

Abstract
It is widely accepted that anti-apoptotic Bcl-2 family members promote cancer cell survival by binding to their pro-apoptotic counterparts, thereby preventing mitochondrial outer membrane permeabilization (MOMP) and cytotoxic caspase activation. Yet, these proteins do not only function as guardians of mitochondrial permeability, preserving it, and maintaining cell survival in the face of acute or chronic stress, they also regulate non-apoptotic functions of caspases and biological processes beyond MOMP from diverse subcellular localizations and in complex with numerous binding partners outside of the Bcl-2 family. In particular, some of the non-canonical effects and functions of Bcl-2 homologs lead to an interplay with E2F-1, NFκB, and Myc transcriptional pathways, which themselves influence cancer cell growth and survival. We thus propose that, by feedback loops that we currently have only hints of, Bcl-2 proteins may act as rulers of survival signaling, predetermining the apoptotic threshold that they also directly scaffold. This underscores the robustness of the control exerted by Bcl-2 homologs over cancer cell survival, and implies that small molecules compounds currently used in the clinic to inhibit their mitochondrial activity may be not always be fully efficient to override this control.
Keywords: Bcl-2 family, E2F-1, Myc, NFκB, caspase
Canonical Function of Bcl-2 Homologs: Direct Inhibition of “Death by MOMP”
Apoptosis is a form of cell death that relies on the activation of executioner caspases (such as caspase 3) downstream of two interconnected signaling pathways. The “extrinsic pathway” is initiated when death ligands engage receptors of the TNFR family and promote activation of initiator caspases upstream of the executioner ones. The “intrinsic pathway” (or mitochondrial pathway) is triggered by internal stress signals (in addition to these induced by death receptors), that lead to mitochondrial outer membrane permeabilisation (MOMP, characterized by the release of proteins such as cytochrome c from the mitochondrial intermembrane space) and subsequent activation of executioners caspases as the result of the formation of the apoptosome, a multi-protein complex encompassing the adaptor protein Apaf-1.1
MOMP is critically regulated by members of the Bcl-2 family. This family is composed of pro- and anti-apoptotic proteins sharing at least one Bcl-2 Homology (BH) domain in common with Bcl-2 (Fig. 1) and the balance between the antagonistic activities of these proteins determines MOMP and cell death decisions. This balance is frequently tipped toward survival in cancer cells, and switching it back by interfering with the survival function(s) of Bcl-2 family members is a highly promising anticancer approach.2 There is thus a need to understand how Bcl-2 homologs regulate survival, and how shrewd inhibition of their activities promotes cell death, on a mechanistic level.

Figure 1. Regulation of transcriptional pathways as an output of non canonical effects and functions of Bcl-2 homologs. (A) Bcl-2 homologs (Bcl-2 hom.) exert their well recognized, canonical anti-apoptotic function by preventing BH3 only protein activation of multidomain proteins (such as Bax), and subsequent MOMP and induction of cell death following caspase activation. These proteins also exert other biological effects (B), due to non apoptotic effects of caspase activity, and/or regulation, among others, of mitochondrial calcium uptake and bioenergetics, ER calcium dynamics, autophagy, or DNA repair by pools of proteins that localize at distinct subcellular compartments and that interact with numerous factors (BF, binding factors). Some of these non canonical effects allow Bcl-2 homologs to regulate transcriptional activities that themselves impact on survivial signaling, enforcing the control exerted by Bcl-2 homologs over cell death.
There is considerable evidence that the main mechanism through which anti-apoptotic proteins (herein after named Bcl-2 homologs, of which Bcl-2, Bcl-xL, and Mcl-1 are the best studied members) promote survival is by interacting with their proapoptotic counterparts. Pro-apoptotic BH3-only proteins act as sensors of death signals, and they integrate them into the activation of pro-apoptotic multidomain proteins (that harbor BH1, 2, and 3) such as Bax and Bak, which triggers MOMP by inserting into mitochondrial membranes. Anti-apoptotic proteins directly inhibit MOMP by preventing Bax/Bak activation and activity, due to the ability of a hydrophobic groove formed at their surface by their BH1, BH2, and BH3 domains to bind to the BH3-domain of pro-apoptotic proteins.2
In most cases, BH3 dependent complexes localize at mitochondrial membranes. It is thus conceived that Bcl-2 and its homologs are anti-apoptotic proteins because they maintain survival as “mitochondrial proteins that bind to BH3 domains” (a phrase which defines their canonical function) and that, reciprocally, inhibition of their BH3 binding activity triggers a all-or-nothing cell death response downstream of MOMP (Fig. 1A). Very good evidence support this view. The occupation state of Bcl-2 homologs by pro-apoptotic counterparts at the mitochondrial membrane of cancer cells correlates with their sensitivity to a variety of pro-apoptotic stimuli. Assessing this occupation state by measuring MOMP in response to an array of distinct BH3 peptides (that inhibit specific anti/pro-apoptotic interactions and/or activate Bax/Bak directly) by a BH3-profiling assay allows evaluation of the reliance of cancer cells on specific Bcl-2 homologs for survival, measurement of their apoptotic threshold, and prediction of their response to therapy.3,4 Moreover, small molecule inhibitors of the BH3 binding activity of Bcl-2, Bcl-xL, and Mcl-1 (BH3-mimetics such as ABT-199 that inhibits Bcl-2, WEHI-539 that inhibits Bcl-xL, ABT-737, and ABT-263 that dually inhibit Bcl-2 and Bcl-xL and MIM-1 that inhibits Mcl-1) have been reported (see ref. 2 for a review). When added to cells that are dependent on their target(s) for survival, these compounds trigger MOMP dependent death by on-target effects. Yet, numerous data, as exposed below, indicate that the biological effects of Bcl-2 homologs extends beyond their regulation of BH3-dependent, MOMP-induced cell death. In particular, some of the non canonical effects and functions of Bcl-2 homologs lead them to interplay with E2F-1, NF-κB, and Myc transcriptional pathways through which Bcl-2 homologs exert feedback control over cancer cell growth and survival.
Beyond the Regulation of Apoptotic Mitochondrial Permeabilization: General Rules for Non Canonical Functions
The generally accepted (and most likely veracious) view of regulation of survival by Bcl-2 homologs needs to be refined on some aspects. There is increasing evidence that Bcl-2 homologs do not only localize to mitochondrial outer membranes, and that they modulate numerous non apoptotic processes that involve caspases and/or direct interactions with proteins that do not belong to the Bcl-2 family (Fig. 1B).
Caspase activation downstream of MOMP does not always kill
The first, and presumably less intuitive aspect, is that features of mitochondrial permeabilisation may not implacably lead to cell demise. MOMP implies a series of steps that range from alterations of outer membrane permeability, actual release of proteins in the cytosol and activation of the apoptosome. Each of these steps may be reached without the cell being committed to die, as long as metabolic pathways are not irreversibly altered below a critical threshold.1 Tait, Green, and colleagues showed that subpopulations of mitochondria may not undergo MOMP in a cell receiving apoptotic stimuli.5 Importantly, inhibition of caspases contributes to the lack of a complete MOMP, which itself correlates with long-term survival. Thus, a first wave of partial mitochondrial “damage” may lead to caspase activation, which will itself kill cells only when more mitochondria have undergone permeabilisation. By inference, caspases may regulate biological processes beyond cell death itself in a mammalian cell that is not committed to die (Fig. 1B).
There is evidence that independently from induction of apoptosis, low levels of caspase 3 activity, for instance, regulate T and B cell homeostasis, cell motility, diverse differentiations processes and even lead to the activation of the survival kinase Akt (ref. 6 and references therein). The data of Tait, Green, and colleagues raise the possibility that such functions may be triggered downstream of inhibition of Bcl-2 homologs, and/or of a partial MOMP. Recent reports by Kraft and colleagues and by our own laboratory bring support to this view. Song, Kraft and colleagues showed that treatment with ABT-737 of cells that are resistant to induction of cell death by the compound lead to the induction of senescence instead.7 This phenotype of senescence resulted from low level of caspase activity that was insufficient to kill the cells but that triggered DNA damage (and NF-κB activity as discussed below). As reactive oxygen species (ROS) were shown to contribute to caspase activation under these conditions, mild mitochondrial damage might play a role even if the involvement of a partial MOMP, and the contribution of the apoptosome, in this process of caspase activation was not directly established. Moreover, we ourselves recently demonstrated that caspase dependent transcriptional induction of the BH3-only protein Noxa (by E2F-1 as discussed below) in response to ABT-737 accounted for the delayed induction of cell death by the compound.8 Induction of such a transcriptional program relied on an on-target effect of ABT-737, as it was recapitulated by the knock down of both Bcl-2 and Bcl-xL. This process was Bax dependent, strongly suggesting (if not demonstrating) that it might ensue from MOMP. Taken together, these data imply that an elaborate, caspase dependent cell response may be triggered by inhibition of some Bcl-2 homologs, in response to acute stress or when BH3 mimetics are used in a clinical setting. It is still uncertain whether such a caspase-induced response is constitutively active under physiological conditions (in the absence of overt stress), and if increasing levels of Bcl-2 homologs (as happens in cancer cells) would shut it down.
