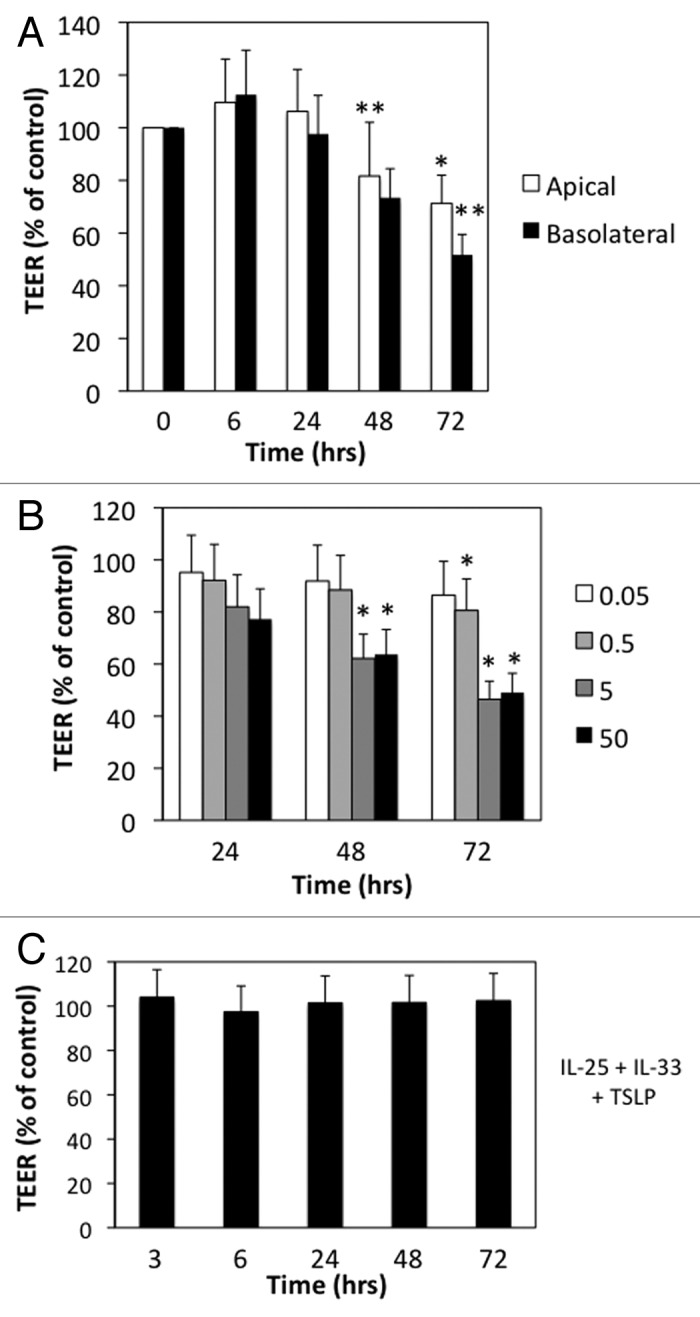
Figure 1. IL-4 selectively increases permeability of model airway epithelial cell monolayers. (A) 16 HBE cells were incubated with IL-4 (50 ng/ml) applied to the apical (open bars) or basolateral (closed bars) surface for different time points. (B) Dose response for the indicated concentrations of IL-4 (ng/ml) over time. (C) 16HBE cells were incubated with a cocktail of TSLP+IL-25+IL-33 (50 ng/ml each) for the indicated time points followed by analysis of TEER. All data are expressed relative to TEER measured in control cells (typical values > 550 Ω × cm2), which was set as 100%, and are the mean ± SEM of n = 5−12 experiments; *p < 0.05, **p < 0.001.
