

Abstract
Nitric oxide (NO) is a key factor in inflammation as it regulates microvascular permeability, leukocyte adhesion and wound healing. This mini-review addresses mainly spatial and temporal requirements of NO regulatory mechanisms, with special emphasis on S-nitrosation. Endothelial nitric oxide synthase (eNOS)-derived NO induces S-nitrosation of p120 and β-catenin, particularly in response to platelet-activating factor (PAF), and through traffic and interactions at the adherens junction promotes endothelial hyperpermeability. S-nitrosation is a determinant in vascular processes such as vasodilation and leukocyte-endothelium interactions. Interestingly, NO decreases leukocytes adhesion to endothelium, but the mechanisms are unknown. Advances in NO molecular biology and regulation may serve as a basis for the development of new therapeutic strategies in the treatment of diseases characterized by inflammation such as ischemia-reperfusion injury, stroke, cancer and atherosclerosis.
Keywords: eNOS, microvascular permeability, S-nitrosation, adherens junction, nitric oxide
NO-Mediated Regulatory Mechanisms of Microvascular Permeability
Regulation of vascular permeability to macromolecules occurs in vivo primarily at postcapillary venules through activation of endothelial nitric oxide synthase (eNOS) and production of NO.1 The main target of the regulation is the structural and functional configuration of the endothelial adherent junctions. The main protein identified at the endothelial adherent junctions is VE-cadherin, which forms a complex with the cytosolic proteins α-catenin, β-catenin, plakoglobin and p120-catenin (p120).1-3 Adherens junctions constitute a paracellular transport pathway which is stimulated by pro-inflammatory agents [such as histamine, bradykinin and platelet-activating factor (PAF)]. Pro-inflammatory agonists stimulate a cascade of processes that phosphorylate junctional proteins, dissociate the junctional complex, cause internalization of junctional proteins and increase paracellular permeability.4,5 These conformational changes probably operate in synchrony with centripetal forces generated by the cytoskeleton, particularly via actin and are likely associated with diminished adhesiveness of integrins to focal adhesions.6 It is worth noting that ultrastructural evidence shows that PAF-stimulated hyperpermeability is independent of actin-associated contractile events.7 The observation that pro-inflammatory agents such as PAF, histamine, bradykinin and thrombin stimulate production of eNOS-derived NO supports the concept that NO signaling regulates endothelial/microvascular permeability.
eNOS and S-nitrosation
Nitric oxide is an important regulator of vascular homeostasis. We demonstrated that eNOS-derived NO is essential for the onset of agonist-induced hyperpermeability using eNOS knockout mice and eNOS depleted coronary postcapillary venular endothelial cells.8,9 In both models, basal permeability was normal while the response to PAF, a pro-inflammatory agent, was nearly abolished. Endothelial NOS localizes primarily to caveolae in the plasma membrane where its interactions with caveolin-1 (cav1) keep eNOS activity at basal levels.10 Upon agonist stimulation, eNOS is activated by phosphorylation, released from the inhibitory interaction with caveolin-1, and internalized.10-13 Because of potency and broad spectrum of the cellular actions of NO and its brief half-life, the mechanisms that regulate NO synthesis, with respect to time and space, are crucial in determining the biological functions of NO. We proposed and established that location of eNOS in the cytosol was a requirement for the onset of increased permeability.9,12 Definitive proof was obtained using eNOS mutants that target and locate eNOS to the cytosol (eNOSG2A) or to the plasma membrane (eNOSCAAX).14
Further research is still required to elucidate the fundamental need for translocation of eNOS to specific locations. Translocation would appear to be unnecessary for a gas with high diffusion coefficient. We and others have argued that NO production must occur near the intended specific functional target because of the abundance of NO scavengers in the intracellular environment. Other arguments include retardation of NO transport in lipid milieu. The overall issue of NO transport has been the subject of consideration through mathematical analysis.15 However, no definitive proof for the biological basis of this credible argument can be found in the literature.