Bcl-2 proteins regulate more than MOMP
Bcl-2 homologs do not only regulate apoptotic mitochondrial permeabilisation, but other cellular functions. The best example is the regulation of intracellular calcium homeostasis by Bcl-2 proteins, that has expected consequences on many processes relying on calcium dependent proteins, such as lymphocyte activation, embryonic development, neural plasticity, cell migration or cell invasion.9 This might account for the effects of Bcl-2 proteins on the transcriptional activity of NFAT, which is regulated by the calcium dependent phosphatase calcineurin.10 The regulation by Bcl-2 homologs of calcium homeostasis result from complex, and interdependent effects on mitochondrial calcium uptake and on calcium exchange at the ER membranes9,11 via the regulations (among others) of: VDAC1 (a porin that exchanges metabolites and ATP between cytosol and mitochondria and Ca2+ ions entry from ER into the mitochondria) ; the mitochondrial Na+/Ca2 + exchanger; the sarco/ER Ca2+-ATPase (SERCA); the inositol 1,4,5-trisphosphate receptor (IP3R) and IP3 induced calcium release (IICR); Bax Inhibitor 1 (an ER-localized protein thought to control ER Calcium homeostasis by its H+/Ca2+ antiporter activity)9,12,13 (Fig. 1B).
Bcl-2 homologs may exert a general effect on bioenergetics by regulating constitutive IP3R dependent calcium exchanges between the ER and the mitochondria11 that are essential for the production of ATP through oxidative phosphorylation.14 Yet they may also regulate bioenergetics independently from calcium homeostasis. Bcl-xL prevents Bad from exerting metabolic functions as part of a mitochondrial multiprotein complex involving active glucokinase.15 By modulating VDAC opening, Bcl-xL may promote the exchange of metabolites, including ADP, across the outer membrane. Even more directly, Bcl-xL might increase the activity of the inner membrane F1F0 ATP synthase, thereby stabilizing the inner membrane potential and optimizing mitochondrial ATP production, an effect it shares with Mcl-1.16-18 Global analysis of the metabolic effects of Bcl-xL, by NMR and mass spectrometry, identified a reduction of intracellular levels of acetyl-Coa, and of glucose-derived citrate in Bcl-xL overexpressing cells. The reason for this decrease is unknown but it may owe to modifications of mitochondrial membrane permeability and is independent of Bax/Bak. As acetyl-coA is required for protein acetylation, including N-α-acetylation, Bcl-xL may by this effect influence the post-translational modifications of a large number of cellular proteins, including that of caspase 2, 3, and 919.
Bcl-2 homologs also influence catabolic pathways that rely on autophagy. As they prevent Beclin from contributing to the formation of a multicomplex that controls class III PI3K activity, they interfere with the induction of autophagy by numerous stresses.20 Moreover, they might also regulate autophagy as a consequence of their ability to control ER/mitochondrial exchanges, since suppressing these exchanges causes the activation of AMP-activated kinase (AMPK) and subsequent induction of autophagy.14 Reduction of mTOR signaling downstream of activated AMPK may have important consequences on protein expression profiles and on Bcl-2 family members themselves: Mcl-1 expression is exquisitely sensitive to changes in cap-dependent translation downstream of mTOR activity.21
Bcl-2 proteins do not interact with mitochondrial membranes only
Some of the effects described above are exerted by pools of Bcl-2 that are not localized at the mitochondrial outer membrane (Fig. 1B). Bcl-2 homologs carry an hydrophobic C-terminal end that, when exposed (otherwise, Bcl-2 proteins remain in the cytosol) functions as a membrane anchor and allows insertion within subcellular membranes, such as outer mitochondrial membranes but also ER or perinuclear membranes.22 Regulation of Beclin, SERCA and IP3R activities, is understood to be exerted by Bcl-2 proteins that localize at the ER. In fact, localization of Bcl-2 proteins at the mitochondria-associated ER membranes (MAMs) would seem logical, as these structures that physically link mitochondria and ER are involved in calcium exchanges between the two organelles and contain interorganellar multi-protein complexes that precisely involve, among other proteins, IP3Rs, and VDACs.23
Proper translocation of Bcl-2 and Bcl-xL appears to result from the activity of molecular chaperones such as the inherent calcineurin inhibitor FKBP38,24 whose absence results in Bcl-2/Bcl-xL mislocalization and affects cell survival.25 It has been suggested that, when free from FKBP38, Bcl-2 is redirected to the nuclear membrane and block transcription factors transport into the nucleus.26,27 This would predict a general decrease in transcriptional activities when Bcl-2 accumulates at the nuclear membranes, which has not been confirmed experimentally in subsequent studies.
Other, more intriguing, subcellular localizations of Bcl-2 proteins have been reported. The regulation of F1FO ATP synthase activity has been ascribed to pools of Bcl-xL and Mcl-1 residing inside mitochondria. Matrix localization of Mcl-1 might be due to the presence of a mitochondrial targeting sequence at its N-terminal end,18 but the mechanisms that would allow Bcl-xL to share this localization remain elusive. The presence of Bcl-2 proteins inside the nucleus has also been reported (see below). This would involve specific interactions with nuclear targeted cofactors, as Bcl-2 homologs do not, to the best of our knowledge, express a nuclear localization signal themselves.
Bcl-2 proteins do not interact with proapoptotic Bcl-2 family members only
Bcl-2 proteins not only have various subcellular localizations, but also diverse binding partners outside of the Bcl-2 family. Interactions of Bcl-2 and Bcl-xL with long lists of proteins have been documented,28 even though it should be noted that these interactions have not always been validated by multiple groups, and that analogous interactions with homologs such as Bcl-w, Mcl-1 or Bfl-1 have not been systematically investigated. Many of these interactions have been studied on the basis of their effects on Bcl-2 homologs, modulating their subcellular localization and/or their canonical functions by post-translational modifications, including protease cleavage, phosphorylation /dephopshorylation, ubiquitinylation (a critical regulatory step for Mcl-1 which is a very labile protein) and conformational changes in the ternary structure of the proteins. Some of them, however, account for non canonical effects of Bcl-2 proteins in that they reciprocally impact on the function of the binding partner (Fig. 1B). With the exception of the regulation of mitochondrial glucokinase activity (which occurs via binding to the BH3-only protein Bad), most of the effects reported in the paragraph above might ensue from reverse regulation of their binding partners by Bcl-2.
The question of the binding interface(s) involved under these situations is apposite as it is critical to understand whether such reverse regulations compete with canonical regulation of apoptosis by Bcl-2 proteins, whether they can be modulated by BH3 mimetics used in the clinic or whether they occur independently from the BH3 binding activity of Bcl-2 proteins. Interaction with Beclin relies on a domain within this protein that has clearly been validated as a BH3 domain.20 This feature might extend to the interaction with the cell cycle checkpoint protein Rad9. Likewise, Bcl-xL’s effect on IP3R sensitization to IP3 was suggested to result from its binding to the C-terminal end of IP3R, a region that displays motifs similar to that a BH3 domain.11 Additional interactions were suggested to rely on the existence of a cryptic BH3 domain in the binding partners of Bcl-2 homologs.28 Yet, this is not systematical and there clearly are additional binding interfaces at the surface of Bcl-2 proteins. Proteolytic cleavage of these proteins, their phosphorylation, and their interaction with the corresponding kinases involve a flexible loop that links their first helix (which contains the BH4 domain) to the rest of the protein (including its BH3 binding cleft and its C-terminal membrane anchor). The BH4 domain itself has been involved in numerous interactions. Bcl-2 binds to the central, modulatory domain of the IP3R via its BH4 domain to regulate its opening. This is not recapitulated by the BH4 domain of Bcl-xL, underscoring the structural differences between Bcl- 2 and Bcl-XL for IP3R binding and regulation.29 BH4 domains were also involved in regulatory interactions with VDACs and Bax inhibitor 1 and are thus considered to play a role in the general effects of Bcl-2 proteins on calcium homeostasis.11 This BH4 domain is also involved in the interaction between Bcl-2 and Bcl-xL with FKBP38, c-MYC, and NF-κB subunits and therefore may contribute to their ability to regulate transcriptional events (see below).