We next examined airway epithelial permeability to macromolecules, which was also enhanced in IL-4 exposed cells as determined by measuring flux of FITC-conjugated dextran (3 kDa) (Fig. 2). By contrast, no effect of IL-25, IL-33 and TSLP on transepithelial dextran flux was observed (data not shown), which is consistent with TEER data. In non-hematopoietic cells, IL-4 binds to a high affinity receptor composed of IL-4Rα/IL-13Rα1 subunits, which stimulates recruitment and phosphorylation of Janus kinases (JAK) 1 and 3. IL-4R signal transduction involves phosphatidylinositide-3 kinase (PI3K)/AKT-dependent signaling as well as Stat6-dependent stimulation of gene expression.16,17 To examine the roles of these signaling events in IL-4-dependent barrier disruption, we used pharmacological inhibitors of JAK (broad spectrum as well as selective Jak3 antagonist) and PI3K. TEER values in cells incubated with IL-4 alone (50 ng/ml, 72 h) were 66 ± 6% of control, which was effectively inhibited in cells co-incubated with a selective JAK3 antagonist (102 ± 7% of control) as well as a pan-Jak antagonist (128 ± 15% of control) but not by wortmannin (73 ± 13% control, mean ± SEM, n = 2−3). Interestingly, however, only the pan-JAK inhibitor reversed the increased transepithelial dextran flux in IL-4 stimulated airway epithelial cells (Fig. 2).
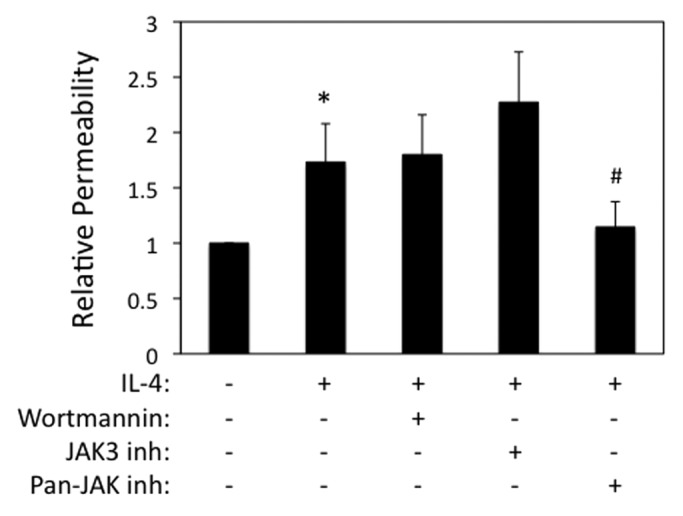
Figure 2. IL-4 enhances paracellular permeability in a Jak-dependent manner. 16HBE cells were grown to confluence and then incubated with IL-4 (50 ng/ml for 72 h), followed by analysis of paracellular flux to 3-kDa-FITC-dextran added to the apical chamber. Cells were also incubated with wortmannin or different JAK inhibitors as indicated. Data are the mean ± SEM of n = 3−4 experiments. *p < 0.05 compared with control cells. #p < 0.05 compared with cells treated with IL-4 alone.
To gain additional insights into mechanisms of IL-4 dependent disruption of airway epithelial barriers, we used immunofluorescence labeling and confocal microscopy to analyze AJ and TJ structure. Control 16HBE cells demonstrated a predominant localization of ZO-1, occludin, E-cadherin, β-catenin and claudin-4 at the areas of cell-cell contacts which is indicative of intact AJC (Fig. 3, arrows). This normal labeling pattern of AJ and TJ protein appears to be perturbed in IL-4-treated epithelial cell monolayers, which was manifested by the decreased AJ/TJ protein labeling of cell-cell contacts, appearance of intercellular gaps and accumulation of claudin-4 in cytoplasmic vesicles (Fig. 3, arrowheads). Several mechanisms can underlie altered structure and function of the epithelial AJC. They include decreased expression of different AJ/TJ proteins or upregulation of so called “leaky” components of the AJC such as claudin-2, which is thought to disrupt homotypic interaction between barrier-forming claudin-s.12 However, immunoblotting analysis of total cell lysates did not detect altered decreased expression of major AJ/TJ proteins and claudin-2 was not induced under these experimental conditions (Fig. 4). Collectively this data suggests that IL-4 disrupts the airway epithelial barrier by altering normal structure of epithelial AJs and TJs via mechanisms not involving changes in junction protein expression.
