

Abstract
Mixed lineage leukemia 1 (MLL1) is a gene that is disrupted by chromosomal translocation characteristically in a large proportion of infant leukemia and also in a fraction of childhood and adult leukemia. MLL1 encodes a chromatin regulatory protein related to the Drosophila Trithorax protein, a well-studied epigenetic factor that functions during development to maintain expression of its target genes. Although tremendous progress has been made understanding the downstream targets of MLL1 fusion oncoproteins and how manipulation of those targets impacts leukemogenesis, very little is known regarding how the initial expression of an MLL1 fusion protein impacts on that cell’s behavior, particularly how the cell cycle is affected. Here, we focused on the function of endogenous MLL1 in the stem and progenitor cell types that are likely to be transformed upon MLL1 translocation. Our studies reveal a differential response of stem or progenitor populations to acute loss of MLL1 on proliferation and survival. These data suggest that the effects of MLL1 fusion oncoproteins will initiate the leukemogenic process differentially depending on the differentiation state of the cell type in which the translocation occurs.
Keywords: leukemia, HSC, proliferation, quiescence, MLL, trithorax, progenitors
Introduction
Regenerating tissues such as the hematopoietic system are characterized by transient differentiation intermediates in which limited self-renewal capacity coupled to differentiation. More specifically, the adult hematopoietic system is wired in such a way as to provide: (1) a reserve pool of the self-renewing hematopoietic stem cells (HSCs), (2) differentiated cell types that are continuously generated in the proper proportions during homeostasis, and (3) adjustability to modulate the final output in response to loss of a particular cell type without automatically generating the entire repertoire of differentiated cells. The mechanisms ensuring the balanced output of differentiated cell types undoubtedly involves the regulation of proliferation, cell death, and cell fate; however, the specific mechanisms remain to be fully elucidated.
Leukemia represents a hematologic disorder in which the balance of self-renewal and proliferation is disrupted. Leukemia frequently results from chromosomal rearrangements that either produce hybrid oncogenic transcriptional regulators, haploinsufficiency, or loss of transcriptional regulators that act as tumor suppressors.1,2 These genetic alterations likely remodel the balance between self-renewal and proliferation; thus to study how these processes are affected in leukemia, we focused on the functions of mixed lineage leukemia 1 (MLL1) protein in hematopoietic stem and progenitor cells. MLL1 is disrupted by chromosomal rearrangement in acute leukemia, predominantly in young children. Most MLL1 rearrangements encode hybrid fusion oncoproteins that have altered functions relative to the larger endogenous MLL1 protein.3,4 Understanding the molecular mechanisms by which MLL1 oncogenic fusion proteins transform cells will be particularly insightful, since this leukemia is characterized by exceedingly few other genomic alterations, in contrast to other cancers.5
Our recent studies6,7 showed that loss of Mll1 using the inducible Mx1-cre transgene resulted in an increase in proliferation within the most HSC-enriched populations, and yet ultimately animals died with anemia and overall pan-cytopenia of the bone marrow 2–3 wk after Mll1 deletion. Further characterization revealed that HSC-enriched populations failed to remain in G0 and began a phase of proliferation and differentiation apparently without self-renewal. In contrast, myelo-erythroid progenitors in the same animals exhibited reduced proliferation. Thus, we concluded that endogenous MLL1 played a critical role in restraining proliferation and promoting self-renewal in HSCs, whereas it promoted proliferation in myelo-erythroid progenitors.6
A common finding across several model systems is that MLL1 fusion oncoproteins act through a gain-of-function mechanism, resulting in overexpression of some endogenous MLL1 target genes.3,4 Given this gain-of-function activity, one prediction incorporating our loss-of-function results described above is that MLL1 fusion oncoproteins that arise within HSCs would result in enhanced quiescence in this cell type, whereas production of the same oncoprotein in myelo-erythroid cells would result in increased proliferation. In contrast to HSCs, myelo-erythroid progenitors are already rapidly dividing, thus the effect of MLL fusion oncoproteins may be to simply lock-in a pre-existing genetic/epigenetic proliferation program and resist further differentiation. Experiments demonstrating similarities and distinctions between leukemia arising from HSCs vs. granulocyte-macrophage progenitors (GMPs) have come to different conclusions regarding the similarity of the resulting leukemia;8-10 however, the immediate consequence on proliferation was not analyzed.
