

Abstract
By means of an unbiased, automated fluorescence microscopy-based screen, we identified the epidermal growth factor receptor (EGFR) inhibitors erlotinib and gefitinib as potent enhancers of the differentiation of HL-60 acute myeloid leukemia (AML) cells exposed to suboptimal concentrations of vitamin A (all-trans retinoic acid, ATRA) or vitamin D (1α,25-hydroxycholecalciferol, VD). Erlotinib and gefitinib alone did not promote differentiation, yet stimulated the acquisition of morphological and biochemical maturation markers (including the expression of CD11b and CD14 as well as increased NADPH oxidase activity) when combined with either ATRA or VD. Moreover, the combination of erlotinib and ATRA or VD synergistically induced all the processes that are normally linked to terminal hematopoietic differentiation, namely, a delayed proliferation arrest in the G0/G1 phase of the cell cycle, cellular senescence, and apoptosis. Erlotinib potently inhibited the (auto)phosphorylation of mitogen-activated protein kinase 14 (MAPK14, best known as p38MAPK) and SRC family kinases (SFKs). If combined with the administration of ATRA or VD, the inhibition of p38MAPK or SFKs with specific pharmacological agents mimicked the pro-differentiation activity of erlotinib. These data were obtained with 2 distinct AML cell lines (HL-60 and MOLM-13 cells) and could be confirmed on primary leukemic blasts isolated from the circulation of AML patients. Altogether, these findings point to a new regimen for the treatment of AML, in which naturally occurring pro-differentiation agents (ATRA or VD) may be combined with EGFR inhibitors.
Keywords: BCR-ABL, PML-RARα, cancer, dasatinib, imatinib, monocyte-specific esterase
Introduction
Leukemia constitutes a type of hematological malignancy characterized by a prominent increase in circulating immature white blood cells named “blasts.” Leukemogenesis involves a compendium of cell-autonomous genetic and epigenetic alterations that interfere with the homeostatic regulation of the hematopoietic (stem cell) compartment. Generally, this results in the accumulation of leukemic (stem) cells that gradually take over normal hematopoiesis as they acquire all hallmarks of cancer.1,2 Thus, curative antileukemic therapies may be conceived to achieve at least 1 out of 3 distinct purposes. First, cytotoxic agents can be designed to selectively kill leukemic (stem) cells, mostly by stimulating mitochondrial apoptosis. Second, cytostatic agents may be used to block the unrestrained proliferation of leukemic cells, thereby reducing their relative occupancy of the bone marrow. Third, differentiation-stimulating agents should stimulate the transition of leukemic (stem) cells to a mature phenotype, thereby abolishing their proliferative potential and allowing them to undergo terminal differentiation-associated cell death.3-5 A prominent example of such a differentiation therapy is all-trans retinoic acid (ATRA), the biologically active variant of vitamin A, which has been successfully employed for decades in the treatment of acute promyelocytic leukemia (APL).6 Similarly, 1α,25-hydroxycholecalciferol, the active form of vitamin D3 (VD) also known as calcifediol, and many of its analogs can stimulate the terminal differentiation of leukemic cell lines as well as primary myeloid precursors, and their therapeutic value has been tested in different clinical trials.7,8 However, the clinical development of VD as an antileukemic agent appears to stand at an impasse, for 2 reasons. First, the high doses of VD that are required to stimulate myeloid differentiation can cause moderate to severe adverse effects related to Ca2+ metabolism. Second, the administration of VD has been associated (at least in specific settings) with the rapid development of resistance.9,10 Thus, no differentiation therapies are currently approved for the clinical management of leukemias other than APL (French-American-British subtype M3).
Acute myeloid leukemia (AML) is a heterogeneous clonal disorder of hematopoietic progenitors and represents one of the most common forms of acute leukemia affecting adults.11 Although AML is a relatively rare disease, accounting for slightly over 1% of cancer-related deaths in the western world, its incidence is expected to augment as the population ages.12 AML develops along a complex, multistep course characterized by the progressive accumulation of a variety of genetic defects that either confer a proliferative/survival advantage to myeloid progenitors (e.g., FLT3 or KIT mutations) or contribute to the failure of these cells to differentiate into mature granulocytes or monocytes (e.g., CEBPA or NPM1 mutations).13,14 The clinical management of AML patients younger than 60 y is based on high-dose chemotherapy and, upon relapse, bone marrow transplantation.15 However, the use of cytotoxic chemotherapy in the elderly is associated with high rates of morbidity and mortality.16,17 Novel antileukemic drugs have brought about a few improvements in disease outcome among elderly patients.18
Because the incidence of AML affecting old patients augments (along with the progressive increase in life expectancy of the general population), novel therapeutic paradigms for the clinical management of leukemia in this patient subset are urgently awaited. Differentiation therapies may represent a valuable alternative to cytotoxic agents in this setting, as they are generally associated with comparatively less severe side effects. However, most chemicals agents with a pro-differentiation activity described in the last 2 decades do not target a disease-specific lesion such as ATRA, which selectively modulates the activity of PML-RARα (the etiological determinant of APL),19 and generally are not potent enough to promote terminal differentiation. Recently, several groups, including ours, have proposed epidermal growth factor receptor (EGFR) inhibitors, such as gefitinib20,21 and erlotinib,22-24 as potential candidates for the treatment of AML, although the expression of EGFR by AML cells is a subject of controversy.24,25 Both gefitinib and erlotinib have been reported to exert a mild differentiation-inducing effect in vitro,24,26,27 which, however, has not been confirmed in vivo.
In the present study, we addressed the question as to whether the maturation of AML cells exposed to suboptimal doses of ATRA and VD may be exacerbated by the concomitant administration of other therapeutically relevant agents. We report that erlotinib and gefitinib synergistically interact with ATRA and VD to stimulate the terminal differentiation of AML cells.
Results and Discussion
EGFR inhibitors stimulate the differentiation of AML cells in synergy with ATRA and VD
To identify novel agents that may induce or favor the differentiation of AML cells, we developed an automated screening system involving the immunofluorescence microscopy-based detection of one particular marker of myeloid maturation, CD11b,28 in human HL-60 (Fig. 1) and MOLM-13 (data not shown) cells. We then employed this system to screen the US Drug Collection (which encompasses most FDA-approved drugs, plus a large amount of compounds that have reached clinical development), finding ATRA (employed at a final concentration of 1 µM) as the most effective differentiation-inducing agent of the library (Fig. 1A and B). To identify possible synergistic interactions between anticancer agents that are currently employed in the clinic and known differentiation-inducing chemicals, we repeated such screen on compounds from the Oncology Drug Set (which comprises most FDA-approved chemotherapeutics; final concentration = 10 µM), alone or in the presence of a suboptimal dose of ATRA (100 nM) or VD (50 nM). These screens revealed that 2 EGFR inhibitors, erlotinib and gefitinib, are capable of stimulating the differentiation of HL-60 cells when combined with ATRA or VD, yet have limited, if any, differentiation-inducing activity when employed as single agents (Fig. 1C and D). To validate these findings, we performed cytofluorometric assays for the detection of the differentiation markers CD11b and CD14 on HL-60 cells exposed to suboptimal doses of ATRA or VD, alone or combined with optimal concentrations of several FDA-approved tyrosine kinase inhibitors (TKIs) (Fig. 2A and B). A number of TKIs stimulated the expression of CD11b on the surface of HL-60 cells, if co-administered with ATRA or VD (Fig. 2C). Moreover, several TKIs, notably erlotinib, favored the acquisition of the monocyte marker CD14 by HL-60 cells exposed to VD, but not to ATRA (Fig. 2D).