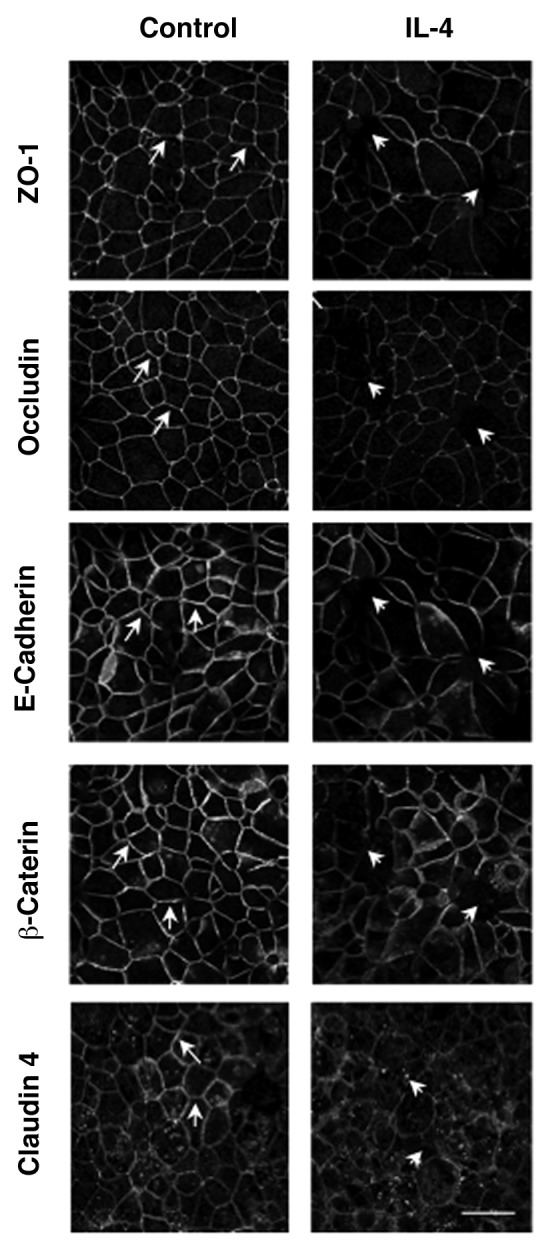
Figure 3. IL-4 disrupts apical junctional complex structures. 16HBE cells were incubated with IL-4 (50 ng/ml for 72 h) followed by analysis of AJC structure using immunofluorescence and antibodies directed against ZO-1, Occludin, E-Cadherin, β-Catenin or Claudin- 4. Results are from one experiment representative of n = 2. Scale bar = 30 microns.
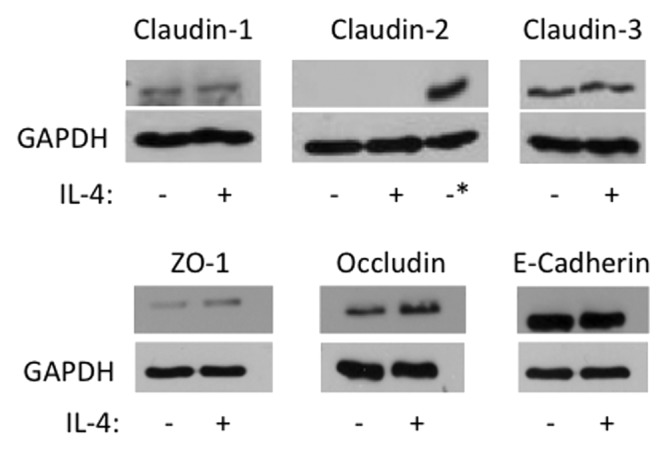
Figure 4. IL-4 does not induce claudin- 2 expression or major changes in AJC protein expression. 16HBE cells were incubated without (-) or with (+) IL-4 (50 ng/ml) for 72 h, and then whole cell lysates were analyzed by western blot for expression of the indicated junctional proteins. GAPDH was used as lane loading control. The asterisk in the Claudin-2 immunoblot represents lysates from Caco-2 cells used as a positive control. Results are from one experiment in representative of n = 2−3.