Taken altogether, available data (that are impossible to list exhaustively here) indicate that numerous binding interfaces in Bcl-2 proteins allow them to regulate calcium dynamics, metabolic pathways but also DNA repair30 and synthesis,31 mitotic progression,32 or response to proinflammatory stimuli (by negatively regulating NLRP1, a key component of the inflammasome required for caspase-1 activation, via their flexible loop33,34).
Implications
The pleiotropic effects of Bcl-2 family members evoked above suggest that they would influence transcriptional programs exquisitely sensitive to intracellular calcium or ATP changes, or to protein modifications that Bcl-2 proteins regulate. Superimposed on this, Bcl-2 proteins may directly impact on transcriptional activities by impairing caspase dependent regulation of these transcriptional activities and/or by specifically interacting with upstream kinases and with transcription factors themselves. This is the case for the E2F-1, NF-κB, and Myc pathways. Bcl-2 homologs are connected by numerous, and tight, links to these pathways (see below) as a consequence of their non canonical effects (on the regulation of sublethal caspase activity) and functions. This seems to us of particular importance as they allow Bcl-2 proteins to exert feedback control on signals involved in cancer cell survival and growth. Of note, the functional relationship between p53 and Bcl-2 proteins35 does not seem to fall into the same category. p53 is a tumor suppressor activated by DNA damage conditions, redox and oncogenic stress that functions both as a transcription factor and as a cytoplasmic protein. p53 was reported to interact with Bcl-2 proteins at the mitochondria. Functionally, this interaction is understood to influence the canonical function of Bcl-2 proteins but reversely, Bcl-2 proteins may interfere with the effects on bioenergetics and redox metabolism of cytoplasmic p53.36 Mitochondrial interactions may also contribute to sequester active p53 away from the nucleus,37 yet, and to the best of our knowledge, there is no firm evidence to date that Bcl-2 proteins regulate the transcriptional activity of p53 (and/or its ability to regulate necrotic cell death38) as they regulate that of E2F-1, Myc or NF-κB .
Specific Regulation of Transcriptional Pathways by Bcl-2 Proteins
Inhibition of E2F-1 activity
E2F-1 belongs to the E2F family of transcription factors that play a key role in cell proliferation by inducing the transcription of genes crucially involved in the transition from G0/G1 phases into S phase and in S phase progression.39 The transactivation of cell cycle genes by E2F-1 is tightly regulated by the phosphorylation of the retinoblastoma protein pRb by the cyclin dependant kinases (Cdks). Binding of the hypophosphorylated pRb to E2F1 inhibits its ability to induce the expression of cell cycle genes while the phosphorylation of pRb by the Cdks releases E2F-1 that transcriptionally activate the corresponding promoters. E2F-1 differs from other E2F family members as it also regulates apoptosis by directly inducing the expression of proapoptotic genes (e.g., caspase 3 and 7, some pro-apoptotic Bcl-2 family members, p73) 40 and by, adversely, favoring that of genes involved in anticancer drug resistance (ABC transporters, Bcl-2 itself) via a p73/DNp73-miR-205 pathway 41. In contrast to what happens with cell cycle related genes, pRb (whether it is phosphorylated or not) participates to a transcriptionally active complex with E2F-1 to regulate the expression of pro-apoptotic genes, and thus favors the expression of these genes together with E2F-1.42
The first link between the pRb/E2F-1 pathway and Bcl-2 homologs was inferred from early reports of cell cycle effects of the latter proteins.43,44 Cell cycle entry was found delayed in Bcl-2 and Bcl-xL overexpressing cells45,46 while lack of Bcl-2 in lymphocytes resulted in a shortened G1 phase.47 Consistently, hematopoietic tumors overexpressing Bcl-2 were found to contain a high percentage of cells in a resting prolonged G1 phase. The cell cycle function of Bcl-2 may be evolutionary conserved, as ectopic expression of the pro-survival Bcl-2 related protein Buffy in Drosophila similarly results in a G1/early S phase arrest.48
There is no clear consensus about the mechanisms involved in the cell cycle effects of Bcl-2 homologs, and distinct mechanisms might intervene. Yet, they arguably converge toward the pRb/E2F-1 pathway (Fig. 2A). Bcl-2 overexpression was associated with a dephosphorylation of pRb44 and a modification of the composition of E2F/pRb family protein complexes.49 Mechanistically, Bcl-2 was shown to increase expression of the cyclin dependent kinase inhibitor (CKI) p2747 and it was shown neither to impact on the cell cycle progression of p27−/− T cells50 nor on that of p27−/− mouse embryonic fibroblasts51 (Fig. 2A). The modulation of cell cycle in Bcl-2 overexpressing fibroblasts was also reported to rely on the induction of the p130 protein, in addition to that of p27, leading to the formation of repressive E2F4-p130 complexes that may inhibit E2F150 (Fig. 2A). The mechanisms by which Bcl-2 and Bcl-xL lead to the induction of p27 and to p130 still remain to be clarified. The negative regulation of NFAT by Bcl-2/Bcl-xL was suggested to play a role in the induction of p27.47 Other data have suggested that Bcl-2 and Bcl-xL might control p27 level by inducing indirectly its phosphorylation at Ser10 promoting its stabilization.52
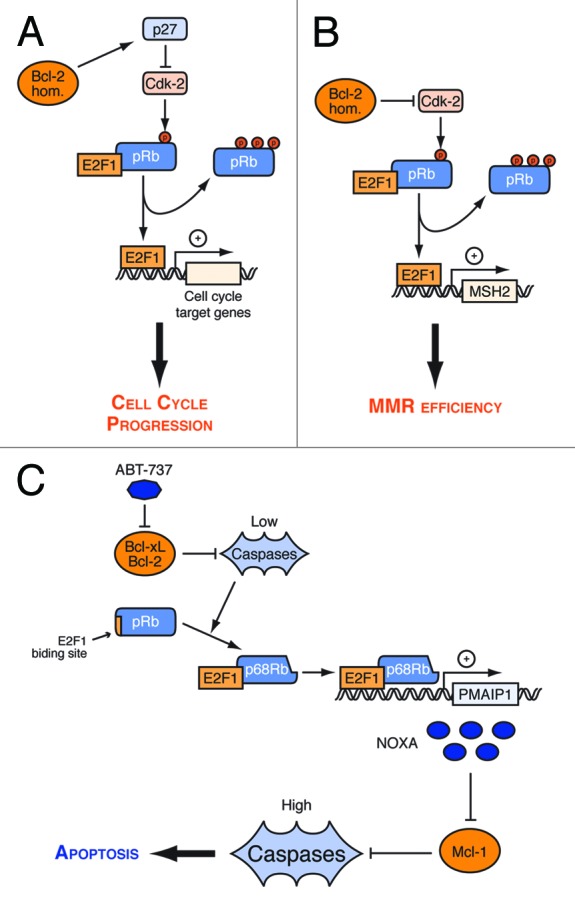
Figure 2. Regulation by Bcl-2 homologs of cell cycle entry, MMR, and apoptosis through the pRb/E2F1 pathway. (A) Bcl-2 homologs (Bcl-2 hom) dependent p27 upregulation inhibits Cdk2. Thus, they prevent pRb phosphorylation and the induction E2F-1 cell cycle targets, thereby delaying cell cycle progression. (B) Bcl-2 directly interacts with Cdk2 and inhibits it, causing increased levels of E2F-pRb complexes, and decrease in hMSH2 expression, thereby mediating suppression of MMR activity. (C) Inhibition of Bcl-2/Bcl-xL induces caspase cleavages of pRb that contributes with E2F-1 to enhance the expression of Noxa, an inhbitor of Mcl-1. As a result, inhibition of Mcl-1 is coupled to that of Bcl-2/Bcl-xL, providing a caspase amplificatory loop that enhances apoptosis.
Bcl-2 may also impact on E2F-1 activity by interacting directly with the catalytic subunit of Cdk2, leading to the inhibition of its activity, to the reduction of pRb phosphorylation and to the formation of inhibitory E2F1-pRb complexes (Fig. 2B). Under these conditions, the regulation of E2F-1 activity by Bcl-2 (and Bcl-xL that also interacts with Cdk2) not only impacts cell cycle progression but also impairs DNA mismatch repair (MMR)53 (Fig. 2). The binding of E2F-1 on the hMSH2 promoter was indeed described to be reduced in cells that overexpressed Bcl-2 or Bcl-xL, resulting to a decrease of hMSH2 expression, a key actor of DNA mismatch repair. Thus, the inhibition of E2F-1 transcriptional activity by Bcl-2/Bcl-xL via inhibition of Cdk2 may contribute to the inhibition of MMR pathways and increase genomic instability. Of note Bcl-xL was also reported to play a role in DNA damage induced G2 checkpoint by directly interacting with Cdk1 and locally inhibiting its activity in DNA damaged cells 54. Inhibition of Cdk1 by Bcl-xL has expected consequences on the Bcl-2 network (and on cell death decisions), as Cdk1 phosphorylates the BH3-only protein Bim 55.