Figure 1. Automated screening platform for the identification of differentiation-inducing agents. (A–D). Human acute myeloid leukemia HL-60 cells were exposed to DMSO (control conditions) (A–D), compounds from the US Drug Library (final concentration = 1 µM) (A and B), or compounds from the NCI Oncology Drug Set (final concentration = 10 µM) (C and D), alone or in combination with 100 nM all-trans retinoic acid (ATRA) or 50 nM 1α,25-hydroxycholecalciferol (VD), for 3 d, and the processed for the automated fluorescence microscopy-assisted detection of CD11b. Representative images (scale bar = 20 µm) and quantitative data on CD11b granularity (AU, means ± SD, n = 3 parallel replicates) upon intra- and inter-plate normalization are reported in (A) and (B–D), respectively. In panels (CandD), dashed lines delimit the zone of statistical non-significance (90% prediction interval).

Figure 2. Effects of tyrosine kinase inhibitors on myeloid differentiation. (A–D). Human acute myeloid leukemia HL-60 cells were treated with 10 μM erlotinib (ERLO, E), 10 μM gefitinib (GEFI), 100 nM dasatinib (DASA), 5 μM lapatinib (LAPA), 5 μM imatinib (IMA), 5 μM sorafenib (SORA), 500 nM nilotinib (NILO), 500 nM sunitinib (SUNI), or an equal volume of DMSO, alone or combined with 100 nM all-trans retinoic acid (ATRA) or 50 nM 1α,25-hydroxycholecalciferol (VD), for 3 d, and then processed for the cytofluorometry-assisted detection of CD11b (A–C) or CD14 (B–D) expression. Panels (Aand B) report representative CD11b and CD14 expression profiles, respectively. ISO, isotype control. Panels (C and D) illustrate quantitative data on the percentage of CD11b- and CD14 -expressing cells, respectively (means ± SEM; n = 2–3). *P < 0.05, **P < 0.01, ***P < 0.001 (ANOVA plus Dunnett post-hoc test) as compared with DMSO-treated cells; ###P < 0.001 (ANOVA plus Bonferroni post-hoc test), as compared with ATRA-treated cells; +P < 0.05, ++P < 0.01 +++P < 0.001 (ANOVA plus Bonferroni post-hoc test), as compared with VD-treated cells.
The capacity of erlotinib to promote the ATRA- or VD-induced differentiation of AML cells was confirmed by 3 additional techniques, namely (1) the morphological evaluation of cells upon May–Grünwald–Giemsa staining, revealing the differentiation-associated decrease in cytoplasmic basophilia accompanied with increased cytoplasmic granularity and reduced nucleus/cytoplasm ratio29 (Fig. 3A and B), (2) the measurement of the respiratory burst that characterizes mature myeloid cells, based on the conversion of nitroblue tetrazolium chloride (NBT) to a blue insoluble product (formazan) by NADPH oxidases30 (Fig. 3C and D), and (3) the cytochemical assessment of monocyte-specific esterase activity, based on the conversion of 1-naphthyl acetate into 1-naphthol (which forms an insoluble red-brown dye in the presence of a diazonium salt)31 (Fig. 3E and F). All these methods confirmed that erlotinib and gefitinib fail to induce the differentiation of HL-60 cells on their own, yet exacerbate the pro-differentiation activity of suboptimal doses of ATRA or VD. Similar findings were obtained when the ability of erlotinib and gefitinib to accentuate differentiation as induced by ATRA and VD was tested on MOLM-13 cells (Fig. S1).

Figure 3. Morphological and functional assessment of the differentiation-inducing activity of EGFR inhibitors. (A–F). Human acute myeloid leukemia HL-60 cells were treated with 10 μM erlotinib (ERLO), 10 μM gefitinib (GEFI), or an equal volume of DMSO, alone or in combination with 100 nM all-trans retinoic acid (ATRA), or 50 nM 1α,25-hydroxycholecalciferol (VD), for 3 d, then cytospun and processed for the cytochemical assessment of myeloid differentiation upon May–Grünwald–Giemsa staining (Aand B) or the colorimetric detection of NADPH oxidase (C and D) or α-naphtyl acetate esterase (E and F) enzymatic activities. Panels (A, C, and E) depict representative images (scale bars = 10 µm), whereas quantitative data on the percentage of cells exhibiting morphological signs of differentiation (decreased cytoplasmic basophilia, nuclear lobulation and cytoplasmic granulation), nitroblue tetrazolium chloride (NBT)-reducing activity and α-naphtyl acetate esterase activity are reported in (B, D, and F), respectively (means ± SEM; n = 3 with at least 100 cells/condition). The inset in (E) depicts a megakaryocyte (M) exhibiting intense α-naphtyl acetate esterase activity (positive control). *P < 0.05, **P < 0.01, ***P < 0.001 (ANOVA plus Dunnett post-hoc test), as compared with DMSO-treated cells; n.s., not significant, ###P < 0.001 (ANOVA plus Bonferroni post-hoc test), as compared with ATRA-treated cells; ++P < 0.01, +++P < 0.001 (ANOVA plus Bonferroni post-hoc test), as compared with VD-treated cells.
We conclude that TKIs, and in particular erlotinib, can synergize with ATRA or VD in promoting the differentiation of AML cells.
Antiproliferative and pro-apoptotic effects of EGFR inhibitors combined with ATRA or VD
Physiologically, the terminal differentiation of myeloid precursors toward the granulocytic or monocytic lineage is coupled to a progressive reduction in cell proliferation followed by apoptosis.32,33 We therefore sought to determine the potential antiproliferative and pro-apoptotic effects of EGFR inhibitors combined with ATRA or VD. For the quantitative assessment of cell number, a fixed amount of fluorescent beads was added to differentiating cell cultures, followed by the cytofluorometric determination of the cell/bead ratio (Fig. 4A and B). In addition, we quantified cell proliferation using 5-(and-6)-carboxyfluorescein diacetate succinimidyl ester (CFSE), a plasma membrane-permeant fluorescent dye that, upon stable incorporation into the cytoplasm, dilutes by a factor of 2 with each successive round of mitosis (Fig. 4C and D).34 Both these experimental settings revealed the capacity of erlotinib and gefitinib to exacerbate the antiproliferative effects of ATRA and VD (Fig. 4A–D). Such a cytostatic activity was particularly pronounced when erlotinib and gefitinib were combined with VD rather than with ATRA, which per se exerted cytostatic effects (Fig. 4A–D). In line with previous reports,21,35 erlotinib also had an antiproliferative activity, yet the combination of erlotinib plus ATRA or VD was more efficient at inhibiting the proliferation of HL-60 cells than any of these agents employed alone (Fig. 4A–D).