The classic dogma in NO signaling established that all the actions of NO are mediated via soluble guanylate cyclase (sGC) and protein kinase G (PKG). This NO-driven signaling for changes in permeability are likely complementary to the classical contraction-associated mechanisms involving RhoA and Rho associated kinase (ROCK), which act also on junctional proteins.16 These contraction-associated mechanisms depend largely on phosphorylation of specific proteins as the biochemical regulatory steps.
Recently, S-nitrosation has emerged as an important NO-dependent posttranslational modification that alters the function of proteins and requires proximity between eNOS and the target proteins for appropriate NO delivery.17,18 S-nitrosation consists in the coupling of an NO moiety to a reactive cysteine thiol to form an S-nitrosothiol.19,20 S-nitrosation can affect the interactions between proteins, protein phosphorylation and intracellular transport processes.21,22 It is likely that phosphorylation and S-nitrosation are complementary signaling mechanisms. We discuss, in the next section, evidence demonstrating that S-nitrosation is a mechanism that contributes to regulation of endothelial permeability.
Adherens Junction Complex, NO and Microvascular Permeability
The adherens junction complex links the actin cytoskeleton with the plasma membrane. It is formed by VE-cadherin, β-catenin, p120-catenin and γ- catenin (also named plakoglobin). VE-cadherin is necessary for the organization of vascular structures and for maintaining the integrity of the vascular endothelial wall.23 The significance of VE-cadherin in vivo is underscored by the report that VE-cadherin-knockout mice exhibit greatly impaired endothelial barrier function.24 VE-cadherin is a single-pass transmembrane protein, with the N-terminal in the extracellular domain and the carboxy terminal in the intracellular domain. The N-terminal is the site of homotypic adhesion, whereas the carboxy terminal regulates cell growth25 and associates with several cytoplasmic proteins, including β-catenin, p120-catenin and γ-catenin. The catenin complex stabilizes VE-cadherin at the junctions by preventing its clathrin-mediated endocytosis.26-28 In particular, β-catenin links the cadherin complex to α-catenin, which in turn binds to the actin cytoskeleton. This linkage appears to be important for maintaining homotypic binding between VE-cadherin molecules of adjacent endothelial cells. VEGF and histamine-stimulated endothelial hyperpermeability has been associated with elevated tyrosine phosphorylation of VE-cadherin, β-catenin and p120-catenin4,5,29,30 and changes in the organization of VE-cadherin at intercellular junctions.4,31-33
S-nitrosation has been recently implicated in the regulation of microvascular/endothelial permeability.34,35 Administration of vascular endothelial growth factor (VEGF) for 15 min caused S-nitrosation of β-catenin and changes in permeability.34 Cysteine 619 was identified as the responsible amino acid undergoing S-nitrosation. Because Cys 619 is located at the interaction site of β-catenin with VE-cadherin its S-nitrosation causes dissociation of the complex and an increase in microvascular permeability.34 We, using PAF and TNF as proinflammatory agonists,35 advanced knowledge demonstrating that S-nitrosation occurs at times that are consistent with the onset of hyperpermeability (1–5 min). We also identified p120-catenin as a target of S-nitrosation35 and we specifically identified, by mass spectroscopy, Cys 579 as the main S-nitrosated- residue. Cys 579 is also located in the interaction site with VE-cadherin. These data point out that S-nitrosation regulates protein-protein interactions and promotes the dissociation of the adherens junction complex. As far as we are currently aware, no other proteins from the adherens junction complex has been identified as a target for S-nitrosation. In this framework, we have documented a role for NO - perhaps by S-nitrosation - in the internalization of VE-cadherin in response to PAF. Our experiments demonstrate that inhibition of eNOS (using L-NMA as eNOS inhibitor) blocks VE-cadherin internalization in response to PAF (Fig. 1).

Figure 1. Inhibition of eNOS inhibits PAF-induced VE-cadherin internalization. EAhy926 cells were pretreated with 300uM L-NMA (eNOS inhibitor) for 1 h before being stimulated with 10−7 M PAF. (A) western blot for VE-cadherin. L-NMA inhibits the internalization of VE-cadherin induced by PAF. (B) Bar graph showing the quantification of the western blots for VE-cadherin. * p < 0.05 compared with control; n = 3.