A point mutation in the BH4-domain of Bcl-2 at the residue tyrosine 28 affects its effects on cell cycle and on MMR while sparing its anti-apoptotic function53,56 Thus, Bcl-2 and Bcl-xL may impact on E2F-1 activity independently from their role in cell survival. However, our recent data indicate that Bcl-2 and Bcl-xL also modulate E2F1 activity by negatively regulating caspase activity.8 In glioblastoma and breast cancer cells that express pRb, we showed that inhibition of the BH3 binding activity of Bcl-2 and Bcl-xL by ABT-737 treatment induced the expression of Noxa, as had been reported previously in another study.7We showed that this induction was Bax and caspase dependent, and that it relied on an increase of E2F-1 activity on the promoter of the PMAIP1 gene (encoding Noxa). pRb was cleaved by caspases upon ABT-737, giving rise to two truncated forms, p48Rb and p68Rb. Cleavages of pRb by caspase 3 and 7 were previously reported to trigger apoptosis but were thought to promote E2F-1 transcriptional activity by inducing the release of E2F-1 from pRb.57 We found that, instead, p68Rb kept its ability to bind E2F-1 and was present in the chromatin fraction of ABT-737 treated cells. Moreover, caspase inhibition prevented the recruitment of pRb on the noxa promoter upon ABT-737 treatment, while ectopic expression of p68Rb was sufficient to induce Noxa, arguing for an active role of p68Rb in the E2F-1 dependent induction of Noxa by Bcl-2/Bcl-xL inhibition. Noxa promotes apoptosis by selectively inhibiting Mcl-1, and in cells that express Bcl-2, Bcl-xL and Mcl-1, inhibition of these three survival proteins is necessary for full blown apoptosis. This may account for the requirement for Noxa induction for efficient induction of cell death by ABT-737 in our studies (Fig. 2C). They imply that, via their caspase dependent regulation of E2F-1 activity, two Bcl-2 homologs (Bcl-2 and Bcl-xL) control the activity of a third one (Mcl-1), thereby reinforcing survival. Conversely, as the coordination between inhibition of Mcl-1 and that of Bcl-2/Bcl-xL put forth by these studies is expected to render cells particularly sensitive to stress, it might increase the selective pressure to loose expression of pRb (which is a major actor of this coordination) in cancer cells, and uncouple regulation of Mcl-1 activity from that of Bcl2/Bcl-xL.
Equivocal regulation of NF-κB
NF-κB was initially described as a major regulator both in adaptive and innate immunity systems58.Five proteins compose the NF-κB subunit family in mammals: RELA (p65), RELB, REL (c-Rel) NF-κB1 (p105 that is processed in active DNA-binding form, p50) and NF-κB2 (p100 that is processed in active DNA-binding form, p52). NF-κB commonly refers to a p50-RelA heterodimer, which is one of the most avidly forming dimers and is the major Rel/NF-κB complex in most cells59 (Fig. 3A). To ensure rapid NF-κB activation, NF-κB complexes exist as a pre-synthesized form, ready for activation yet sequestered in the cytoplasm by binding to Inhibitors of NF-κB proteins (IκBα, IκBβ and IκBε). The rapid and transient activation of NF-κB is induced by the phosphorylation of IκBs, promoting their ubiquitylation and proteasome-mediated degradation with consequent NF-κB nuclear localization and activation of transcriptional activity.60 IKKβ is largely responsible for signal-induced phosphorylation and the subsequent degradation of IκBα, leading to the induction of the canonical pathway of p50-RELA complexes.

Figure 3. Context dependent regulation of NF-κB activity by Bcl-2 homologs. (A) Bcl-2 homologs (Bcl-2 hom.) contribute to the survival of growth factor deprived cells and to that of cells stimulated by ligands of the TNFR family (such as FasL and TNFα) by inhibiting caspase cleavage of IKKβ, IκBα and NF-κB subunits. (B) Bcl-2 homologs also favor constitutive NF-κB signaling, by activating IKKβ and/or promoting the phosphorylation-induced degradation of IκBα independenlty from caspases. This favors tumor progression as it results in the induction of pro-metastatic and pro-angiogenic genes. (C) In stressed cells, Bcl-2 homologs prevent the induction of pro-apoptotic and pro-senescence genes by NF-κB activated by ROS, sublethal caspase activities and DNA damage. They do so by inhibiting these events, as revealed upon ABT-737 treatment, or by binding to NF-κB subunits. See text for further details.
NF-κB exerts an anti-apoptotic effect as it induces many anti-apoptotic genes including some of the Bcl-2 family. Nuclear translocation of RELA/p65 is associated with increased expression of prosurvival Bcl-2 family members such as BCL2, BLC2L1, or BCL2A1.61 There is evidence that Bcl-2 family proteins reversely regulate NF-κB transcriptional activity, by mediating caspase cleavage of NF-κB, by modulating NF-κB localization or of its regulators or by favoring phosphorylation of NF-κB regulators.
Caspases activated downstream of Bcl-2 homologs inhibition cannot only cleave pRb but also, numerous transcription factors including the p65 and the p50 subunits of NF-κB.62,63 Apoptosis of activated T cells in response to stimulation of CD95/Fas was associated with the repression of NF-κB by caspase cleavage of these subunits.62 p65 was also reported to be cleaved by caspases in response to growth factor deprivation63. In that case, caspase cleavage gives rise to a truncated form that acts as a dominant negative inhibitor, thereby constituting a feedback mechanism between caspase activation and NF-κB inhibition that could amplify the pro-apoptotic effects of otherwise sublethal caspase activity. Moreover, IKKβ and IκBα can both be cleaved by caspases and their cleavages lead to the inactivation of NF-κB transcriptional activity (Fig. 3A). This process tips the balance toward death upon TNF-α treatment (which induces both caspase activities and compensatory NF-κB activation).64
The above data imply that Bcl-2 homologs might contribute to maintain the survival activity NF- κB exerts in cells treated with ligands of the TNFR family and/or upon growth factor deprivation (Fig. 3A) by regulating caspases. Bcl-2 homologs may also favor constitutive NF-κB activity independently from caspases (Fig. 3B). This has important consequences with respect to tumor progression, as NF-κB is understood to influence proliferation, cell adhesion and the cellular microenvironment under these conditions65,66. Bcl-xL overexpression was reported to contribute to the angiogenesis of glioblastoma and melanoma by impacting on NF-κB DNA binding to the promoter of IL8.67,68 Likewise, Bcl-2 overexpression was reported to decrease the expression of IκBα, enhancing NF-κB dependent expression of the metalloproteinanse MMP9 in human MCF-7 breast cancer cells and contributing to human breast cancer-cell metastatic potential.69 Such regulation also occurs in primary human dermal microvascular endothelial cells (HDMEC), where Bcl-xL overexpression induces vascular endothelial growth factor (VEGF) secretion, through activation of MAPK, promoting CXCL1, and IL8 expression and that of Bcl-2.70 The mechanisms involved remain elusive. Bcl-xL overexpression in glioblastoma cells was found to increase IκBα phosphorylation.68 The kinase involved was not identified but the identification of an interaction between Bcl-2 and the Raf-1 kinase suggest a possible role of the Ras/Raf-1/ERK pathway.71 Bcl-2-induced loss of IκBα and increase of NF-kB DNA binding was impaired by a kinase-defective Raf-1 and a kinase-defective MEKK1.72 The deletion of the BH4 domain, by which Bcl-2 interacts with the Raf-1 kinase, abolishes the ability of Bcl-2 to induce degradation of IκBα.73 Altogether these data suggest that Bcl-2 homologs may signal the Raf-1/MEKK pathway via its N-terminal BH4-domain activating IKKβ leading to the phosphorylation-induced degradation of IκBα and thereby to the activation of NF-κB. Further characterization of the molecular interactions between Bcl-2 homologs and elements of the Ras/Raf-1/ERK pathway, and investigations of their consequences on cellular signaling are required to understand how Bcl-2 homologs favor NF-κB signals in more detail.