Figure 4. Antiproliferative response associated with myeloid differentiation. (A–F). Human acute myeloid leukemia HL-60 cells were treated with 10 μM erlotinib (ERLO), 10 μM gefitinib (GEFI), or an equal volume of DMSO, alone or in combination with 100 nM all-trans retinoic acid (ATRA) or 50 nM 1α,25-hydroxycholecalciferol (VD), for the indicated number of days (7, where not specified), followed by (A, B, E, and F) or alongside with (C and D) the assessment of proliferation (A–D) or cell cycle distribution (E and F) based on the bead-assisted cytofluorometric quantification of cell number (A and B), the CFSE-dependent assessment of replication (C and D), or the analysis of DNA content upon propidium iodide staining (E and F). Representative dot plots, CFSE emission profiles and cell cycle distributions are reported in (A, C, andE), respectively. In (A), the cell:bead (C/B) ratio is reported. In (E), numbers refer to the percentage of cells with a DNA content >2n and <4n (compatible with the S phase of the cell cycle). Quantitative data on the C/B ratio, CFSE fluorescence and distribution in different phases of the cell cycle are reported in (B, D, and F), respectively (means ± SEM; n = 3). ***P < 0.001 (ANOVA plus Dunnett post-hoc test), as compared with DMSO-treated cells; #P < 0.05, ##P < 0.01 (ANOVA plus Bonferroni post-hoc test), as compared with ATRA-treated cells; +++P < 0.001 (ANOVA plus Bonferroni post-hoc test), as compared with VD-treated cells.
Cytofluorometric analyses confirmed the capacity of EGFR inhibitors to block HL-60 cells in the G0/G1 phase of the cell cycle when combined with ATRA or VD (Fig. 4E and F). HL-60 cell cultures exposed to ATRA displayed multiple hallmarks of apoptosis, including the accumulation of cell corpses with a subdiploid DNA content (Fig. 4E and F), the dissipation of the mitochondrial transmembrane potential (Δψm) (Fig. 5A and B), the exposure of phosphatidylserine on the cell surface (Fig. 5C and D), as well the permeabilization of plasma membranes36,37 (Fig. 5A–D). Conversely, erlotinib and VD alone had negligible pro-apoptotic effects in this setting, yet caused consistent levels of apoptotic cell death when combined (Figs. 4E and F and 5D). Clonogenic assays confirmed these findings. The combination of erlotinib plus ATRA or VD near-to-completely abolished the clonogenic potential of HL-60 cells, while neither of these agents alone achieved such dramatic effects (Fig. 5E and F).

Figure 5. Apoptotic response of AML cells undergoing terminal differentiation. (A–D). Human acute myeloid leukemia (AML) HL-60 cells were treated with 10 μM erlotinib (ERLO), 10 μM gefitinib (GEFI), or an equal volume of DMSO, alone or in combination with 100 nM all-trans retinoic acid (ATRA), or 50 nM 1α,25-hydroxycholecalciferol (VD), for 7 d (A and B) or 14 d (C and D), followed by the determination of apoptosis-related parameters upon DiOC6(3)/propidium iodide (PI) (A and B) or AnnexinV/PI (C and D) co-staining. Representative dot plots are depicted in (A and C) (numbers refer to the percentage of PI+ cells). In (B and D), black and white column illustrate the percentage of dead (PI+) or dying (PI-DiOC6(3)low or PI-AnnV+) cells, respectively (means ± SEM; n = 3). (E and F) Alternatively, HL60 cells were cultured in solid media in the presence of 10 μM ERLO or an equivalent volume of DMSO, alone Oncology Drug Set (which comprises most FDA-approved chemotherapeutics) or combined with 100 nM ATRA or 50 nM VD for 14 d, followed by the assessment of clonogenicity. Representative images (scale bar = 1 cm) and quantitative data (means ± SEM; n = 3) are reported in (E and F), respectively. In (B, D, and F), ***P < 0.001 (ANOVA plus Dunnett post-hoc test), as compared with DMSO-treated cells; #P < 0.05, ##P < 0.01, ###P < 0.001 (ANOVA plus Bonferroni post-hoc test), as compared with ATRA-treated cells; +++P < 0.001 (ANOVA plus Bonferroni post-hoc test), as compared with VD-treated cells.
These results indicate that erlotinib synergizes with ATRA and VD in inducing the terminal differentiation of HL-60 cells, a process that is coupled to a proliferative arrest and to the induction of apoptosis.
Mechanisms of action underlying the pro-differentiation activity of erlotinib
Several protein kinases have been involved in myeloid cell differentiation, including mitogen-activated protein kinase 1 (MAPK1, best known as extracellular signal-regulated kinase, ERK), which positively regulates the process,38,39 as well as MAPK14 (best known as p38MAPK) and multiple SRC family kinases (SFKs), which inhibit it.40,41 Immunoblotting revealed that erlotinib and gefitinib inhibit the activating phosphorylation of p38MAPK and SFKs, yet fail to suppress that of ERK and MAPK8 (best known as c-JUN N-terminal kinase 1, JNK1) (Fig. 6A). Rather erlotinib (but less so gefitinib) appeared to stimulate ERK phosphorylation (Fig. 6A).

Figure 6. Molecular mechanisms underlying the differentiation-inducing effects of EGFR inhibitors. (A). Serum-starved human acute myeloid leukemia HL-60 cells were treated with 10 μM erlotinib (ERLO), 10 μM gefitinib (GEFI) or an equal volume of DMSO for 1 h, and then processed for the immunoblotting-assisted assessment of the phosphorylation status of the indicated proteins. Representative immunoblots are reported. β actin levels were monitored to ensure equal lane loading. (B–D). HL60 cells were exposed to 10 μM ERLO, 10 μM SB203580 (SB203), 10 µM SP600165 (SP600), 5 µM U0126, 10 µM PP2, 100 nM rapamycin (RAPA), or an equivalent volume of DMSO, alone or in combination with 100 nM all-trans retinoic acid (ATRA) or 50 nM 1α,25-hydroxycholecalciferol (VD), for 72 h, then processed for the cytofluorometric assessment of CD11b expression, upon immunostaining with a CD11b-specific antibody (B), cell cycle distribution, upon propidium iodide (PI) staining (C), or apoptosis-related parameters, upon DiOC6(3)/PI co-staining (D). Quantitative data on the percentage of cells expressing CD11b and distribution in different phases of the cell cycle are reported in (B and C), respectively (means ± SEM; n = 3). In (D), black and white column illustrate the percentage of dead (PI+) or dying (PI-DiOC6(3)low) cells (means ± SEM; n = 3). In (B andD), *P < 0.05,**P < 0.01,***P < 0.001 (ANOVA plus Dunnett post-hoc test), as compared with DMSO-treated cells; ###P < 0.001 (ANOVA plus Bonferroni post-hoc test), as compared with ATRA-treated cells; ++P < 0.01, +++P < 0.001 (ANOVA plus Bonferroni post-hoc test), as compared with VD-treated cells.