We propose a new model to explain how NO induces increase in permeability (Fig. 2). In the model, PAF-induced NO causes S-nitrosylation of β-catenin and p120 and destabilizes the adherens junction complex. The dissociation of the junctional complex, in turn, leads to hyperpermeability. Our results demonstrating that S-nitrosylation of β-catenin and p120 lead to changes in the localization and interaction between these proteins at the adherens junction are the first data linking NO signaling directly to adherens junction in response to PAF.
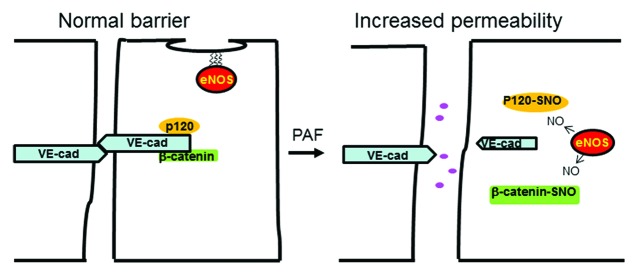
Figure 2. Schematic model showing the spatial regulation of endothelial permeability to macromolecules by eNOS-derived NO. The adherens junction complex at the endothelial plasma membrane establishes a normal barrier that restricts the diffusion of macromolecules from the luminal to the interstitial side of the endothelium. In the presence of agonist (PAF) that activates eNOS, the enzyme translocates to the cytosol, where by releasing NO it S-nitrosates β-catenin and p120-catenin. S-nitrosation of the junctional proteins disturbs the interactions between them, destabilizes the adherens junction complex and leads to increased permeability. Dark pink circles represent macromolecules.
The on-off regulation of S-nitrosation is a subject of discussion and merits direct research efforts. The onset of S-nitrosation is usually attributed to the redox sate of the cells, but there is no specific information on the levels of oxidation-reduction that enhance S-nitrosation. The off signal (denitrosation) is due to the activity of the thioredoxin/thioredoxin reductase enzymes.18,36 The biochemical foundation for the specificity of the S-nitrosation/de-nitrosation regulatory mechanisms is an attractive field for further investigation.
The relationships between phosphorylation and S-nitrosation of junctional proteins in the onset of hyperpermeability remain to be elucidated. The available data show that inhibition of S-nitrosation of β-catenin does not affect the phosphorylation of β-catenin on Tyr 654, a site that is involved in the dissociation from VE-cadherin.34 These observations suggest that S-nitrosation and phosphorylation are signaling processes that may regulate microvascular permeability in an independent way at the junctional proteins.
S-nitrosation and Vascular Function
Changes in microvascular permeability are events associated with different tissue and/or cellular functions. In fact, many substances that induce vasodilation cause an associated increase in vascular permeability [i.e., histamine, bradykinin and vascular endothelial growth factor (VEGF)]. For this reason, we will consider here changes in vascular function induced by S-nitrosation.