The relationship between Bcl-2 homologs and NF-κB is ambiguous. Indeed, NF-κB promotes apoptosis or senescence in response to stress74 and under these conditions, Bcl-2 homologs might prevent its activity (Fig. 3C). Hour and colleagues reported that the overexpression of Bcl-2 delays the degradation of IκBα in the cytoplasm, affects the DNA-binding of NF-κB and inhibits the expression of its downstream target c-Myc to prevent cell death in S-nitrosoglutathione treated cells.75 In response to peroxidative damage, Bcl-2 was found associated with the inactive NF-κB complex (p65, p50, IκBα) in the cytoplasm and bound to the p50 subunit in the nuclei, even though the mechanism involved in these interactions remain obscure. In the same line, high levels of Bcl-2 were reported to lead to its accumulation at the nuclear membrane and result in a decrease in the activity of several transcription factors including NF-κB.26 This effect relied on the C-terminal end of Bcl-2, suggesting the involvement of subcellular membrane anchoring, but not on its BH4 domain. Finally, Bcl-2 may inhibit NF-κB activation by sublethal caspase activity. Kraft and colleagues reported NF-κB dependent upregulation of the expression of the Death Receptor 5 (DR5) upon ABT-737 treatment (and thus sensitization to induction of apoptosis by its cognate ligand TRAIL)76 by a process that involves induction of ROS. The same authors reported that activated NF-κB contributed to modify the secretory phenotype of cancer cells undergoing caspase-induced senescence in response to ABT-737.7 Several approaches have shown that NF-κB is involved in the secretion of pro-inflammatory mediators and by cells undergoing senescence.77,78 This senescence-associated secretory phenotype (SASP) can be deleterious or beneficial to the cell as it can either enforce senescence and recruit immune cells to clear senescent cells or instead promote malignancy by favoring cell proliferation, angiogenesis, epithelial-to-mesenchymal transition (EMT) and invasiveness.79 This implies that inhibition of the canonical function of Bcl-2 homologs (as regulators of caspases) may exert detrimental effects when it fails to induce full blown apoptosis in cells that can escape from senescence.
These last observations underscore the complexity of the functional interplay between Bcl-2 homologs and NF-κB. While Bcl-2 homologs appear to prevent the deleterious effects of acute NF-κB activation by oxidative and DNA damage, they enforce the beneficial effects of its chronic activation downstream of oncogenic signaling. We propose that the consequences might be particularly perverse regarding cancer cell survival and the establishment of a tumorogenic microenvironment. Since Bcl-2 and Bcl-xL can regulate NF-κB transcriptional activity and since NF-κB activates BCL2 and BCL2L1 promoters Bcl-2 and Bcl-xL might regulate their own expression by a feedback loop that amplifies their pro-tumoral effects. Moreover, by non canonical BH3 mimetic resistant mechanisms, Bcl-2 homologs may enhance NF-κB dependent induction of oncogenic soluble factors (Fig. 3B), which might be further enhanced by non lethal treatment with BH3 mimetics (Fig. 3C).
Enhancement of Myc activity
Myc is a basic-helix-loop-helix-leucine zipper protein that, as a heterodimer with Max, binds preferentially to a palindromic E-box element CACGTG in DNA. Expression of Myc protein and mRNA (which are short-lived) are continuously dependent upon mitogenic signals in normal proliferating somatic cells. By contrast, Myc expression in cancer cells is typically deregulated and elevated, as a result of genomic amplifications or downstream of oncogenic signals that enhance the translation of its mRNA and/or stabilize the protein. Myc selectively enforces proliferation and dedifferentiation but specific targets genes involved in these effects (and a specific Myc induced gene expression signature) have remained elusive, as Myc appears to act as a contingent amplifier of active transcriptional programs, modulating up to one third of the transcriptome. The generally held notion is that cells sense differential Myc levels, and that high Myc in cancer cells contribute to oncogenesis by impacting on cell proliferation but also on, non exhaustively, metabolism, miRNA expression, angiogenesis or inflammation.
High Myc levels also promote apoptosis,80 and this constitutes a build-in restrain to the propagation of cells with deregulated proliferation.81 The exact mechanisms involved, which remain elusive, seem to rely on direct effects on the expression of Bcl-2 family members, and on indirect effects modulating their activity. In all cases, available data indicate that Myc promotes apoptosis by promoting MOMP.82-85 This is understood to be the mechanistic basis for the strong oncogenic cooperation between c-Myc and Bcl-2, as Bcl-2 abrogates apoptotic cell death induced by c-Myc and thus favors the expansion of cells with high Myc levels. This mechanistic cooperation seems all the more efficient as Bcl-2 might not affect Myc mitogenic function, and as Myc might override the cell cycle inhibitory effects exerted by Bcl-2 homologs (discussed above). It should be noted, however, that, in one study, the ability of Bcl-2 and Bcl-xL to delay cell cycle was reported to result in inhibition of c-Myc activity due to an upregulation of p27 as shown for E2F1.51
While it is classically assumed that Bcl-2 homologs may simply block one consequence of Myc activation, the latter observation suggests that they may more directly impact on the activity of this transcription factor. Work by Deng and colleagues further support this notion. These authors, indeed, initially reported a direct binding of Bcl-2 to c-MYC in the nucleus, as well as on the outer mitochondrial membrane, of cells treated with Nitrosamine 4-(methylnitrosamine)-14-(3-pyridyl)-1-butanone (NKK, the most potent carcinogen contained in cigarette smoke).86 The BH4-domain of Bcl-2 was identified as essential for nuclear Bcl-2 to interact with c-Myc, as well as phosphorylation of Bcl-2 at the Ser70 in its flexible loop induced by NKK.
Mechanistically, the binding of Bcl-2 to c-Myc was suggested to favor Myc transcriptional activity, and even though the nuclear events involved remain particularly obscure, it is striking to mention that downregulation of endogenous Bcl-2 was shown to decrease constitutive Myc activity.86 In the context of NKK treated cells more specifically studied by Deng and colleagues, this leads to a decrease in DNA repair and might thus contribute to genetic instability. Whether or not Bcl-2, and its homologs, generally influences other Myc induced effects by its interaction with this transcription factor, or as a result of its effects on cellular homeostasis, remains to be validated by more studies. Importantly, the implication of Bcl-2 in vascular smooth muscle cells migration and invasion was also ascribed to its ability to interact (in a phosphorylation dependent manner) with c-Myc and to its regulation of the expression of a c-Myc target, the metalloproteinase gene (mmp-2).87 The fact that Bcl-2 homologs may favor the transcription of pro-apoptotic genes by Myc remains, to date, theoretical, but it might contribute to the addiction of cancer cells to these proteins.
Concluding Remarks
Compared with the numerous studies that mechanistically investigate the regulation of MOMP by the BH3 binding activities of Bcl-2 homologs and that validate these BH3 binding activities as critical targets in cancer therapy (see ref. 2 for a review), studies of additional effects exerted by these proteins in the context of cancer appear to be underrepresented.
There is however cumulating evidence that Bcl-2 homologs modulate many more biological processes due to their ability to inhibit non apoptotic functions of caspases, their multiple distinct subcellular localizations and the diversity of their interactome. This underscores the complexity of the effects of Bcl-2 proteins on the survival, expansion and therapeutic response of cancer cells. By mechanistically interfering with the processes herein described, Bcl-2 proteins should indeed regulate cell survival beyond the sole blocking of MOMP and of apoptosis stricto sensu, shaping the cell’s responses to insults by fine tuning of ion exchanges, energy production or DNA repair, and influencing (at least theoretically) the immune response to therapy-induced cell death by regulating the inflammasomes88. Moreover, by their functional interplay with the E2F-1, NF-κB, and Myc transcription factors, Bcl-2 homologs might favor genomic instability and enforce survival (in part by initiating autoregulatory loops) in cancer cells and favor a pro-tumoral microenvironment by modifying their secretome. Such a role for Bcl-2 homologs, as rulers of survival signaling, implies that small molecules compounds currently used in the clinic to inhibit their activity at the mitochondria may be insufficiently well designed, and/or that the effects of these inhibitors may be insufficiently well circumscribed.
Acknowledgments
We apologize for our inability to cite all the contributing literature. We wish to thank Prof O Coqueret and Dr B Barré for fruitful discussions, and all members of our laboratory for their support. PJ is supported by Institut de Recherche Servier, ARC, Ligue Nationale contre le Cancer and Institut National du Cancer.