Of note, the pharmacological inhibition of p38MAPK with SB203580 and SB202190 (data not shown) or that of SFKs with PP2—but neither that of MAPK kinase 1 and 2 (MAP2K1 and MAP2K2) with U01326, nor that of JNK1 with SP600125, nor that of the mammalian target of rapamycin (mTOR) with rapamycin—favored the maturation of HL-60 cells in the presence of ATRA or VD (but not in their absence), hence mimicking the effect of erlotinib (Fig. 6B). Thus, the inhibition of p38MAPK and SFKs may account for the differentiation-inducing activity of erlotinib. Next, we wondered whether SB203580 and PP2 might also mimic the cytostatic and pro-apoptotic effects of erlotinib. Indeed, SB203580 and (less so) PP2 were able to enhance the cell cycle-arresting effect of ATRA and VD (Fig. 6C) and to synergize with the latter (but not with the former, as ATRA induced per se consistent degrees of cell death) in the killing of HL-60 cells (Fig. 6D). When combined with VD, SB203580 and PP2 also mimicked erlotinib in its ability to elicit 2 hallmarks of cellular senescence, namely (1) an arrest in the G0/G1 phase of the cell cycle (defined by a diploid DNA content and quantified by cytofluorometry upon Hoechst 33342 staining) coupled to reduced RNA synthesis (assessed by cytofluorometry upon pyronin Y staining)42 (Fig. 7A and B), and (2) the acquisition of senescence-associated β-galactosidase activity43 (Fig. 7C and D).

Figure 7. Metabolic aspects of myeloid differentiation. (A–D). Human acute myeloid leukemia HL-60 cells were treated with 10 μM erlotinib (ERLO), 10 µM PP2, 10 μM SB203580 (SB203), or an equivalent volume of DMSO, alone or in combination with 50 nM 1α,25-hydroxycholecalciferol (VD), for 7 d, then processed for the cytofluorometric detection of DNA and RNA content, upon Hoechst 33342/Pyronin Y co-staining (A and B) or the colorimetric detection of senescence-associated β-galactosidase activity (C and D). Representative dot plots and images are reported in (A) (numbers refer to the percentage of cells exhibiting a 2n DNA content and reduced RNA levels) and (C) (scale bar = 5 µM; arrows indicate senescent cells), respectively. Panels (BandD) report quantitative data on the percentage of Pyronin Ylow cells in the G0/G1 phase of the cell cycle or cells expressing senescence-associated β-galactosidase, respectively (means ± SEM; n = 3). **P < 0.01, ***P < 0.001 (ANOVA plus Dunnett post-hoc test), as compared with DMSO-treated cells; +++P < 0.001 (ANOVA plus Bonferroni post-hoc test), as compared with VD-treated cells.
Altogether, these results are compatible with the hypothesis that erlotinib stimulates the differentiation of myeloid cells by inhibit p38MAPK and SFKs.
Differentiation-inducing effects of erlotinib on primary AML blasts
The results presented above underscore the capacity of erlotinib and gefitinib combined with either ATRA or VD to drive the differentiation of HL-60 (Figs. 1–7) and MOLM-13 cells (Fig. S1). We next investigated whether a similar effect might be observed on primary leukemic cells isolated from the peripheral blood of AML patients (Table S1). Indeed, a fraction of AML samples acquired the differentiation marker CD11b more robustly (difference in the percentage of CD11b+ cells > 10%) upon treatment with erlotinib plus ATRA or VD than upon exposure to any of these agents alone (Fig. 8). This applied to 4 out of 24 AML samples treated with ATRA plus erlotinib and 4 out of 9 AML samples treated with VD plus erlotinib. Thus, erlotinib appears to exacerbate the capacity of ATRA and VD to promote the differentiation not only of AML cell lines, but also of primary leukemic cells from AML patients, at least in a fraction of cases.
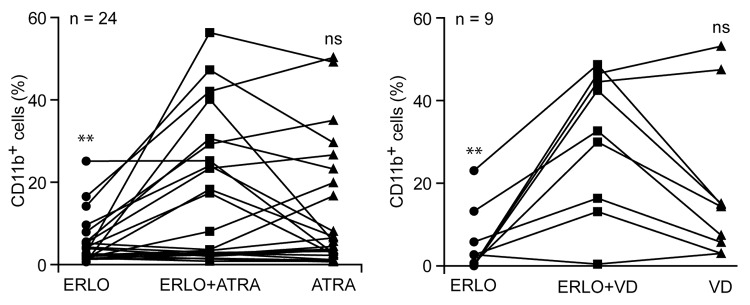
Figure 8. Effects of differentiation-inducing agents on primary leukemic cells from AML patients. CD34+ cells isolated from the peripheral blood of acute myeloid leukemia (AML) patients were treated with 10 μM erlotinib (ERLO), 100 nM all-trans retinoic acid (ATRA), 50 nM 1α,25-hydroxycholecalciferol (VD) or an equivalent volume of DMSO, alone or in combination, for 3 d, then processed for the cytofluorometric assessment of CD11b expression upon immunostaining with a CD11b-specific antibody. Results are expressed as differences between the percentage of CD11b+ cells upon treatment and that recorded in control conditions (i.e., among DMSO-treated cells). n.s., not significant, *P < 0.05, **P < 0.01 (ANOVA plus Student post-hoc t test), as compared with cells treated with ERLO plus ATRA or ERLO plus VD.
Concluding Remarks
Our results reveal the capacity of erlotinib to favor the differentiation of primary or cultured AML cells as triggered by ATRA or VD. In particular, erlotinib stimulated the acquisition of multiple differentiation markers by HL-60 cells exposed to suboptimal doses of ATRA (CD11b expression on the cell surface and NADPH oxidase activity) or VD (CD14 expression on the cell surface and monocyte-specific esterase activity). Moreover, in the presence of ATRA or VD, the pharmacological inhibition of EGFR with erlotinib promoted several processes that normally accompany terminal differentiation, including a proliferation arrest in the G0/G1 phases of the cell cycle, cellular senescence, and apoptosis. Of note, erlotinib (and gefitinib) alone exerted limited pro-differentiation activity, yet strongly synergized with VD (and less so with ATRA) in inducing the maturation of AML cells, confirming previous results by Miranda and colleagues.44 Along similar lines, gefitinib alone reportedly fails to induce the differentiation of APL NB4 cells, yet synergizes with arsenic trioxide (As2O3) in doing so.45 Moreover, the SFK inhibitor PP2 has recently been shown to rescue inducible differentiation in emergent ATRA-resistant myeloblastic leukemia cells, confirming the important role of SFKs in this process.46
The detailed molecular mechanisms whereby erlotinib exerts differentiation-inducing effects remain largely elusive. However, it appears plausible that this activity involves the inhibition of p38MAPK and/or SFKs, as (1) several distinct EGFR-targeting agents are known to interfere with the enzymatic functions of these kinases,47,48 and (2) the pharmacological inhibition of p38MAPK or SFKs mimicked the differentiation-inducting effects of EGFR inhibitors combined with ATRA of VD.40,44,49 p38MAPK phosphorylates serine and threonine (rather than tyrosine) residues and hence is unlikely to be directly inhibited by erlotinib or gefitinib. A recent report suggests that SRC is not a direct target of erlotinib either.50 Thus, the erlotinib/gefitinib-mediated inhibition of p38MAPK and SFKs must involve an indirect signal transduction cascade that deserves further scrutiny. Interestingly, a large panel of TKIs (some of which exert a biological activity overlapping with that of erlotinib) also favored the differentiation of AML cells in the presence of ATRA and/or VD. These encompass alternative EGFR inhibitors (such as lapatinib),51 as well as imatinib (targeting BCR-ABL, KIT and platelet-derived growth factor receptor, PDGFR)52 and dasatinib,53,54 which has a quite distinct inhibitory spectrum55 including the SFK LYN.56 The molecular effects of these TKIs remain to be precisely elucidated.