Since the first experiments that led to the discovery of NO as vascular regulator, its function has been associated with vasodilation.37 The classical biochemical pathway consists of activation of eNOS and production of NO, followed by stimulation of soluble guanylyl cyclase (sGC) and production of cyclic GMP. In accordance with this classical view, vascular dysfunction in several diseases reflected by failure to produce competent vasodilation, particularly in diabetes mellitus, hypertension and angina, has been interpreted as the result of deficiency in NO bioavailability. Interestingly, administration of NO donors fails to restore adequate function; thus, raising questions as to whether NO availability per se is the responsible factor. Recently, an attractive hypothesis, fundamentally based on S-nitrosation of sGC, has been advanced with appropriate experimental in vitro and in vivo support by Beuve et al. Basically, they demonstrated that S-nitrosation of sCG impairs the response of the microvasculature to nitroglycerin, an NO donor used in the treatment of angina.38 Similarly, S-nitrosation of sGC - due to nitrosative stress caused by chronic administration of angiotensin II, is an important factor in the development of hypertension.39 The latter study identified Cys 516 as the site of S-nitrosation responsible for sGC desensitization to NO.39
S-nitrosation may also play a role in the regulation of leukocyte - endothelium interactions in inflammation. Indirect evidence for the contribution of S-nitrosation is the demonstration that S-nitrosated tissue plasminogen activator, most likely by inhibition of leukocyte adhesion to endothelium, reduces the tissue damage associated with ischemia-reperfusion in cat hearts.40
Leukocyte-endothelium interactions are important determinants of the inflammatory reactions associated with ischemia-reperfusion injury. Nitric oxide is considered an anti-inflammatory agent by many investigators due to its ability to decrease leukocyte adhesion to endothelium. A potential anti-inflammatory endothelial mechanism is the inhibition of Weibel-Palade body (WPB) exocytosis and release of von Willebrandt factor (vWF). These effects were demonstrated by inhibiting NOS with L-NAME for 16 h and subsequent identification of the target cysteine (21, 91 and 264) in N-ethylmaleimide-sensitive factor (NSF).41
Indirect evidence shows that S100A8 (calgranulin A, a calcium-binding protein), which is abundant in neutrophils and endothelial cells, may have anti-inflammatory properties. Administration of S-nitrosated S100A8 by local superfusion prevents degranulation of mast cells and reduces leukocyte adhesion and extravasation in the mesenteric microcirculation.42 How S-nitrosation of S100A8 may occur in vivo is unknown.
Because the evidence linking NO and release of vWF and WP bodies was obtained after inhibiting NOS for 16 h, and that S-nitrosation of S100A8 was achieved after prolonged stimulation, it is possible to speculate that the NO release was the result of activation of the inducible iNOS, rather than eNOS. The precise mechanisms by which S-nitrosation enhances or reduces function are beginning to be addressed. Nitric oxide may serve as a negative feedback regulator of eNOS (and potentially other NOS) activity by S-nitrosation of the enzyme.36 While the evidence in eNOS knockout mouse shows that eNOS-derived NO is fundamental for the onset of hyperpermeability,8,43 the relative significance of eNOS and iNOS as pro-inflammatory or anti-inflammatory enzymes and their involvement via S-nitrosation vs. cGMP pathways in different regulatory phases of the inflammatory response, including the release of vWF and WP bodies may be re-investigated in specific eNOS and iNOS knockout mice.
In conclusion, S-nitrosation is a current focus of intense investigation as an emergent regulatory mechanism of vascular function. It is important to establish, particularly in the field of microvascular permeability, the precise biochemical pathways as well as the location of the source of the effective NO.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Footnotes