IL-13 shares similar receptor components with IL-4 and is a central mediator of airway inflammation and asthma.18,19 In parallel experiments, we found that IL-13 also induced AEC barrier dysfunction as assessed by reduction in TEER and enhanced macromolecular permeability (Fig. 5). The combined effects of IL-4 and IL-13 were not synergistic, consistent with these two cytokines acting via shared receptors and signaling modules. In parallel immunofluorescence labeling and confocal microscopy experiments, we found that AJC structure was disrupted following exposure to IL-13 (data not shown). Interestingly, contrary to its effects in the intestinal epithelium (10) IL-13 (similar to IL-4) did not induce claudin-2 expression in human AEC (data not shown).
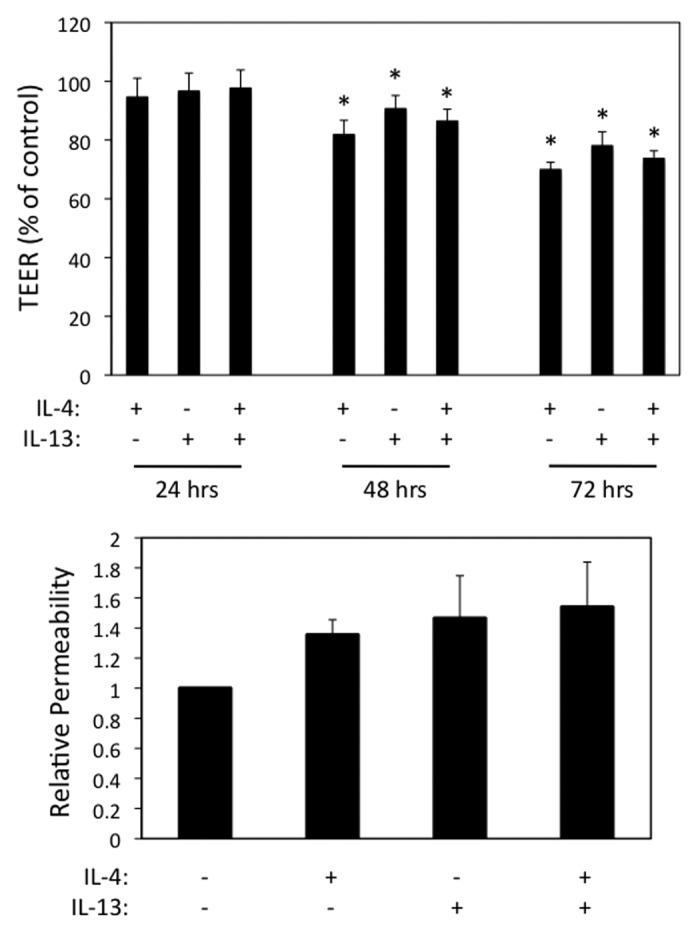
Figure 5. IL-13 induces airway epithelial barrier dysfunction similar to IL-4. (A) 16HBE cells were incubated with IL-4 or IL-13 (50 ng/ml each) alone or in combination followed by analysis of TEER over time (A) or paracellular permeability to 3-kDa dextran after 72 h (B). Results in (A) are normalized to control TEER, which was set as 100. Data are the mean ± SEM of n = 3 experiments. * indicates p < 0.05 compared with the control cells.