The prediction of enhanced quiescence upon expression of MLL fusion oncoproteins in HSCs is counterintuitive, given that the ultimate consequence of MLL1 fusion protein expression is leukemia. However, one hypothesis to accommodate this HSCs prediction is as follows: MLL1 fusion proteins initially induce enhanced quiescence when expressed in HSCs, but stochastic events result in a few MLL fusion protein-expressing HSCs escaping quiescence via differentiation. Once slightly differentiated, the MLL1 fusion oncoprotein encounters a distinct cellular context that responds by increased and sustained proliferation and ultimately, leukemia. Interestingly, this hypothesis also suggests that a pool of quiescent, MLL1 fusion oncoprotein-expressing cells may be maintained in G0, thus contributing to a chemotherapy-resistant reservoir of cells for relapse. In this study, we assessed the role of endogenous MLL1 in regulating proliferation in HSCs vs. differentiating progeny. We performed single cell proliferation studies in defined cytokine signaling environments, either those that support HSC maintenance or those supporting differentiation. Our results demonstrate that the proliferation kinetics of Mll1-deficient HSCs respond to the extracellular environment within 1–2 cell divisions, switching cells to conditions that support differentiation results in adopting the proliferation kinetics observed in vivo for myelo-erythroid progenitors. These data reveal the difference in the wiring of the MLL1 pathway as a function of differentiation, and suggest that MLL fusion oncoproteins will affect these pathways in a distinct manner during leukemogenesis.
Results and Discussion
Two observations prompted us to analyze further the effect of Mll1 loss as a function of the cell’s differentiation state. In vivo bromo-deoxy uridine (BrdU) pulse-chase experiments demonstrated that Mll1-deficient lineage-negative/Sca-1+/c-Kit+ (LSK) cells proliferated more rapidly than wild-type controls.6 Yet when these cells were purified from animals and cultured in methylcellulose to support differentiation, smaller colonies with fewer of all differentiated cell types were observed (Fig. 1). Thus, we hypothesized that the culture conditions designed to support myelo-erythroid progenitor differentiation resulted in reduced proliferation as cells differentiated, despite the fact that they were initially proliferating more than control LSK cells.6

Figure 1.Mll1-deficient LSK cells plated in methylcellulose produce smaller colonies. LSK cells were sorted from pI:pC-treated animals of the genotype indicated as described in the “Materials and Methods”.(A) Representative colonies produced after 7 d growth on methylcellulose containing myelo-erythroid supportive cytokines (M3434 GF).(B) Colony enumeration after 7 d growth, and(C) total cell number from pooled duplicate plates, harvested from individual plates after scoring. Bars represent averages ± 95% CI of cell yield per donor animal.