Previously published online: www.landesbioscience.com/journals/tissuebarriers/article/23896
References
- 1.Durán WN. The double-edge sword of TNF-alpha in ischemia-reperfusion injury. Am J Physiol Heart Circ Physiol. 2008;295:H2221–2. doi: 10.1152/ajpheart.01050.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bazzoni G, Dejana E. Endothelial cell-to-cell junctions: molecular organization and role in vascular homeostasis. Physiol Rev. 2004;84:869–901. doi: 10.1152/physrev.00035.2003. [DOI] [PubMed] [Google Scholar]
- 3.Weis WI, Nelson WJ. Re-solving the cadherin-catenin-actin conundrum. J Biol Chem. 2006;281:35593–7. doi: 10.1074/jbc.R600027200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Esser S, Lampugnani MG, Corada M, Dejana E, Risau W. Vascular endothelial growth factor induces VE-cadherin tyrosine phosphorylation in endothelial cells. J Cell Sci. 1998;111:1853–65. doi: 10.1242/jcs.111.13.1853. [DOI] [PubMed] [Google Scholar]
- 5.Shasby DM, Ries DR, Shasby SS, Winter MC. Histamine stimulates phosphorylation of adherens junction proteins and alters their link to vimentin. Am J Physiol Lung Cell Mol Physiol. 2002;282:L1330–8. doi: 10.1152/ajplung.00329.2001. [DOI] [PubMed] [Google Scholar]
- 6.Yuan A, Mills RG, Chia CP, Bray JJ. Tubulin and neurofilament proteins are transported differently in axons of chicken motoneurons. Cell Mol Neurobiol. 2000;20:623–32. doi: 10.1023/A:1007090422866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Adamson RH, Zeng M, Adamson GN, Lenz JF, Curry FE. PAF- and bradykinin-induced hyperpermeability of rat venules is independent of actin-myosin contraction. Am J Physiol Heart Circ Physiol. 2003;285:H406–17. doi: 10.1152/ajpheart.00021.2003. [DOI] [PubMed] [Google Scholar]
- 8.Hatakeyama T, Pappas PJ, Hobson RW, 2nd, Boric MP, Sessa WC, Durán WN. Endothelial nitric oxide synthase regulates microvascular hyperpermeability in vivo. J Physiol. 2006;574:275–81. doi: 10.1113/jphysiol.2006.108175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sánchez FA, Kim DD, Durán RG, Meininger CJ, Durán WN. Internalization of eNOS via caveolae regulates PAF-induced inflammatory hyperpermeability to macromolecules. Am J Physiol Heart Circ Physiol. 2008;295:H1642–8. doi: 10.1152/ajpheart.00629.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Dimmeler S, Fleming I, Fisslthaler B, Hermann C, Busse R, Zeiher AM. Activation of nitric oxide synthase in endothelial cells by Akt-dependent phosphorylation. Nature. 1999;399:601–5. doi: 10.1038/21224. [DOI] [PubMed] [Google Scholar]
- 11.García-Cardeña G, Martasek P, Masters BS, Skidd PM, Couet J, Li S, et al. Dissecting the interaction between nitric oxide synthase (NOS) and caveolin. Functional significance of the nos caveolin binding domain in vivo. J Biol Chem. 1997;272:25437–40. doi: 10.1074/jbc.272.41.25437. [DOI] [PubMed] [Google Scholar]
- 12.Sánchez FA, Rana R, Kim DD, Iwahashi T, Zheng R, Lal BK, et al. Internalization of eNOS and NO delivery to subcellular targets determine agonist-induced hyperpermeability. Proc Natl Acad Sci U S A. 2009;106:6849–53. doi: 10.1073/pnas.0812694106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sánchez FA, Savalia NB, Durán RG, Lal BK, Boric MP, Durán WN. Functional significance of differential eNOS translocation. Am J Physiol Heart Circ Physiol. 2006;291:H1058–64. doi: 10.1152/ajpheart.00370.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sánchez FA, Rana R, González FG, Iwahashi T, Durán RG, Fulton DJ, et al. Functional significance of cytosolic endothelial nitric-oxide synthase (eNOS): regulation of hyperpermeability. J Biol Chem. 2011;286:30409–14. doi: 10.1074/jbc.M111.234294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chen K, Pittman RN, Popel AS. Nitric oxide in the vasculature: where does it come from and where does it go? A quantitative perspective. Antioxid Redox Signal. 