Of note, erlotinib has previously been shown to synergize with other anticancer agents, namely azacytidine and etoposide, in reducing the proliferation and inducing the apoptotic demise of AML cells.57,58 These synergistic effects mainly reflect the ability of erlotinib to exacerbate the intracellular accumulation of azacytidine and etoposide as it inhibits multiple transmembrane transporters of the ATP-binding cassette (ABC) family.57,58 However, at least under some circumstances, PP2 and SB203580 exacerbate the antiproliferative and cytotoxic response of HL-60 cells exposed to azacytidine. Moreover, the pharmacological inhibition of ABC transporters also promotes to some extent the differentiation-inducing effects of VD (Lainey et al., unpublished observations). Thus, there may be a link between the pharmacokinetic effects of erlotinib and its ability to inhibit p38MAPK and SFKs that warrants further investigation.
In synthesis, our results reveal an antileukemic cooperation between 2 groups of therapeutic agents, (1) relatively non-toxic natural compounds like ATRA and VD, which are well known for their differentiation-inducing activity, and (2) TKIs such as erlotinib, which can amplify the biological effects of the former. Erlotinib and gefitinib may normalize multiple signal transduction pathways that are deregulated in leukemic cells, including those centered around MAPKs and SFKs, and hence may be useful as part of combinatorial therapeutic regimens. Further clinical studies are needed to fully elucidate the antineoplastic potential of these EGFR inhibitors and determine which specific subsets of AML patients may obtain therapeutic benefits from the administration of erlotinib and gefitinib.
Materials and Methods
Chemical, cell lines, and culture conditions
Unless otherwise indicated, media and supplements for cell culture were obtained by Gibco®-Life Technologies™ and plasticware from Corning. Erlotinib hydrochloride, gefitinib, imatinib mesylate, sorafenib, and sunitinib were purchased from LC Laboratories; dasatinib, lapatinib ditosylate, and nilotinib from Selleck Chemicals LLC; 1α,25-hydroxycholecalciferol (VD), all-trans retinoic acid (ATRA) and rapamycin from Sigma-Aldrich; 4-amino-5-(4-chlorophenyl)-7-(dimethylethyl)pyrazolo[3,4-α]pyrimidine (PP2), SB203580, SB202190, and U0126 from Calbiochem; and SP600125 from InvivoGen. All chemicals were dissolved in DMSO and stored at −20 °C. Human AML HL-60 and MOLM-13 cells were purchased from the Deutsche Sammlung von Mikroorganismen und Zellkulturen (DSMZ) and cultured in RPMI 1640 medium supplemented with 20% fetal calf serum (FCS). Cells in the logarithmic phase of growth seeded at a density of 5 × 104 cells/mL were used for most experiments. For the long-term evaluation of cell death or senescence (at day 14), cells were seeded at the concentration of 1 × 104 cell/mL.
Patient samples and CD34+ cell selection
Patient samples were studied after obtaining informed consent, as per the Declaration of Helsinki. AML was diagnosed by the morphological examination of peripheral blood and bone marrow biopsies, according to the World Health Organization (WHO) 2008 and French–American–British (FAB) classifications. Relevant clinicopathological features of the patients included in this study are reported in Table S1. To obtain CD34+ cells, peripheral blood mononuclear cells (PBMCs) were isolated on a Ficoll–Paque PLUS density gradient (Amersham Biosciences) and the subjected to positive selection with the MiniMacs system (Miltenyi Biotec), according to the manufacturer’s instructions. Patient-derived CD34+ cells were cultured in standard conditions (37 °C, 5% CO2) in Iscove modified Dulbecco medium (IMDM) supplemented with 1% L-glutamine, 100 units/mL penicillin sodium, 100 μg/mL streptomycin sulfate, 10 ng/mL interleukin-3 (Peprotech), 10 ng/mL interleukin-6 (Peprotech), 50 ng/mL thrombopoietin (Peprotech), 100 ng/mL FLT3-ligand (Miltenyi Biotec), 50 ng/mL stem cell factor (SCF) (Miltenyi Biotec), and 20% BIT 9500 serum substitute (200 µg/mL transferrin, 10 µg/mL insulin, 2% bovine serum albumin; StemCell Technologies).
High-throughput screening for myeloid cell differentiation
Ten thousand HL-60 or MOLM-13 cells were seeded in V-shaped 96-well plates in 100 µL complete medium and treated with compounds from the US Drug Collection (Microsource Discovery Systems; final concentration = 1 µM) or the Oncology Drug Set (NCI/DTP Open Chemical Repository Collection; final concentration = 10 µM), alone or combined with 100 nM ATRA or 50 nM VD, for 3 d. Thereafter, cells were washed twice with PBS and stained with PBS supplemented with 10 µg/mL Hoechst 33342 (Molecular Probes®-Life Technologies™), to visualize nuclear morphology; 1:2000 Far Red Live/Dead® Fixable Dead Cell Stain (Molecular Probes®-Life Technologies™), a vital dye that only incorporates into dead cells owing to permeabilized plasma membranes; 1:200 Human FcR Blocking Reagent (Miltenyi Biotec), to minimize unspecific antibody binding; and 1:25 fluorescein isothiocyanate (FITC)-conjugated anti-CD11b antibodies (clone Bear1, Beckman Coulter), for assessing granulo-monocytic differentiation, for 20 min at 4 °C. Cells were then washed, transferred into poly-L-lysine pre-treated Black/Clear 96-well Imaging Plates (Becton Dickinson), spun for 5 min at 350 g and fixed with 4% paraformaldehyde (PFA) for 20 min at room temperature (RT). Four view fields per well were acquired by means of a BD Pathway 855 automated imaging station (Becton Dickinson) equipped with a 20× objective (Olympus) and coupled with a robotized Twister II plate handler (Caliper Life Sciences). Finally, image were segmented and analyzed for nuclear area, Live/Dead® Fixable Dead Cell Stain uptake, FITC intensity and FITC granularity by means of the BD AttoVision™ software (Becton Dickinson). Data were invariably normalized to take into account inter- and intra-plate signal variations.
Assessment of cellular differentiation by conventional approaches
For the morphological/cytochemical analysis of differentiation, AML cells were cytospun onto slides, fixed with absolute methanol for 15 min at RT, and then sequentially stained with the May–Grünwald and Giemsa reagents, as previously described.29 Morphological and cytochemical hallmarks of differentiation (i.e., decreased cytoplasmic basophilia, decreased nucleus/cytoplasm ratio, appearance of granulation, nuclear lobulation) were then quantified among at least 100 cells for experimental condition on a Primo Star microscope (Zeiss) equipped with a Plan-Achromat 100×/1.25 oil immersion objective and an AxioCam EEc 5s CCD camera (both from Zeiss).