Glossary
Abbreviations:
- MOMP
mitochondrial outer membrane permeabilization
- TNFR
tumor necrosis factor receptor
- BH
Bcl-2 homology
- ROS
reactive oxygen species
- ER
endoplasmic reticulum
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Footnotes
Previously published online: www.landesbioscience.com/journals/cc/article/25972
References
- 1.Tait SWG, Green DR. Mitochondria and cell death: outer membrane permeabilization and beyond. Nat Rev Mol Cell Biol. 2010;11:621–32. doi: 10.1038/nrm2952. [DOI] [PubMed] [Google Scholar]
- 2.Juin P, Geneste O, Gautier F, Depil S, Campone M. Decoding and unlocking the BCL-2 dependency of cancer cells. Nat Rev Cancer. 2013;13:455–65. doi: 10.1038/nrc3538. [DOI] [PubMed] [Google Scholar]
- 3.Vo T-T, Ryan J, Carrasco R, Neuberg D, Rossi DJ, Stone RM, Deangelo DJ, Frattini MG, Letai A. Relative mitochondrial priming of myeloblasts and normal HSCs determines chemotherapeutic success in AML. Cell. 2012;151:344–55. doi: 10.1016/j.cell.2012.08.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ni Chonghaile T, Sarosiek KA, Vo T-T, Ryan JA, Tammareddi A, Moore VdelG, Deng J, Anderson KC, Richardson P, Tai YT, et al. Pretreatment mitochondrial priming correlates with clinical response to cytotoxic chemotherapy. Science. 2011;334:1129–33. doi: 10.1126/science.1206727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Tait SWG, Parsons MJ, Llambi F, Bouchier-Hayes L, Connell S, Muñoz-Pinedo C, Green DR. Resistance to caspase-independent cell death requires persistence of intact mitochondria. Dev Cell. 2010;18:802–13. doi: 10.1016/j.devcel.2010.03.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Khalil H, Peltzer N, Walicki J, Yang JY, Dubuis G, Gardiol N, Held W, Bigliardi P, Marsland B, Liaudet L, et al. Caspase-3 protects stressed organs against cell death. Mol Cell Biol. 2012;32:4523–33. doi: 10.1128/MCB.00774-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Song JH, Kandasamy K, Zemskova M, Lin Y-W, Kraft AS. The BH3 mimetic ABT-737 induces cancer cell senescence. Cancer Res. 2011;71:506–15. doi: 10.1158/0008-5472.CAN-10-1977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bertin-Ciftci J, Barré B, Le Pen J, Maillet L, Couriaud C, Juin P, Braun F. pRb/E2F-1-mediated caspase-dependent induction of Noxa amplifies the apoptotic effects of the Bcl-2/Bcl-xL inhibitor ABT-737. Cell Death Differ. 2013;20:755–64. doi: 10.1038/cdd.2013.6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bonneau B, Prudent J, Popgeorgiev N, Gillet G. Non-apoptotic roles of Bcl-2 family: the calcium connection. Biochim Biophys Acta. 2013;1833:1755–65. doi: 10.1016/j.bbamcr.2013.01.021. [DOI] [PubMed] [Google Scholar]
- 10.Li C, Fox CJ, Master SR, Bindokas VP, Chodosh LA, Thompson CB. Bcl-X(L) affects Ca(2+) homeostasis by altering expression of inositol 1,4,5-trisphosphate receptors. Proc Natl Acad Sci U S A. 2002;99:9830–5. doi: 10.1073/pnas.152571899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Monaco G, Vervliet T, Akl H, Bultynck G. The selective BH4-domain biology of Bcl-2-family members: IP3Rs and beyond. Cell Mol Life Sci. 2013;70:1171–83. doi: 10.1007/s00018-012-1118-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zhu L, Yu Y, Chua BH, Ho YS, Kuo TH. Regulation of sodium-calcium exchange and mitochondrial energetics by Bcl-2 in the heart of transgenic mice. J Mol Cell Cardiol. 2001;33:2135–44. doi: 10.1006/jmcc.2001.1476. [DOI] [PubMed] [Google Scholar]
- 13.Xu C, Xu W, Palmer AE, Reed JC. BI-1 regulates endoplasmic reticulum Ca2+ homeostasis downstream of Bcl-2 family proteins. J Biol Chem. 2008;283:11477–84. doi: 10.1074/jbc.M708385200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cárdenas C, Miller RA, Smith I, Bui T, Molgó J, Müller M, Vais H, Cheung KH, Yang J, Parker I, et al. Essential regulation of cell bioenergetics by constitutive InsP3 receptor Ca2+ transfer to mitochondria. Cell. 2010;142:270–83. doi: 10.1016/j.cell.2010.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Danial NN, Gramm CF, Scorrano L, Zhang CY, Krauss S, Ranger AM, Datta SR, Greenberg ME, Licklider LJ, Lowell BB, et al. BAD and glucokinase reside in a mitochondrial complex that integrates glycolysis and apoptosis. Nature. 2003;424:952–6. doi: 10.1038/nature01825. [DOI] [PubMed] [Google Scholar]
- 16.Chen Y-B, Aon MA, Hsu Y-T, Soane L, Teng X, McCaffery JM, Cheng WC, Qi B, Li H, Alavian KN, et al. Bcl-xL regulates mitochondrial energetics by stabilizing the inner membrane potential. J Cell Biol. 2011;195:263–76. doi: 10.1083/jcb.201108059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Alavian KN, Li H, Collis L, Bonanni L, Zeng L, Sacchetti S, Lazrove E, Nabili P, Flaherty B, Graham M, et al. Bcl-xL regulates metabolic efficiency of neurons through interaction with the mitochondrial F1FO ATP synthase. Nat Cell Biol. 2011;13:1224–33. doi: 10.1038/ncb2330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Perciavalle RM, Stewart DP, Koss B, Lynch J, Milasta S, Bathina M, Temirov J, Cleland MM, Pelletier S, Schuetz JD, et al. Anti-apoptotic MCL-1 localizes to the mitochondrial matrix and couples mitochondrial fusion to respiration. Nat Cell Biol. 2012;14:575–83. doi: 10.1038/ncb2488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Yi CH, Pan H, Seebacher J, Jang IH, Hyberts SG, Heffron GJ, Vander Heiden MG, Yang R, Li F, Locasale JW, et al. Metabolic regulation of protein N-alpha-acetylation by Bcl-xL promotes cell survival. Cell. 2011;146:607–20. doi: 10.1016/j.cell.2011.06.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Maiuri MC, Le Toumelin G, Criollo A, Rain JC, Gautier F, Juin P, Tasdemir E, Pierron G, Troulinaki K, Tavernarakis N, et al. Functional and physical interaction between Bcl-X(L) and a BH3-like domain in Beclin-1. EMBO J. 2007;26:2527–39. doi: 10.1038/sj.emboj.7601689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hsieh AC, Costa M, Zollo O, Davis C, Feldman ME, Testa JR, Meyuhas O, Shokat KM, Ruggero D. Genetic dissection of the oncogenic mTOR pathway reveals druggable addiction to translational control via 4EBP-eIF4E. Cancer Cell. 2010;17:249–61. doi: 10.1016/j.ccr.2010.01.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Petros AM, Olejniczak ET, Fesik SW. Structural biology of the Bcl-2 family of proteins. Biochim Biophys Acta. 2004;1644:83–94. doi: 10.1016/j.bbamcr.2003.08.012. [DOI] [PubMed] [Google Scholar]
- 23.Rizzuto R, Marchi S, Bonora M, Aguiari P, Bononi A, De Stefani D, Giorgi C, Leo S, Rimessi A, Siviero R, et al. Ca(2+) transfer from the ER to mitochondria: when, how and why. Biochim Biophys Acta. 2009;1787:1342–51. doi: 10.1016/j.bbabio.2009.03.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Choi B-H, Yoon HS. FKBP38-Bcl-2 interaction: a novel link to chemoresistance. Curr Opin Pharmacol. 2011;11:354–9. doi: 10.1016/j.coph.2011.04.013. [DOI] [PubMed] [Google Scholar]
- 25.Shirane M, Nakayama KI. Inherent calcineurin inhibitor FKBP38 targets Bcl-2 to mitochondria and inhibits apoptosis. Nat Cell Biol. 2003;5:28–37. doi: 10.1038/ncb894. [DOI] [PubMed] [Google Scholar]
- 26.Massaad CA, Portier BP, Taglialatela G. Inhibition of transcription factor activity by nuclear compartment-associated Bcl-2. J Biol Chem. 2004;279:54470–8. doi: 10.1074/jbc.M407659200. [DOI] [PubMed] [Google Scholar]