2008;10:1185–98. doi: 10.1089/ars.2007.1959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Breslin JW, Sun H, Xu W, Rodarte C, Moy AB, Wu MH, et al. Involvement of ROCK-mediated endothelial tension development in neutrophil-stimulated microvascular leakage. Am J Physiol Heart Circ Physiol. 2006;290:H741–50. doi: 10.1152/ajpheart.00238.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Iwakiri Y, Satoh A, Chatterjee S, Toomre DK, Chalouni CM, Fulton D, et al. Nitric oxide synthase generates nitric oxide locally to regulate compartmentalized protein S-nitrosylation and protein trafficking. Proc Natl Acad Sci U S A. 2006;103:19777–82. doi: 10.1073/pnas.0605907103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ozawa K, Whalen EJ, Nelson CD, Mu Y, Hess DT, Lefkowitz RJ, et al. S-nitrosylation of beta-arrestin regulates beta-adrenergic receptor trafficking. Mol Cell. 2008;31:395–405. doi: 10.1016/j.molcel.2008.05.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Stamler JS, Simon DI, Jaraki O, Osborne JA, Francis S, Mullins M, et al. S-nitrosylation of tissue-type plasminogen activator confers vasodilatory and antiplatelet properties on the enzyme. Proc Natl Acad Sci U S A. 1992;89:8087–91. doi: 10.1073/pnas.89.17.8087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Stamler JS, Jaraki O, Osborne J, Simon DI, Keaney J, Vita J, et al. Nitric oxide circulates in mammalian plasma primarily as an S-nitroso adduct of serum albumin. Proc Natl Acad Sci U S A. 1992;89:7674–7. doi: 10.1073/pnas.89.16.7674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Whalen EJ, Foster MW, Matsumoto A, Ozawa K, Violin JD, Que LG, et al. Regulation of beta-adrenergic receptor signaling by S-nitrosylation of G-protein-coupled receptor kinase 2. Cell. 2007;129:511–22. doi: 10.1016/j.cell.2007.02.046. [DOI] [PubMed] [Google Scholar]
- 22.Pei DS, Song YJ, Yu HM, Hu WW, Du Y, Zhang GY. Exogenous nitric oxide negatively regulates c-Jun N-terminal kinase activation via inhibiting endogenous NO-induced S-nitrosylation during cerebral ischemia and reperfusion in rat hippocampus. J Neurochem. 2008;106:1952–63. doi: 10.1111/j.1471-4159.2008.05531.x. [DOI] [PubMed] [Google Scholar]
- 23.Vittet D, Prandini MH, Berthier R, Schweitzer A, Martin-Sisteron H, Uzan G, et al. Embryonic stem cells differentiate in vitro to endothelial cells through successive maturation steps. Blood. 1996;88:3424–31. [PubMed] [Google Scholar]
- 24.Carmeliet P, Lampugnani MG, Moons L, Breviario F, Compernolle V, Bono F, et al. Targeted deficiency or cytosolic truncation of the VE-cadherin gene in mice impairs VEGF-mediated endothelial survival and angiogenesis. Cell. 1999;98:147–57. doi: 10.1016/S0092-8674(00)81010-7. [DOI] [PubMed] [Google Scholar]
- 25.Caveda L, Martin-Padura I, Navarro P, Breviario F, Corada M, Gulino D, et al. Inhibition of cultured cell growth by vascular endothelial cadherin (cadherin-5/VE-cadherin) J Clin Invest. 1996;98:886–93. doi: 10.1172/JCI118870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Davis MA, Ireton RC, Reynolds AB. A core function for p120-catenin in cadherin turnover. J Cell Biol. 2003;163:525–34. doi: 10.1083/jcb.200307111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Xiao K, Allison DF, Kottke MD, Summers S, Sorescu GP, Faundez V, et al. Mechanisms of VE-cadherin processing and degradation in microvascular endothelial cells. J Biol Chem. 2003;278:19199–208. doi: 10.1074/jbc.M211746200. [DOI] [PubMed] [Google Scholar]
- 28.Xiao K, Garner J, Buckley KM, Vincent PA, Chiasson CM, Dejana E, et al. p120-Catenin regulates clathrin-dependent endocytosis of VE-cadherin. Mol Biol Cell. 2005;16:5141–51. doi: 10.1091/mbc.E05-05-0440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Andriopoulou P, Navarro P, Zanetti A, Lampugnani MG, Dejana E. Histamine induces tyrosine phosphorylation of endothelial cell-to-cell adherens junctions. Arterioscler Thromb Vasc Biol. 1999;19:2286–97. doi: 10.1161/01.ATV.19.10.2286. [DOI] [PubMed] [Google Scholar]