To establish a system in which proliferation, cell death, and cell fate could be analyzed over time on a per-cell basis, we modified the method of Uchida et al.11 In this system, high stem cell factor (SCF) is critical for the maintenance of HSC function as cells proliferate, determined by single-well transplantation experiments. Using these conditions, we tracked the proliferation and survival of HSC-enriched LSK/CD48neg cells from Mll1-deficient or control wild-type bone marrow. We found that Mll1-deficient HSCs proliferated more rapidly than their wild-type counterparts,7 consistent with our in vivo observations.6 To test whether altering the cytokine conditions to those of the methylcellulose cultures (Fig. 1) altered the proliferation kinetics, we performed this single-cell assay in the serum and cytokine concentrations found in M3434 growth factor (GF) medium (Fig. 2A). We again sorted LSK/CD48neg cells to start with the same population as tested in HSC maintenance conditions. As shown in Figure 2B, Mll1-deficient cells initially proliferate more rapidly than their wild-type counterparts, likely reflecting the HSC-enriched sorted population. However, as cells divide, they rapidly change behavior, such that by 72 h there is no longer a difference in the number of divisions achieved between wild-type and Mll1-deficient cells (Fig. 2C and D). In the HSC maintenance conditions, we found that as many Mll1-deficient clones survived and proliferated as the wild-type control clones.7 To examine overall survival in the differentiation-supporting medium, we scored the number of clones that ultimately divided vs. those that either underwent cell death or simply stopped dividing (see “Materials and Methods”). In contrast to our findings in HSC maintenance medium, Mll1-deficient cells did not survive as well as their wild-type counterparts (Fig. 3). Together, these data account for our observations of smaller colonies and fewer total cells upon growth in methylcellulose (Fig. 1). Furthermore, this single cell analysis demonstrates that after 1–2 cell divisions, Mll1-deficient LSK/CD48neg cells adopt the reduced proliferation characteristic of Mll1-deficient myelo-erythroid progenitors in vivo.6

Figure 2.Mll1-deficient LSK/CD48neg cells slow down upon growth in differentiation-supporting culture conditions.(A) Scheme for purifying control and Mll1-deficient HSC-enriched cells. Cells were generated from Mll1F/F or Mx1-cre;Mll1F/F animals injected with pI:pC, sorted 6 d later, then resorted as single cells into wells; n = 4–5 donor animals per genotype. One-hundred-forty-two (Mll1F/F) or 338 Mx1-cre;Mll1F/F cells clones were observed. Percentages of responding cells (cells that divided at least once) are shown at 24 h (B) 48 h (C), or 72 h (D). Cells that were categorized as dead included two groups: those that were no longer refractile and exhibited signs of membrane blebbing or degradation, and cells that appeared refractile and large but failed to divide for the duration of the observation period.
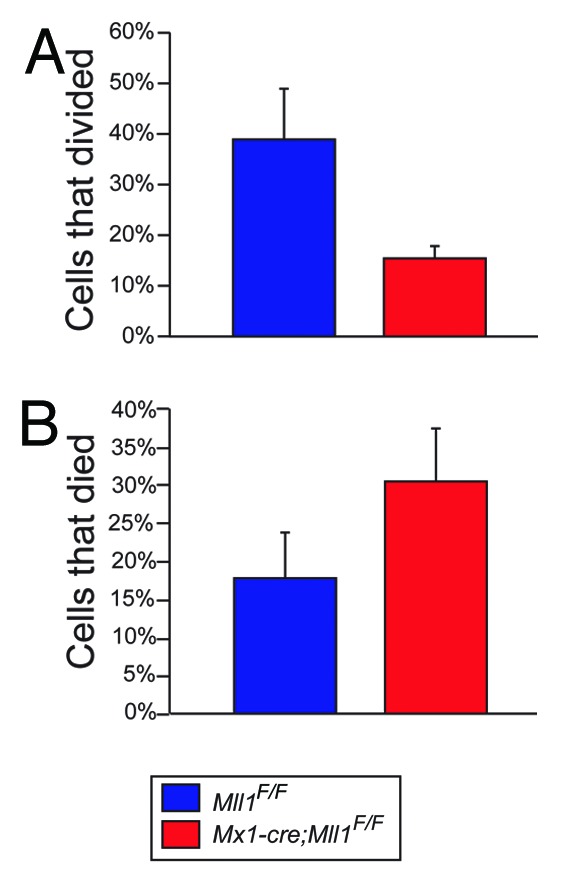
Figure 3. Overall response of wild-type (blue) and Mll1-deficient (red) LSK/CD48neg cells in differentiation medium.(A) Percentage of cells that ultimately divided at least once during the 72-h observation period.(B) Percentage of cells scored as dead/non-dividing during the observation period (see “Materials and Methods”).