We conclude that the Th2 cytokines IL-4 and IL-13 cause substantial reduction in AEC junctional complex structure and function in a JAK-dependent manner. Th2-cytokine dependent barrier disruption may underly the observed defects in barrier function seen in allergic asthma,2 which is a prototypic Th2-type disease. In addition to enhancing airway inflammation, disrupted barrier function may promote allergen sensitization by facilitating the uptake of inhaled allergens by subepithelial dendritic cells. Taken together with the results of Weber et al.,12 our data suggest that Th2 cytokines induce barrier dysfunction in intestinal and aiwary epithelial cells via distinct mechanisms. Whereas intestinal barrier disruption involves induction of claudin- 2, AEC barrier disruption occurs in the absence of claudin-2 induction via mechanisms that require further study. We speculate that cytoskeletal rearrangement and altered vesicle trafficking underlies IL-4/IL-13-induced AJC disassembly, but further studies will be needed in this area.8 Our results build on recent studies and suggest that Th2 cytokine-dependent epithelial barrier dysfunction is an important but previously overlooked contributor to airway inflammation in asthma.20,21
Methods
Bronchial epithelial cell culture
16HBE14o- human bronchial epithelial cells (a gift from Dr D. C. Gruenert, University of California) were grown in minimum essential medium containing 10% FBS, 10 mmol/L HEPES and glutamine on collagen-coated, permeable polycarbonate filters of 0.4 µm or 5.0 µm in pore size (Costar). Cells treated with or without cytokines were incubated at 37⁰C in a 95% air / 5% CO2 atmosphere up to 72 h. Recombinant human cytokines were purchased from Peprotech and used at (0.5−50 ng/ml, IL-4 and IL-13) or 50 ng/ml (IL-25, IL-33 and TSLP). In some experiments cells were pretreated with the PI3 kinase inhibitor wortmannin (100 nM, Calbiochem), a JAK3 inhibitor or a pan-JAK inhibitor (100 nM each, Calbiochem) for 30 min prior to addition of IL-4 or IL-13.
Transepithelial electrical resistance
16HBE14o- human bronchial epithelial cells were grown on permeable polycarbonate filters of 0.4 µm for Transepithelial electrical resistance (TEER) measurement with an EVOMX volt-ohm-meter (World Precision Instruments). The resistance of cell-free collagen-coated filters was subtracted from each experimental point, and the data were presented either as absolute values (Ω × cm2) or changes relative to the control group.
Paracellular FITC-Dextran flux assay
16HBE14o- human bronchial epithelial cells were grown on permeable polycarbonate filters of 5 µm. Paracellular flux of fluorescent markers was investigated by measuring passage of apically added markers across epithelial monolayers. The sodium fluorescence and dextran apical-basal passage was measured using a Fluoroskan Ascent FL2.5 reader (Thermo Fisher). FITC-Dextran 3 KDa (Sigma-Aldrich) was added to apical surface of 16HBE cells at the concentration 10 mg/ml and samples were taken from the basolateral chamber at the indicated time points.
Immunofluorescence staining of junctional proteins
Control and cytokine-exposed 16HBE14o-cell monolayers were fixed in cold methanol and subjected by dual-immunolabeling and confocal microscopy as previously described.3 Briefly, AJ/TJ proteins were visualized using the following primary polyclonal (p) or monoclonal (m) antibodies: anti ZO-1 pAb, occludin and claudin–4 mAbs (Invitrogen), E-cadherin and β-catenin mAb (BD Bioscience). After exposure to primary antibodies, cells were incubated with AlexaFluor 488 and 568 labeled secondary antibodies and mounted with Prolong Gold antifade medium (Invitrogen). Immunofluorescently labeled cell monolayers were examined using Olympus FV1000 laser scanning confocal microscope (Olympus America) through the University of Rochester Medical Center Confocal and Conventional Microscopy Core. A 100 × U Plan S Apo 1.4 NA (oil) objective was used and images were taken at 512 × 512 resolution with a Kalman setting of 14. All images were optimized so that fluorescence intensity remained in the linear range. Images were processed with Adobe Photoshop software.