For the cytofluorometric assessment of differentiation, AML cells were harvested, washed with PBS, incubated with the Human FcR Blocking Reagent for 15 min at RT, and then stained with allophycocyanin (APC)-conjugated anti-CD11b (5 µL/sample; clone D12, Becton Dickinson) and phycoerythrin (PE)-conjugated anti-CD14 antibodies (10 µL/sample; clone RMO52, Beckman Coulter) for 20 min at 4 °C (under protection from light) in the presence of 10 µg/mL 4',6-diamidino-2-phenylindole (DAPI, Molecular Probes®-Life Technologies™). Isotypic APC-conjugated mouse IgG2a and PE-conjugated IgG1 antibodies (both from Beckman Coulter) were used to determine threshold levels. Samples were run on a FACSCalibur™ (BD Biosciences) or a Gallios™ cytometer (Beckman Coulter) and first-line statistical evaluations were performed on live (DAPI −) cells by means of the CellQuest™ (BD Biosciences) or Kaluza™ (Beckman Coulter) software.
The capacity of mature myeloid cells to undergo respiratory bursts was assessed by measuring their nitroblue tetrazolium chloride (NBT)-reducing activity, as previously described.59 Briefly, AML cells were harvested and incubated for 30 min at 37 °C with Hanks buffer freshly supplemented with 1 µg/mL phorbol-12-myristate-13-acetate and 2 mg/mL NBT (both from Sigma-Aldrich). Samples were then cytospun on slides for 5 min at 350 g and the percentage of blue cells (containing insoluble formazan) was quantified on a Primo Star microscope (Zeiss) equipped with a Plan-Achromat 100× oil immersion objective. At least 100 cells per experimental condition were counted. Monocyte-specific α-naphtyl acetate esterase activity was evaluated with the LEUCOGNOST® EST kit (Merck Millipore), according to the manufacturer’s instructions.
Cytofluorometric assessments of cell proliferation
Cell number was quantified by adding a predefined amount of fluorescent AlignFlow™ Plus Alignment Beads (Molecular Probes®-Life Technologies™), emitting at 488 nm, to 500 µL of cell suspensions. Thereafter, samples were evaluated on a Gallios™ cytometer (Beckman Coulter) for the number of cells acquired together with 3000 beads (both identified based on forward and side scatter parameters), allowing for the estimation of cell:bead ratios. Alternatively, cell proliferation was assessed by means of the CellTrace™ CFSE Cell Proliferation Kit (Molecular Probes®-Life Technologies™), as previously described.34,60
Clonogenic assays
For the assessment of clonogenicity, 2 × 103 HL-60 cells were seeded in MethoCult® H4434 classic methylcellulose-based medium (StemCell Technologies) and treated with 2.5 µM erlotinib, 0.1 µM ATRA, 50 nM VD, or an equivalent volume of DMSO, alone or in combination. Fourteen days later, dishes were imaged by means of a G:BOX imaging system (Syngene) and colonies were quantified with the GeneTools software (Syngene).
Cytofluorometric assessment of apoptosis and cell cycle distribution
For apoptosis determinations, cells were co-stained with 1 µg/mL propidium iodide (PI) (Sigma-Aldrich), which identifies cells with ruptured plasma membranes, and either 20 nM 3,3′dihexiloxalocarbocyanine iodide (DiOC6(3); Molecular Probes®-Life Technologies™), which measures mitochondrial transmembrane potential (Δψm), or FITC-conjugated AnnexinV (Beckton Dickinson), which evaluate phosphatidylserine exposure, as previously reported.61,62 For the assessment of cell cycle distribution, 5 × 105 cells were collected, washed once with ice-cold PBS and permeabilized with 100 µL 0.5% Triton X-100. Thereafter, cells were stained with 50 µg/mL PI in 200 µL PBS supplemented with 20 μg/mL (w/v) RNase A (Molecular Probes®-Life Technologies™) for 1 h at 4 °C. Cytofluorometric acquisitions were performed on a FACSCalibur™ or a Gallios™ cytometer at a low flow rate mode. First-line analysis was performed with the CellQuest™, DIVA 6.1 (BD Biosciences) or Kaluza™ software, upon gating on the events characterized by normal forward and side scatter parameters and discrimination of doublets in a FSC-A vs. FSC-H bivariate plot.
Assessment of quiescence and senescence
To precisely distinguish the G0 and G1 phases of the cell cycle, cells were harvested and incubated with 8 µM Hoechst 33342 in complete culture medium for 30 min at 37 °C, followed by staining with and 100 µg/mL Pyronin Y (Sigma-Aldrich), which specifically labels RNA, in the presence of 10 µM Hoechst 33342, for additional 15 min at 37 °C. Tubes were then kept on ice until the acquisition on a FACSVantage flow cytometer (BD Biosciences) equipped with blue (488 nM) and UV (350 nM) lasers. The percentage of resting cells (in the G0 phase of the cell cycle) was estimated based on events having a diploid DNA content and low RNA levels on Hoechst 33342 vs. Pyronin Y dot plots. Cell senescence was evaluated using the Senescence β-Galactosidase Staining Kit (Cell Signaling Technology), according to the manufacturer’s instructions.
Immunoblotting
Immunoblotting was performed according to conventional procedures.63,64 Briefly, cells were lysed on ice in a buffer containing 1% NP40, 20 mM HEPES pH 7.9, 10 mM KCl, 1 mM EDTA, 10% glycerol, 1 mM orthovanadate, 1 mM PMSF, 1 mM dithiothreitol, and 10 µg/mL aprotinin, leupeptin, pepstatin. Total protein extracts were then separated on pre-cast NuPAGE® Novex® 4–12% Bis-Tris Gels (Invitrogen®-Life Technologies™) and subjected to standard immunoblotting procedures based on primary antibodies specific for phospho-p38MAPK (Thr180/Tyr182) (rabbit monoclonal, #9215; Cell Signaling Technology); p38MAPK (rabbit monoclonal, #2371; Cell Signaling Technology); phospho-p44/42MAPK (Thr202/Tyr204) (rabbit monoclonal, #4377; Cell Signaling Technology); p44/42MAPK (rabbit monoclonal, #4695; Cell Signaling Technology); phospho-SAPK/JNK (Thr183/Tyr185) (mouse monoclonal, #9255; Cell Signaling Technology); SAPK/JNK (rabbit antiserum, #9252; Cell Signaling Technology); phospho-SRC (Tyr416) (rabbit antiserum, #2101; Cell Signaling Technology); total SRC (mouse monoclonal, #05–184; Upstate Biotechnology-Millipore); and β actin (mouse monoclonal, #MAB1501; Chemicon-Millipore). Primary antibodies were revealed with appropriate horseradish peroxidase-conjugated secondary antibodies (Amersham) combined with an enhanced chemiluminescence detection system (Amersham) on an ImageQuant LAS 4000 camera (GE Healthcare).
Data treatment and statistical analyses
Unless otherwise specified, all experiments were performed in triplicate parallel instances and independently repeated at least 2 times. Data were analyzed with Excel (Microsoft) and Prismv.5 (GraphPad Software Inc). Statistical significance was invariably assessed by means of ANOVA followed by Dunnett, Bonferroni, or Student t post-hoc tests, as appropriate. P values < 0.05 were considered statistically significant.
