

Scientists have been investigating the critical role type 1 interferons (IFN-I) play in controlling viral infections for more than 50 y. Common pathogen motifs present on viral particles trigger intracellular and membrane-associated pattern recognition receptors, leading to the production of IFN-I and rapid expression of interferon stimulated genes (ISIGs).1 ISIGs encode proteins with direct antiviral activity, as well modulating multiple parameters of the immune response. However, in situations of persistent viral infections, such as HIV, although produced, IFN-Is are not able to quench the initial infection, which eventually establishes life-long persistence. In this situation, the chronic IFN-I mediated immune activation/inflammation that accompanies persistent HIV infection is strongly associated with disease progression, even in patients with antiretroviral therapy-suppressed viral titers.2 In fact, in the SIV model of primate infection, immune activation, CD4 T cell depletion, and disease progression are integrally correlated with IFN-I signaling regardless of virus titers.3-5 Thus, while the precise mechanisms driving immune activation in HIV infection still remain to be determined, there is mounting evidence implicating IFN-I. Excitingly, independent work from our laboratory and from Michael Oldstone’s laboratory using a mouse model of persistent virus infection demonstrated that prolonged IFN-I signaling is highly detrimental to the antiviral immune response, and that, astonishingly, quenching IFN-I signals results in control of infection.6,7 These studies indicate the important and somewhat counterintuitive possibility that blocking IFN-I signaling in persistent infection may be an effective therapy for controlling virus replication and halting disease progression.
Lymphocytic choriomeningitis virus (LCMV) in mice has been utilized for decades as an experimental model to explore immune responses to persistent infection. Using the LCMV system, the 2 groups established that a surprising amount of the immune dysfunctions associated with viral persistence are indeed due to prolonged IFN-I signaling. Chronic IFN-I signaling enhanced expression of activation markers on total T cell populations, caused functional suppression/depletion of virus-specific CD4+ T cells, and led to severe disruption of lymphoid tissue architecture. IFN-I also drove expression of multiple factors and cell types that inhibit antiviral immunity, such as the immunoregulatory cytokine IL-10, the inhibitory receptor PDL1, and immunoregulatory APC populations that co-express multiple suppressive factors.6-8 Blockade of IFN-I during persistent LCMV infection reversed the immune defects, diminished the expression of immunosuppressive molecules, and restored lymphoid tissue architecture. Consistent with the ongoing antiviral activity of IFN-I, blockade of IFN-I signaling initially increased virus titers, but then ultimately facilitated the control of the persistent infection. The exact mechanisms underlying the increased control of virus replication through IFN-I blockade are still under investigation and will likely be highly complex and multi-factorial, involving enhancements through the relief of chronic activation and a switch from an immunosuppressive to pro-immune environment, as well as other as-yet-undetermined mechanisms. As a first step in unraveling the mechanisms, initial experiments identified a need for CD4 T cells and IFNγ expression,6,7 but how they individually contribute to immune health and viral clearance in the absence of IFN-I is still unclear. Ultimately, these studies reveal a duality in IFN-I signaling, wherein strong initial interferon responses are necessary to directly inhibit virus replication and establish effective antiviral immune responses; however, prolonged IFN-I signals chronically activate the immune environment and induce tissue pathology and suppression of antiviral immunity. Thus, our studies have identified IFN-I signaling as the critical rheostat in an immunologic surveillance system that measures the duration and magnitude of virus replication (i.e., whether the host or pathogen is winning the battle) and then modulates the balance between pro- and anti-inflammatory immune programs accordingly.
Cumulatively, this data stimulates interest in potentially blocking IFN-I signaling to therapeutically resolve other persistent infections characterized by high levels of chronic immune activation. However, given the critical antiviral affects of IFN-I, future studies should focus on dissecting out the multiple functions of IFN-I (as well as the specific functions of individual IFNα’s and β) to preserve the beneficial antiviral properties while ablating the destructive and immunosuppressive effects. Whether or not this can be achieved remains to be determined, but if successful, it would represent a significant advance in our understanding of antiviral immunity and our potential to develop effective antiviral immunotherapies. (Fig. 1)
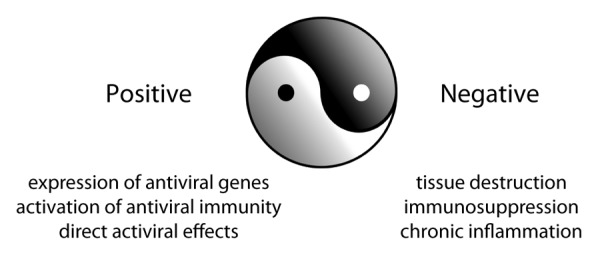
Figure 1. The yin-yang of type I interferon signaling. IFN-Is elicit seemingly opposite yet interrelated positive and negative influences on virus replication and dissemination. Throughout the course of viral infection IFN-I is constantly monitoring the virus: immune battle. Upon initial infection, the antiviral and immune-stimulatory (positive) effects of IFN-I predominate, but when virus replication persists due to the host’s inability to clear the infection the immunosuppressive (negative) effects of IFN-I take over, leading to many of the cellular and tissue dysfunctions associated with persistent virus infections and facilitating viral persistence.
Footnotes
Previously published online: www.landesbioscience.com/journals/cc/article/26175
References
- 1.Pitha PM, et al. Curr Top Microbiol Immunol. 2007;316:41–70. doi: 10.1007/978-3-540-71329-6_4. [DOI] [PubMed] [Google Scholar]
- 2.Moir S, et al. Annu Rev Pathol. 2011;6:223–48. doi: 10.1146/annurev-pathol-011110-130254. [DOI] [PubMed] [Google Scholar]
- 3.Jacquelin B, et al. J Clin Invest. 2009;119:3544–55. doi: 10.1172/JCI40093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bosinger SE, et al. J Clin Invest. 2009;119:3556–72. doi: 10.1172/JCI40115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Estes JD, et al. J Immunol. 2008;180:6798–807. doi: 10.4049/jimmunol.180.10.6798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wilson EB, et al. Science. 2013;340:202–7. doi: 10.1126/science.1235208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Teijaro JR, et al. Science. 2013;340:207–11. doi: 10.1126/science.1235214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wilson EB, et al. Cell Host Microbe. 2012;11:481–91. doi: 10.1016/j.chom.2012.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]