- 27.Portier BP, Taglialatela G. Bcl-2 localized at the nuclear compartment induces apoptosis after transient overexpression. J Biol Chem. 2006;281:40493–502. doi: 10.1074/jbc.M606181200. [DOI] [PubMed] [Google Scholar]
- 28.Beverly LJ. Regulation of anti-apoptotic BCL2-proteins by non-canonical interactions: the next step forward or two steps back? J Cell Biochem. 2012;113:3–12. doi: 10.1002/jcb.23335. [DOI] [PubMed] [Google Scholar]
- 29.Monaco G, Decrock E, Akl H, Ponsaerts R, Vervliet T, Luyten T, De Maeyer M, Missiaen L, Distelhorst CW, De Smedt H, et al. Selective regulation of IP3-receptor-mediated Ca2+ signaling and apoptosis by the BH4 domain of Bcl-2 versus Bcl-Xl. Cell Death Differ. 2012;19:295–309. doi: 10.1038/cdd.2011.97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Laulier C, Lopez BS. The secret life of Bcl-2: apoptosis-independent inhibition of DNA repair by Bcl-2 family members. Mutat Res. 2012;751:247–57. doi: 10.1016/j.mrrev.2012.05.002. [DOI] [PubMed] [Google Scholar]
- 31.Fujise K, Zhang D, Liu J, Yeh ET. Regulation of apoptosis and cell cycle progression by MCL1. Differential role of proliferating cell nuclear antigen. J Biol Chem. 2000;275:39458–65. doi: 10.1074/jbc.M006626200. [DOI] [PubMed] [Google Scholar]
- 32.Barillé-Nion S, Bah N, Véquaud E, Juin P. Regulation of cancer cell survival by BCL2 family members upon prolonged mitotic arrest: opportunities for anticancer therapy. Anticancer Res. 2012;32:4225–33. [PubMed] [Google Scholar]
- 33.Bruey J-M, Bruey-Sedano N, Luciano F, Zhai D, Balpai R, Xu C, Kress CL, Bailly-Maitre B, Li X, Osterman A, et al. Bcl-2 and Bcl-XL regulate proinflammatory caspase-1 activation by interaction with NALP1. Cell. 2007;129:45–56. doi: 10.1016/j.cell.2007.01.045. [DOI] [PubMed] [Google Scholar]
- 34.Faustin B, Chen Y, Zhai D, Le Negrate G, Lartigue L, Satterthwait A, Reed JC. Mechanism of Bcl-2 and Bcl-X(L) inhibition of NLRP1 inflammasome: loop domain-dependent suppression of ATP binding and oligomerization. Proc Natl Acad Sci U S A. 2009;106:3935–40. doi: 10.1073/pnas.0809414106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Vousden KH, Prives C. Blinded by the Light: The Growing Complexity of p53. Cell. 2009;137:413–31. doi: 10.1016/j.cell.2009.04.037. [DOI] [PubMed] [Google Scholar]
- 36.Green DR, Kroemer G. Cytoplasmic functions of the tumour suppressor p53. Nature. 2009;458:1127–30. doi: 10.1038/nature07986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Froesch BA, Aimé-Sempé C, Leber B, Andrews D, Reed JC. Inhibition of p53 transcriptional activity by Bcl-2 requires its membrane-anchoring domain. J Biol Chem. 1999;274:6469–75. doi: 10.1074/jbc.274.10.6469. [DOI] [PubMed] [Google Scholar]
- 38.Montero J, Dutta C, van Bodegom D, Weinstock D, Letai A. p53 regulates a non-apoptotic death induced by ROS. Cell Death Differ. 2013 doi: 10.1038/cdd.2013.52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Polager S, Ginsberg D. E2F - at the crossroads of life and death. Trends Cell Biol. 2008;18:528–35. doi: 10.1016/j.tcb.2008.08.003. [DOI] [PubMed] [Google Scholar]
- 40.Stevens C, La Thangue NB. The emerging role of E2F-1 in the DNA damage response and checkpoint control. DNA Repair (Amst) 2004;3:1071–9. doi: 10.1016/j.dnarep.2004.03.034. [DOI] [PubMed] [Google Scholar]
- 41.Alla V, Kowtharapu BS, Engelmann D, Emmrich S, Schmitz U, Steder M, Pützer BM. E2F1 confers anticancer drug resistance by targeting ABC transporter family members and Bcl-2 via the p73/DNp73-miR-205 circuitry. Cell Cycle. 2012;11:3067–78. doi: 10.4161/cc.21476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ianari A, Natale T, Calo E, Ferretti E, Alesse E, Screpanti I, Haigis K, Gulino A, Lees JA. Proapoptotic function of the retinoblastoma tumor suppressor protein. Cancer Cell. 2009;15:184–94. doi: 10.1016/j.ccr.2009.01.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Zinkel S, Gross A, Yang E. BCL2 family in DNA damage and cell cycle control. Cell Death Differ. 2006;13:1351–9. doi: 10.1038/sj.cdd.4401987. [DOI] [PubMed] [Google Scholar]
- 44.Mazel S, Burtrum D, Petrie HT. Regulation of cell division cycle progression by bcl-2 expression: a potential mechanism for inhibition of programmed cell death. J Exp Med. 1996;183:2219–26. doi: 10.1084/jem.183.5.2219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.O’Reilly LA, Huang DC, Strasser A. The cell death inhibitor Bcl-2 and its homologues influence control of cell cycle entry. EMBO J. 1996;15:6979–90. [PMC free article] [PubMed] [Google Scholar]
- 46.Borner C. Diminished cell proliferation associated with the death-protective activity of Bcl-2. J Biol Chem. 1996;271:12695–8. doi: 10.1074/jbc.271.22.12695. [DOI] [PubMed] [Google Scholar]
- 47.Linette GP, Li Y, Roth K, Korsmeyer SJ. Cross talk between cell death and cell cycle progression: BCL-2 regulates NFAT-mediated activation. Proc Natl Acad Sci U S A. 1996;93:9545–52. doi: 10.1073/pnas.93.18.9545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Quinn LM, Richardson H. Bcl-2 in cell cycle regulation. Cell Cycle. 2004;3:7–9. doi: 10.4161/cc.3.1.602. [DOI] [PubMed] [Google Scholar]
- 49.Lind EF, Wayne J, Wang QZ, Staeva T, Stolzer A, Petrie HT. Bcl-2-induced changes in E2F regulatory complexes reveal the potential for integrated cell cycle and cell death functions. J Immunol. 1999;162:5374–9. [PubMed] [Google Scholar]
- 50.Vairo G, Soos TJ, Upton TM, Zalvide J, DeCaprio JA, Ewen ME, Koff A, Adams JM. Bcl-2 retards cell cycle entry through p27(Kip1), pRB relative p130, and altered E2F regulation. Mol Cell Biol. 2000;20:4745–53. doi: 10.1128/MCB.20.13.4745-4753.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Greider C, Chattopadhyay A, Parkhurst C, Yang E. BCL-x(L) and BCL2 delay Myc-induced cell cycle entry through elevation of p27 and inhibition of G1 cyclin-dependent kinases. Oncogene. 2002;21:7765–75. doi: 10.1038/sj.onc.1205928. [DOI] [PubMed] [Google Scholar]
- 52.Janumyan Y, Cui Q, Yan L, Sansam CG, Valentin M, Yang E. G0 function of BCL2 and BCL-xL requires BAX, BAK, and p27 phosphorylation by Mirk, revealing a novel role of BAX and BAK in quiescence regulation. J Biol Chem. 2008;283:34108–20. doi: 10.1074/jbc.M806294200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Youn CK, Cho HJ, Kim SH, Kim HB, Kim MH, Chang IY, Lee JS, Chung MH, Hahm KS, You HJ. Bcl-2 expression suppresses mismatch repair activity through inhibition of E2F transcriptional activity. Nat Cell Biol. 2005;7:137–47. doi: 10.1038/ncb1215. [DOI] [PubMed] [Google Scholar]
- 54.Wang J, Beauchemin M, Bertrand R. Phospho-Bcl-x(L)(Ser62) plays a key role at DNA damage-induced G(2) checkpoint. Cell Cycle. 2012;11:2159–69. doi: 10.4161/cc.20672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Mac Fhearraigh S, Mc Gee MM. Cyclin B1 interacts with the BH3-only protein Bim and mediates its phosphorylation by Cdk1 during mitosis. Cell Cycle. 2011;10:3886–96. doi: 10.4161/cc.10.22.18020. [DOI] [PubMed] [Google Scholar]
- 56.Janumyan YM, Sansam CG, Chattopadhyay A, Cheng N, Soucie EL, Penn LZ, Andrews D, Knudson CM, Yang E. Bcl-xL/Bcl-2 coordinately regulates apoptosis, cell cycle arrest and cell cycle entry. EMBO J. 2003;22:5459–70. doi: 10.1093/emboj/cdg533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Fattman CL, Delach SM, Dou QP, Johnson DE. Sequential two-step cleavage of the retinoblastoma protein by caspase-3/-7 during etoposide-induced apoptosis. Oncogene. 2001;20:2918–26. doi: 10.1038/sj.onc.1204414. [DOI] [PubMed] [Google Scholar]