Supplementary Material
Acknowledgments
We thank Stéphane De Botton (Gustave Roussy; Villejuif, France) for providing patient samples. EL received a PhD fellowship from Institut National de la Santé et de la recherche Médicale, AW was supported by a fellowship from Association pour la Recherche sur le Cancer (ARC). LG is supported by the LabEx Immuno-Oncology. GK is financed by the Ligue contre le Cancer (équipe labelisée); Agence National de la Recherche (ANR); AXA Chair for Longevity Research; ARC; Cancéropôle Ile-de-France; Institut National du Cancer (INCa); Fondation Bettencourt-Schueller; Fondation de France; Fondation pour la Recherche Médicale (FRM); the European Commission (ArtForce); the European Research Council (ERC); the LabEx Immuno-Oncology; the SIRIC Stratified Oncology Cell DNA Repair and Tumor Immune Elimination (SOCRATE); the SIRIC Cancer Research and Personalized Medicine (CARPEM); and the Paris Alliance of Cancer Research Institutes (PACRI).
Glossary
Abbreviations:
- ABC
ATP-binding cassette
- AML
acute myeloid leukemia
- APC
allophycocyanin
- APL
acute promyelocytic leukemia
- ATRA
all-trans retinoic acid
- CFSE
5-(and-6)-carboxyfluorescein diacetate succinimidyl ester
- DAPI
4',6-diamidino-2-phenylindole
- DiOC6(3)
3,3′-dihexiloxalocarbocyanine iodide
- FAB
French-American-British
- FITC
fluorescein isothiocyanate
- EGFR
epidermal growth factor receptor
- JNK1
c-JUN N-terminal kinase 1
- MAPK
mitogen-activated protein kinase
- MAPK2K
MAPK kinase
- mTOR
mammalian target of rapamycin
- NBT
nitroblue tetrazolium chloride
- PDGFR
platelet-derived growth factor receptor
- PI
propidium iodide
- RT
room temperature
- SFK
SRC family kinase
- TKI
tyrosine kinase inhibitor
- VD
1α,25-hydroxycholecalciferol
Disclosure of Potential Conflicts of Interest
The authors declare no conflicts of interest.
Footnotes
Previously published online: www.landesbioscience.com/journals/cc/article/26016
References
- 1.Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144:646–74. doi: 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- 2.Pandolfi A, Barreyro L, Steidl U. Concise review: preleukemic stem cells: molecular biology and clinical implications of the precursors to leukemia stem cells. Stem Cells Transl Med. 2013;2:143–50. doi: 10.5966/sctm.2012-0109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Fulda S, Galluzzi L, Kroemer G. Targeting mitochondria for cancer therapy. Nat Rev Drug Discov. 2010;9:447–64. doi: 10.1038/nrd3137. [DOI] [PubMed] [Google Scholar]
- 4.Petrie K, Zelent A, Waxman S. Differentiation therapy of acute myeloid leukemia: past, present and future. Curr Opin Hematol. 2009;16:84–91. doi: 10.1097/MOH.0b013e3283257aee. [DOI] [PubMed] [Google Scholar]
- 5.Nowak D, Stewart D, Koeffler HP. Differentiation therapy of leukemia: 3 decades of development. Blood. 2009;113:3655–65. doi: 10.1182/blood-2009-01-198911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Burnett A, Wetzler M, Löwenberg B. Therapeutic advances in acute myeloid leukemia. J Clin Oncol. 2011;29:487–94. doi: 10.1200/JCO.2010.30.1820. [DOI] [PubMed] [Google Scholar]
- 7.Hughes PJ, Marcinkowska E, Gocek E, Studzinski GP, Brown G. Vitamin D3-driven signals for myeloid cell differentiation--implications for differentiation therapy. Leuk Res. 2010;34:553–65. doi: 10.1016/j.leukres.2009.09.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Deeb KK, Trump DL, Johnson CS. Vitamin D signalling pathways in cancer: potential for anticancer therapeutics. Nat Rev Cancer. 2007;7:684–700. doi: 10.1038/nrc2196. [DOI] [PubMed] [Google Scholar]
- 9.Trump DL, Deeb KK, Johnson CS. Vitamin D: considerations in the continued development as an agent for cancer prevention and therapy. Cancer J. 2010;16:1–9. doi: 10.1097/PPO.0b013e3181c51ee6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chadha MK, Tian L, Mashtare T, Payne V, Silliman C, Levine E, Wong M, Johnson C, Trump DL. Phase 2 trial of weekly intravenous 1,25 dihydroxy cholecalciferol (calcitriol) in combination with dexamethasone for castration-resistant prostate cancer. Cancer. 2010;116:2132–9. doi: 10.1002/cncr.24973. [DOI] [PubMed] [Google Scholar]
- 11.Estey EH. Acute myeloid leukemia: 2013 update on risk-stratification and management. Am J Hematol. 2013;88:318–27. doi: 10.1002/ajh.23404. [DOI] [PubMed] [Google Scholar]
- 12.Visser O, Trama A, Maynadié M, Stiller C, Marcos-Gragera R, De Angelis R, Mallone S, Tereanu C, Allemani C, Ricardi U, et al. RARECARE Working Group Incidence, survival and prevalence of myeloid malignancies in Europe. Eur J Cancer. 2012;48:3257–66. doi: 10.1016/j.ejca.2012.05.024. [DOI] [PubMed] [Google Scholar]
- 13.Renneville A, Roumier C, Biggio V, Nibourel O, Boissel N, Fenaux P, Preudhomme C. Cooperating gene mutations in acute myeloid leukemia: a review of the literature. Leukemia. 2008;22:915–31. doi: 10.1038/leu.2008.19. [DOI] [PubMed] [Google Scholar]
- 14.Fröhling S, Scholl C, Gilliland DG, Levine RL. Genetics of myeloid malignancies: pathogenetic and clinical implications. J Clin Oncol. 2005;23:6285–95. doi: 10.1200/JCO.2005.05.010. [DOI] [PubMed] [Google Scholar]
- 15.Roboz GJ. Current treatment of acute myeloid leukemia. Curr Opin Oncol. 2012;24:711–9. doi: 10.1097/CCO.0b013e328358f62d. [DOI] [PubMed] [Google Scholar]
- 16.Yanada M, Naoe T. Acute myeloid leukemia in older adults. Int J Hematol. 2012;96:186–93. doi: 10.1007/s12185-012-1137-3. [DOI] [PubMed] [Google Scholar]
- 17.Pollyea DA, Kohrt HE, Medeiros BC. Acute myeloid leukaemia in the elderly: a review. Br J Haematol. 2011;152:524–42. doi: 10.1111/j.1365-2141.2010.08470.x. [DOI] [PubMed] [Google Scholar]
- 18.Löwenberg B, Ossenkoppele GJ, van Putten W, Schouten HC, Graux C, Ferrant A, Sonneveld P, Maertens J, Jongen-Lavrencic M, von Lilienfeld-Toal M, et al. Dutch-Belgian Cooperative Trial Group for Hemato-Oncology (HOVON) German AML Study Group (AMLSG) Swiss Group for Clinical Cancer Research (SAKK) Collaborative Group High-dose daunorubicin in older patients with acute myeloid leukemia. N Engl J Med. 2009;361:1235–48. doi: 10.1056/NEJMoa0901409. [DOI] [PubMed] [Google Scholar]