Analysis of tight junction proteins by western blot
Cell monolayers grown in cell culture plates were stimulated with and without cytokines and then washed with cold PBS and lysed on ice in RIPA buffer with Protease and Phosphatase Inhibitor Cocktails (Sigma), followed by scraping. Proteins were resolved on SDSPAGE 6–15% and transferred to nitrocellulose membranes. Membranes were incubated in blotting solution (5% non-fat dry milk in TBS/0.1% Tween 20) at room temperature for 1h prior to overnight incubation with primary antibodies. After overnight incubation at 4°C, the blots were washed in TBS/0.1% Tween-20 followed by incubation with horseradish peroxidase-conjugated secondary antibodies. The blots were exposed to ECL (GE Healthcare, RPN 2106) and subjected to autoradiography with Kodak BioMax MR Film.
Statistical analysis
Results are expressed as means ± SEMs, unless otherwise specified. Statistical analysis of the data was performed by using the Student t test. Significance was considered at a P value of less than 0.05.
Acknowledgments
We gratefully acknowledge helpful suggestions from Dr Andrei Ivanov during the course of these studies. Declaration of all sources of funding: NIH R01HL071933 (SG), Pilot Project Funding supported by NIH P30ES01247 (SG), NIH K12 HD068373 (FR), NIH T32 HL066988 (TC) and the University of Rochester Department of Medicine.
Disclosure of Potential Conflicts of Interest
No potential conflict of interest was disclosed.
Footnotes
Previously published online: www.landesbioscience.com/journals/tissuebarriers/article/24333
References
- 1.Trautmann A, Kruger K, Akdis M, Muller-Wening D, Akkaya A, Brocker EB, et al. Apoptosis and loss of adhesion of bronchial epithelial cells in asthma. Int Arch Allergy Immunol. 2005;138:142–50. doi: 10.1159/000088436. [DOI] [PubMed] [Google Scholar]
- 2.Xiao C, Puddicombe SM, Field S, Haywood J, Broughton-Head V, Puxeddu I, et al. Defective epithelial barrier function in asthma. J Allergy Clin Immunol. 2011;128:549–56, e1-12. doi: 10.1016/j.jaci.2011.05.038. [DOI] [PubMed] [Google Scholar]
- 3.Rezaee F, Meednu N, Emo JA, Saatian B, Chapman TJ, Naydenov NG, et al. Polyinosinic:polycytidylic acid induces protein kinase D-dependent disassembly of apical junctions and barrier dysfunction in airway epithelial cells. J Allergy Clin Immunol. 2011;128:1216–24, e11. doi: 10.1016/j.jaci.2011.08.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Niessen CM. Tight junctions/adherens junctions: basic structure and function. J Invest Dermatol. 2007;127:2525–32. doi: 10.1038/sj.jid.5700865. [DOI] [PubMed] [Google Scholar]
- 5.Schulzke JD, Fromm M. Tight junctions: molecular structure meets function. Ann N Y Acad Sci. 2009;1165:1–6. doi: 10.1111/j.1749-6632.2009.04925.x. [DOI] [PubMed] [Google Scholar]
- 6.Meng W, Takeichi M. Adherens junction: molecular architecture and regulation. Cold Spring Harb Perspect Biol. 2009;1:a002899. doi: 10.1101/cshperspect.a002899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Shapiro L, Weis WI. Structure and biochemistry of cadherins and catenins. Cold Spring Harb Perspect Biol. 2009;1:a003053. doi: 10.1101/cshperspect.a003053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ivanov AI, Parkos CA, Nusrat A. Cytoskeletal regulation of epithelial barrier function during inflammation. Am J Pathol. 2010;177:512–24. doi: 10.2353/ajpath.2010.100168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bruewer M, Luegering A, Kucharzik T, Parkos CA, Madara JL, Hopkins AM, et al. Proinflammatory cytokines disrupt epithelial barrier function by apoptosis-independent mechanisms. J Immunol. 2003;171:6164–72. doi: 10.4049/jimmunol.171.11.6164. [DOI] [PubMed] [Google Scholar]