- 58.Vallabhapurapu S, Karin M. Regulation and function of NF-kappaB transcription factors in the immune system. Annu Rev Immunol. 2009;27:693–733. doi: 10.1146/annurev.immunol.021908.132641. [DOI] [PubMed] [Google Scholar]
- 59.Luo J-L, Kamata H, Karin M. The anti-death machinery in IKK/NF-kappaB signaling. J Clin Immunol. 2005;25:541–50. doi: 10.1007/s10875-005-8217-6. [DOI] [PubMed] [Google Scholar]
- 60.Kanarek N, London N, Schueler-Furman O, Ben-Neriah Y. Ubiquitination and degradation of the inhibitors of NF-kappaB. Cold Spring Harb Perspect Biol. 2010;2:a000166. doi: 10.1101/cshperspect.a000166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Lee HH, Dadgostar H, Cheng Q, Shu J, Cheng G. NF-kappaB-mediated up-regulation of Bcl-x and Bfl-1/A1 is required for CD40 survival signaling in B lymphocytes. Proc Natl Acad Sci U S A. 1999;96:9136–41. doi: 10.1073/pnas.96.16.9136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Ravi R, Bedi A, Fuchs EJ, Bedi A. CD95 (Fas)-induced caspase-mediated proteolysis of NF-kappaB. Cancer Res. 1998;58:882–6. [PubMed] [Google Scholar]
- 63.Levkau B, Scatena M, Giachelli CM, Ross R, Raines EW. Apoptosis overrides survival signals through a caspase-mediated dominant-negative NF-kappa B loop. Nat Cell Biol. 1999;1:227–33. doi: 10.1038/12050. [DOI] [PubMed] [Google Scholar]
- 64.Tang G, Yang J, Minemoto Y, Lin A. Blocking caspase-3-mediated proteolysis of IKKbeta suppresses TNF-alpha-induced apoptosis. Mol Cell. 2001;8:1005–16. doi: 10.1016/S1097-2765(01)00380-X. [DOI] [PubMed] [Google Scholar]
- 65.Pahl HL. Activators and target genes of Rel/NF-kappaB transcription factors. Oncogene. 1999;18:6853–66. doi: 10.1038/sj.onc.1203239. [DOI] [PubMed] [Google Scholar]
- 66.Karin M. Nuclear factor-kappaB in cancer development and progression. Nature. 2006;441:431–6. doi: 10.1038/nature04870. [DOI] [PubMed] [Google Scholar]
- 67.Giorgini S, Trisciuoglio D, Gabellini C, Desideri M, Castellini L, Colarossi C, Zangemeister-Wittke U, Zupi G, Del Bufalo D. Modulation of bcl-xL in tumor cells regulates angiogenesis through CXCL8 expression. Mol Cancer Res. 2007;5:761–71. doi: 10.1158/1541-7786.MCR-07-0088. [DOI] [PubMed] [Google Scholar]
- 68.Gabellini C, Castellini L, Trisciuoglio D, Kracht M, Zupi G, Del Bufalo D. Involvement of nuclear factor-kappa B in bcl-xL-induced interleukin 8 expression in glioblastoma. J Neurochem. 2008;107:871–82. doi: 10.1111/j.1471-4159.2008.05661.x. [DOI] [PubMed] [Google Scholar]
- 69.Ricca A, Biroccio A, Del Bufalo D, Mackay AR, Santoni A, Cippitelli M. bcl-2 over-expression enhances NF-kappaB activity and induces mmp-9 transcription in human MCF7(ADR) breast-cancer cells. Int J Cancer. 2000;86:188–96. doi: 10.1002/(SICI)1097-0215(20000415)86:2<188::AID-IJC7>3.0.CO;2-W. [DOI] [PubMed] [Google Scholar]
- 70.Karl E, Zhang Z, Dong Z, Neiva KG, Soengas MS, Koch AE, Polverini PJ, Núñez G, Nör JE. Unidirectional crosstalk between Bcl-xL and Bcl-2 enhances the angiogenic phenotype of endothelial cells. Cell Death Differ. 2007;14:1657–66. doi: 10.1038/sj.cdd.4402174. [DOI] [PubMed] [Google Scholar]
- 71.Kang J, Pervaiz S. Crosstalk between Bcl-2 family and Ras family small GTPases: potential cell fate regulation? Front Oncol. 2012;2:206. doi: 10.3389/fonc.2012.00206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Regula KM, Ens K, Kirshenbaum LA. IKK beta is required for Bcl-2-mediated NF-kappa B activation in ventricular myocytes. J Biol Chem. 2002;277:38676–82. doi: 10.1074/jbc.M206175200. [DOI] [PubMed] [Google Scholar]
- 73.de Moissac D, Zheng H, Kirshenbaum LA. Linkage of the BH4 domain of Bcl-2 and the nuclear factor kappaB signaling pathway for suppression of apoptosis. J Biol Chem. 1999;274:29505–9. doi: 10.1074/jbc.274.41.29505. [DOI] [PubMed] [Google Scholar]
- 74.Kucharczak J, Simmons MJ, Fan Y, Gélinas C. To be, or not to be: NF-kappaB is the answer--role of Rel/NF-kappaB in the regulation of apoptosis. Oncogene. 2003;22:8961–82. doi: 10.1038/sj.onc.1207230. [DOI] [PubMed] [Google Scholar]
- 75.Hour TC, Chen L, Lin JK. Suppression of transcription factor NF-kappaB activity by Bcl-2 protein in NIH3T3 cells: implication of a novel NF-kappaB p50-Bcl-2 complex for the anti-apoptotic function of Bcl-2. Eur J Cell Biol. 2000;79:121–9. doi: 10.1078/S0171-9335(04)70014-X. [DOI] [PubMed] [Google Scholar]
- 76.Song JH, Kandasamy K, Kraft AS. ABT-737 induces expression of the death receptor 5 and sensitizes human cancer cells to TRAIL-induced apoptosis. J Biol Chem. 2008;283:25003–13. doi: 10.1074/jbc.M802511200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Hardy K, Mansfield L, Mackay A, Benvenuti S, Ismail S, Arora P, O’Hare MJ, Jat PS. Transcriptional networks and cellular senescence in human mammary fibroblasts. Mol Biol Cell. 2005;16:943–53. doi: 10.1091/mbc.E04-05-0392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Acosta JC, O’Loghlen A, Banito A, Raguz S, Gil J. Control of senescence by CXCR2 and its ligands. Cell Cycle. 2008;7:2956–9. doi: 10.4161/cc.7.19.6780. [DOI] [PubMed] [Google Scholar]
- 79.Gorgoulis VG, Halazonetis TD. Oncogene-induced senescence: the bright and dark side of the response. Curr Opin Cell Biol. 2010;22:816–27. doi: 10.1016/j.ceb.2010.07.013. [DOI] [PubMed] [Google Scholar]
- 80.Murphy DJ, Junttila MR, Pouyet L, Karnezis A, Shchors K, Bui DA, Brown-Swigart L, Johnson L, Evan GI. Distinct thresholds govern Myc’s biological output in vivo. Cancer Cell. 2008;14:447–57. doi: 10.1016/j.ccr.2008.10.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Lowe SW, Cepero E, Evan G. Intrinsic tumour suppression. Nature. 2004;432:307–15. doi: 10.1038/nature03098. [DOI] [PubMed] [Google Scholar]
- 82.Juin P, Hueber AO, Littlewood T, Evan G. c-Myc-induced sensitization to apoptosis is mediated through cytochrome c release. Genes Dev. 1999;13:1367–81. doi: 10.1101/gad.13.11.1367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Juin P, Hunt A, Littlewood T, Griffiths B, Swigart LB, Korsmeyer S, Evan G. c-Myc functionally cooperates with Bax to induce apoptosis. Mol Cell Biol. 2002;22:6158–69. doi: 10.1128/MCB.22.17.6158-6169.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Campone M, Noël B, Couriaud C, Grau M, Guillemin Y, Gautier F, Gouraud W, Charbonnel C, Campion L, Jézéquel P, et al. c-Myc dependent expression of pro-apoptotic Bim renders HER2-overexpressing breast cancer cells dependent on anti-apoptotic Mcl-1. Mol Cancer. 2011;10:110. doi: 10.1186/1476-4598-10-110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Hoffman B, Liebermann DA. Apoptotic signaling by c-MYC. Oncogene. 2008;27:6462–72. doi: 10.1038/onc.2008.312. [DOI] [PubMed] [Google Scholar]
- 86.Jin Z, May WS, Gao F, Flagg T, Deng X. Bcl2 suppresses DNA repair by enhancing c-Myc transcriptional activity. J Biol Chem. 2006;281:14446–56. doi: 10.1074/jbc.M511914200. [DOI] [PubMed] [Google Scholar]
- 87.Lu Q, Hong W. Bcl2 enhances c-Myc-mediated MMP-2 expression of vascular smooth muscle cells. Cell Signal. 2009;21:1054–9. doi: 10.1016/j.cellsig.2009.02.020. [DOI] [PubMed] [Google Scholar]
- 88.Kroemer G, Galluzzi L, Kepp O, Zitvogel L. Immunogenic cell death in cancer therapy. Annu Rev Immunol. 2013;31:51–72. doi: 10.1146/annurev-immunol-032712-100008. [DOI] [PubMed] [Google Scholar]