Together with the data shown in Artinger et al.,7 we speculate that the loss of Mll1 has a distinct impact on cell proliferation, based on differentiation state of the hematopoietic stem/progenitor. Starting with the same cell population (LSK/CD48neg) we observed that Mll1-deficient cells proliferate faster in defined cytokine conditions that support HSC identity and function, yet switching to growth medium that instead favors differentiation is sufficient to reverse the proliferation phenotype after 1–2 cell divisions. These loss-of-function data serve to delineate the role of endogenous MLL1 in HSCs and multi-potent progenitors, the cell types in which MLL1 translocations likely occur. Moreover, these data provide a framework for understanding how MLL1 fusion oncoproteins influence proliferation behavior. Based on these studies, we hypothesize that the effect of producing an MLL1 fusion oncoprotein in HSCs vs. more differentiated progenitors may result in initially distinct proliferation outcomes and potentially influence the type of secondary alterations/mutations that occur after the initial translocation. For example, alterations that relieve quiescence in the HSC vs. mutations that affect survival in the progenitor may be differentially favored depending on the cell type in which the translocation occurred. This prediction must be tested explicitly, but considering the heterogeneity in clinical outcome in MLL1-rearranged leukemia and recent data illustrating the prognostic value of the inferred cell-of-origin,10 this information may influence treatment strategies.
Materials and Methods
Mice
Mll1F/F mice were described6 and were maintained on a C57BL/6.SJL background (Ptprca Pep3b/BoyJ, stock #002014, The Jackson Laboratory). Mll1 deletion was achieved using 3–4 polyinosinic:polycytidylic acid (pI:pC) injections and the efficiency of gene deletion was assessed by genomic PCR as described.6 Experimental protocols were approved by the Institutional Animal Care and Use Committee of Dartmouth College.
Flow cytometry and cell sorting
Cells were stained on ice in Hank buffered saline solution (HBSS, Mediatech) plus 2% fetal bovine serum (FBS, Gibco Life Technologies) and sorted using a FACSAria (BD Biosciences) using the DartLab core facilities as described.7 Phenotypic analyses were confirmed on a FACSCalibur (BD Biosciences), and data was analyzed using FlowJo software (TreeStar). The purity of sorted cells was >90% based on post-sort analyses.
Single cell and semi-solid medium culture
LSK/CD48neg cells from pI:pC injected control MllF/F or Mx1-cre;MllF/F mice were prepared and sorted as described7 then re-sorted at one cell per well into individual wells of U-bottom 96-well plates (Nunc) containing 100 μL of “differentiation medium” (IMDM containing 20% FBS, 50 ng/mL SCF, 10 ng/mL IL-6, 10 ng/mL IL-3, and 3 U/mL Epo). Cytokines were obtained from R&D Systems, with the exception of Epo (Procrit, kindly provided by Dr Lowrey, Geisel School of Medicine). After sorting, plates were centrifuged briefly at 380 × g, incubated at 37 °C in 5% CO2 for 2 h then scored for the presence of a single cell. The number of live or dead cells in each well was then scored visually every 24 h to determine the number of cell divisions that had occurred by that time. Three to four donor animals were used per genotype and 142 (wild-type) or 388 (Mll1-deficient) cells were scored. The percentage of responding clones in Figure 3 corresponds to the percentage of visually confirmed cells that ultimately divide at least once during 72 h of culture, and in Figure 2 is the percentage of those clones that have divided the number indicated on the x-axis. For the semi-solid cultures in Figure 1, LSK cells were sorted from 3–4 individual pI:pC injected MllF/F or Mx1-cre;MllF/F mice and 5000 sorted cells were plated in duplicate per donor animal on M3434 GF semi-solid medium (StemCell Technologies) in 35 mm dishes. Seven days later, colonies were scored and photographed on a Leica MZ7.5 stereomicroscope fitted with a consumer Cannon Powershot S3 IS camera. Bars represent averages ± 95% confidence interval (CI).
Statistics
Error bars reflect 95% CI unless otherwise indicated. P values in Figure 2 were calculated using the Pearson chi-squared test. Prism (Graphpad) and Excel (Microsoft) were used for graphs and statistical calculations.
Acknowledgments
We thank lab members and David Bryder for discussion and critical comments. We are grateful to Gary Ward for expert help with cell sorting. ELA was partially supported by the Lady Tata Memorial Trust. This work was supported by NIH grant HL090036 and funds from the Gabrielle’s Angel and Lauri Strauss Foundations.