- 10.Utech M, Ivanov AI, Samarin SN, Bruewer M, Turner JR, Mrsny RJ, et al. Mechanism of IFN-gamma-induced endocytosis of tight junction proteins: myosin II-dependent vacuolarization of the apical plasma membrane. Mol Biol Cell. 2005;16:5040–52. doi: 10.1091/mbc.E05-03-0193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Capaldo CT, Nusrat A. Cytokine regulation of tight junctions. Biochim Biophys Acta. 2009;1788:864–71. doi: 10.1016/j.bbamem.2008.08.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Weber CR, Raleigh DR, Su L, Shen L, Sullivan EA, Wang Y, et al. Epithelial myosin light chain kinase activation induces mucosal interleukin-13 expression to alter tight junction ion selectivity. J Biol Chem. 2010;285:12037–46. doi: 10.1074/jbc.M109.064808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Georas SN, Guo J, De Fanis U, Casolaro V. T-helper cell type-2 regulation in allergic disease. Eur Respir J. 2005;26:1119–37. doi: 10.1183/09031936.05.00006005. [DOI] [PubMed] [Google Scholar]
- 14.Barrett NA, Austen KF. Innate cells and T helper 2 cell immunity in airway inflammation. Immunity. 2009;31:425–37. doi: 10.1016/j.immuni.2009.08.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Spits H, Artis D, Colonna M, Diefenbach A, Di Santo JP, Eberl G, et al. Innate lymphoid cells--a proposal for uniform nomenclature. Nat Rev Immunol. 2013;13:145–9. doi: 10.1038/nri3365. [DOI] [PubMed] [Google Scholar]
- 16.Kelly-Welch AE, Hanson EM, Boothby MR, Keegan AD. Interleukin-4 and interleukin-13 signaling connections maps. Science. 2003;300:1527–8. doi: 10.1126/science.1085458. [DOI] [PubMed] [Google Scholar]
- 17.Kuperman DA, Schleimer RP. Interleukin-4, interleukin-13, signal transducer and activator of transcription factor 6, and allergic asthma. Curr Mol Med. 2008;8:384–92. doi: 10.2174/156652408785161032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wills-Karp M, Luyimbazi J, Xu X, Schofield B, Neben TY, Karp CL, et al. Interleukin-13: central mediator of allergic asthma. Science. 1998;282:2258–61. doi: 10.1126/science.282.5397.2258. [DOI] [PubMed] [Google Scholar]
- 19.Corren J, Lemanske RF, Hanania NA, Korenblat PE, Parsey MV, Arron JR, et al. Lebrikizumab treatment in adults with asthma. N Engl J Med. 2011;365:1088–98. doi: 10.1056/NEJMoa1106469. [DOI] [PubMed] [Google Scholar]
- 20.Petecchia L, Sabatini F, Usai C, Caci E, Varesio L, Rossi GA. Cytokines induce tight junction disassembly in airway cells via an EGFR-dependent MAPK/ERK1/2-pathway. Lab Invest. 2012;92:1140–8. doi: 10.1038/labinvest.2012.67. [DOI] [PubMed] [Google Scholar]
- 21.Soyka MB, Wawrzyniak P, Eiwegger T, Holzmann D, Treis A, Wanke K, et al. Defective epithelial barrier in chronic rhinosinusitis: the regulation of tight junctions by IFN-γ and IL-4. J Allergy Clin Immunol. 2012;130:1087–96, e10. doi: 10.1016/j.jaci.2012.05.052. [DOI] [PubMed] [Google Scholar]