- 30.Cohen AW, Carbajal JM, Schaeffer RC., Jr. VEGF stimulates tyrosine phosphorylation of beta-catenin and small-pore endothelial barrier dysfunction. Am J Physiol. 1999;277:H2038–49. doi: 10.1152/ajpheart.1999.277.5.H2038. [DOI] [PubMed] [Google Scholar]
- 31.Alexander JS, Alexander BC, Eppihimer LA, Goodyear N, Haque R, Davis CP, et al. Inflammatory mediators induce sequestration of VE-cadherin in cultured human endothelial cells. Inflammation. 2000;24:99–113. doi: 10.1023/A:1007025325451. [DOI] [PubMed] [Google Scholar]
- 32.Aramoto H, Breslin JW, Pappas PJ, Hobson RW, 2nd, Durán WN. Vascular endothelial growth factor stimulates differential signaling pathways in in vivo microcirculation. Am J Physiol Heart Circ Physiol. 2004;287:H1590–8. doi: 10.1152/ajpheart.00767.2003. [DOI] [PubMed] [Google Scholar]
- 33.Kevil CG, Ohno N, Gute DC, Okayama N, Robinson SA, Chaney E, et al. Role of cadherin internalization in hydrogen peroxide-mediated endothelial permeability. Free Radic Biol Med. 1998;24:1015–22. doi: 10.1016/S0891-5849(97)00433-4. [DOI] [PubMed] [Google Scholar]
- 34.Thibeault S, Rautureau Y, Oubaha M, Faubert D, Wilkes BC, Delisle C, et al. S-nitrosylation of beta-catenin by eNOS-derived NO promotes VEGF-induced endothelial cell permeability. Mol Cell. 2010;39:468–76. doi: 10.1016/j.molcel.2010.07.013. [DOI] [PubMed] [Google Scholar]
- 35.Marín N, Zamorano P, Carrasco R, Mujica P, González FG, Quezada C, et al. S-Nitrosation of β-catenin and p120 catenin: a novel regulatory mechanism in endothelial hyperpermeability. Circ Res. 2012;111:553–63. doi: 10.1161/CIRCRESAHA.112.274548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Erwin PA, Lin AJ, Golan DE, Michel T. Receptor-regulated dynamic S-nitrosylation of endothelial nitric-oxide synthase in vascular endothelial cells. J Biol Chem. 2005;280:19888–94. doi: 10.1074/jbc.M413058200. [DOI] [PubMed] [Google Scholar]
- 37.Furchgott RF, Zawadzki JV. The obligatory role of endothelial cells in the relaxation of arterial smooth muscle by acetylcholine. Nature. 1980;288:373–6. doi: 10.1038/288373a0. [DOI] [PubMed] [Google Scholar]
- 38.Sayed N, Kim DD, Fioramonti X, Iwahashi T, Durán WN, Beuve A. Nitroglycerin-induced S-nitrosylation and desensitization of soluble guanylyl cyclase contribute to nitrate tolerance. Circ Res. 2008;103:606–14. doi: 10.1161/CIRCRESAHA.108.175133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Crassous PA, Couloubaly S, Huang C, Zhou Z, Baskaran P, Kim DD, et al. Soluble guanylyl cyclase is a target of angiotensin II-induced nitrosative stress in a hypertensive rat model. Am J Physiol Heart Circ Physiol. 2012;303:H597–604. doi: 10.1152/ajpheart.00138.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Delyani JA, Nossuli TO, Scalia R, Thomas G, Garvey DS, Lefer AM. S-nitrosylated tissue-type plasminogen activator protects against myocardial ischemia/reperfusion injury in cats: role of the endothelium. J Pharmacol Exp Ther. 1996;279:1174–80. [PubMed] [Google Scholar]
- 41.Matsushita K, Morrell CN, Cambien B, Yang SX, Yamakuchi M, Bao C, et al. Nitric oxide regulates exocytosis by S-nitrosylation of N-ethylmaleimide-sensitive factor. Cell. 2003;115:139–50. doi: 10.1016/S0092-8674(03)00803-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lim SY, Raftery M, Cai H, Hsu K, Yan WX, Hseih HL, et al. S-nitrosylated S100A8: novel anti-inflammatory properties. J Immunol. 2008;181:5627–36. doi: 10.4049/jimmunol.181.8.5627. [DOI] [PubMed] [Google Scholar]
- 43.Fukumura D, Gohongi T, Kadambi A, Izumi Y, Ang J, Yun CO, et al. Predominant role of endothelial nitric oxide synthase in vascular endothelial growth factor-induced angiogenesis and vascular permeability. Proc Natl Acad Sci U S A. 2001;98:2604–9. doi: 10.1073/pnas.041359198. [DOI] [PMC free article] [PubMed] [Google Scholar]