Glossary
Abbreviations:
- MLL
mixed lineage leukemia
- HSC
hematopoietic stem cell
- FBS
fetal bovine serum
- SCF
stem cell factor
- Epo
erythropoietin
- IMDM
Iscove modified Dulbecco medium
- HBSS
Hank buffered saline solution
- LSK
lineage-negative/Sca-1 positive/c-Kit positive
- pI:pC
polyinosinic:polycytidylic acid polymer
- BrdU
bromo-deoxy uridine
- GMPs
granulocyte-macrophage progenitors
- CI
confidence interval
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Footnotes
Previously published online: www.landesbioscience.com/journals/cc/article/26032
References
- 1.Mullighan CG, Willman CL. Advances in the Biology of Acute Lymphoblastic Leukemia-From Genomics to the Clinic. J Adolesc Young Adult Oncol. 2011;1:77–86. doi: 10.1089/jayao.2011.0012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Rubnitz JE, Inaba H. Childhood acute myeloid leukaemia. Br J Haematol. 2012;159:259–76. doi: 10.1111/bjh.12040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Krivtsov AV, Armstrong SA. MLL translocations, histone modifications and leukaemia stem-cell development. Nat Rev Cancer. 2007;7:823–33. doi: 10.1038/nrc2253. [DOI] [PubMed] [Google Scholar]
- 4.Rodriguez-Perales S, Cano F, Lobato MN, Rabbitts TH. MLL gene fusions in human leukaemias: in vivo modelling to recapitulate these primary tumourigenic events. Int J Hematol. 2008;87:3–9. doi: 10.1007/s12185-007-0001-3. [DOI] [PubMed] [Google Scholar]
- 5.Downing JR, Wilson RK, Zhang J, Mardis ER, Pui CH, Ding L, Ley TJ, Evans WE. The Pediatric Cancer Genome Project. Nat Genet. 2012;44:619–22. doi: 10.1038/ng.2287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Jude CD, Climer L, Xu D, Artinger E, Fisher JK, Ernst P. Unique and independent roles for MLL in adult hematopoietic stem cells and progenitors. Cell Stem Cell. 2007;1:324–37. doi: 10.1016/j.stem.2007.05.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Artinger EL, Mishra BP, Zaffuto KM, Li BE, Chung EK, Moore AW, Chen Y, Cheng C, Ernst P. An MLL-dependent network sustains hematopoiesis. Proc Natl Acad Sci U S A. 2013;110:12000–5. doi: 10.1073/pnas.1301278110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cozzio A, Passegué E, Ayton PM, Karsunky H, Cleary ML, Weissman IL. Similar MLL-associated leukemias arising from self-renewing stem cells and short-lived myeloid progenitors. Genes Dev. 2003;17:3029–35. doi: 10.1101/gad.1143403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Arai S, Yoshimi A, Shimabe M, Ichikawa M, Nakagawa M, Imai Y, Goyama S, Kurokawa M. Evi-1 is a transcriptional target of mixed-lineage leukemia oncoproteins in hematopoietic stem cells. Blood. 2011;117:6304–14. doi: 10.1182/blood-2009-07-234310. [DOI] [PubMed] [Google Scholar]
- 10.Krivtsov AV, Figueroa ME, Sinha AU, Stubbs MC, Feng Z, Valk PJ, Delwel R, Döhner K, Bullinger L, Kung AL, et al. Cell of origin determines clinically relevant subtypes of MLL-rearranged AML. Leukemia. 2013;27:852–60. doi: 10.1038/leu.2012.363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Uchida N, Dykstra B, Lyons KJ, Leung FY, Eaves CJ. Different in vivo repopulating activities of purified hematopoietic stem cells before and after being stimulated to divide in vitro with the same kinetics. Exp Hematol. 2003;31:1338–47. doi: 10.1016/j.exphem.2003.09.001. [DOI] [PubMed] [Google Scholar]