- 19.Raelson JV, Nervi C, Rosenauer A, Benedetti L, Monczak Y, Pearson M, Pelicci PG, Miller WH., Jr. The PML/RAR alpha oncoprotein is a direct molecular target of retinoic acid in acute promyelocytic leukemia cells. Blood. 1996;88:2826–32. [PubMed] [Google Scholar]
- 20.Lindhagen E, Eriksson A, Wickström M, Danielsson K, Grundmark B, Henriksson R, Nygren P, Aleskog A, Larsson R, Höglund M. Significant cytotoxic activity in vitro of the EGFR tyrosine kinase inhibitor gefitinib in acute myeloblastic leukaemia. Eur J Haematol. 2008;81:344–53. doi: 10.1111/j.1600-0609.2008.01120.x. [DOI] [PubMed] [Google Scholar]
- 21.Boehrer S, Adès L, Galluzzi L, Tajeddine N, Tailler M, Gardin C, de Botton S, Fenaux P, Kroemer G. Erlotinib and gefitinib for the treatment of myelodysplastic syndrome and acute myeloid leukemia: a preclinical comparison. Biochem Pharmacol. 2008;76:1417–25. doi: 10.1016/j.bcp.2008.05.024. [DOI] [PubMed] [Google Scholar]
- 22.Pitini V, Arrigo C, Altavilla G. Erlotinib in a patient with acute myelogenous leukemia and concomitant non-small-cell lung cancer. J Clin Oncol. 2008;26:3645–6. doi: 10.1200/JCO.2008.17.0357. [DOI] [PubMed] [Google Scholar]
- 23.Chan G, Pilichowska M. Complete remission in a patient with acute myelogenous leukemia treated with erlotinib for non small-cell lung cancer. Blood. 2007;110:1079–80. doi: 10.1182/blood-2007-01-069856. [DOI] [PubMed] [Google Scholar]
- 24.Boehrer S, Adès L, Braun T, Galluzzi L, Grosjean J, Fabre C, Le Roux G, Gardin C, Martin A, de Botton S, et al. Erlotinib exhibits antineoplastic off-target effects in AML and MDS: a preclinical study. Blood. 2008;111:2170–80. doi: 10.1182/blood-2007-07-100362. [DOI] [PubMed] [Google Scholar]
- 25.Sun JZ, Lu Y, Xu Y, Liu F, Li FQ, Wang QL, Wu CT, Hu XW, Duan HF. Epidermal growth factor receptor expression in acute myelogenous leukaemia is associated with clinical prognosis. Hematol Oncol. 2012;30:89–97. doi: 10.1002/hon.1002. [DOI] [PubMed] [Google Scholar]
- 26.Stegmaier K, Ross KN, Colavito SA, O’Malley S, Stockwell BR, Golub TR. Gene expression-based high-throughput screening(GE-HTS) and application to leukemia differentiation. Nat Genet. 2004;36:257–63. doi: 10.1038/ng1305. [DOI] [PubMed] [Google Scholar]
- 27.Stegmaier K, Corsello SM, Ross KN, Wong JS, Deangelo DJ, Golub TR. Gefitinib induces myeloid differentiation of acute myeloid leukemia. Blood. 2005;106:2841–8. doi: 10.1182/blood-2005-02-0488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kansas GS, Muirhead MJ, Dailey MO. Expression of the CD11/CD18, leukocyte adhesion molecule 1, and CD44 adhesion molecules during normal myeloid and erythroid differentiation in humans. Blood. 1990;76:2483–92. [PubMed] [Google Scholar]
- 29.Lainey E, Thépot S, Bouteloup C, Sébert M, Adès L, Tailler M, Gardin C, de Botton S, Baruchel A, Fenaux P, et al. Tyrosine kinase inhibitors for the treatment of acute myeloid leukemia: delineation of anti-leukemic mechanisms of action. Biochem Pharmacol. 2011;82:1457–66. doi: 10.1016/j.bcp.2011.05.011. [DOI] [PubMed] [Google Scholar]
- 30.Breitman TR, Hemmi H, Imaizumi M. Induction by physiological agents of differentiation of the human leukemia cell line HL-60 to cells with functional characteristics. Prog Clin Biol Res. 1986;226:215–33. [PubMed] [Google Scholar]
- 31.Sokol RJ, Hudson G, Wales JM, Goldstein DJ, James NT. Quantitative enzyme cytochemistry during human macrophage development. J Anat. 1993;183:97–101. [PMC free article] [PubMed] [Google Scholar]
- 32.Kroemer G, Galluzzi L, Vandenabeele P, Abrams J, Alnemri ES, Baehrecke EH, Blagosklonny MV, El-Deiry WS, Golstein P, Green DR, et al. Nomenclature Committee on Cell Death 2009 Classification of cell death: recommendations of the Nomenclature Committee on Cell Death 2009. Cell Death Differ. 2009;16:3–11. doi: 10.1038/cdd.2008.150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Galluzzi L, Vitale I, Abrams JM, Alnemri ES, Baehrecke EH, Blagosklonny MV, Dawson TM, Dawson VL, El-Deiry WS, Fulda S, et al. Molecular definitions of cell death subroutines: recommendations of the Nomenclature Committee on Cell Death 2012. Cell Death Differ. 2012;19:107–20. doi: 10.1038/cdd.2011.96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Vitale I, Jemaà M, Galluzzi L, Metivier D, Castedo M, Kroemer G. Cytofluorometric assessment of cell cycle progression. Methods Mol Biol. 2013;965:93–120. doi: 10.1007/978-1-62703-239-1_6. [DOI] [PubMed] [Google Scholar]
- 35.Boehrer S, Adès L, Tajeddine N, Hofmann WK, Kriener S, Bug G, Ottmann OG, Ruthardt M, Galluzzi L, Fouassier C, et al. Suppression of the DNA damage response in acute myeloid leukemia versus myelodysplastic syndrome. Oncogene. 2009;28:2205–18. doi: 10.1038/onc.2009.69. [DOI] [PubMed] [Google Scholar]
- 36.Kepp O, Galluzzi L, Lipinski M, Yuan J, Kroemer G. Cell death assays for drug discovery. Nat Rev Drug Discov. 2011;10:221–37. doi: 10.1038/nrd3373. [DOI] [PubMed] [Google Scholar]
- 37.Galluzzi L, Aaronson SA, Abrams J, Alnemri ES, Andrews DW, Baehrecke EH, Bazan NG, Blagosklonny MV, Blomgren K, Borner C, et al. Guidelines for the use and interpretation of assays for monitoring cell death in higher eukaryotes. Cell Death Differ. 2009;16:1093–107. doi: 10.1038/cdd.2009.44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zhang T, He YM, Wang JS, Shen J, Xing YY, Xi T. Ursolic acid induces HL60 monocytic differentiation and upregulates C/EBPβ expression by ERK pathway activation. Anticancer Drugs. 2011;22:158–65. doi: 10.1097/CAD.0b013e3283409673. [DOI] [PubMed] [Google Scholar]
- 39.Yen A, Roberson MS, Varvayanis S, Lee AT. Retinoic acid induced mitogen-activated protein (MAP)/extracellular signal-regulated kinase (ERK) kinase-dependent MAP kinase activation needed to elicit HL-60 cell differentiation and growth arrest. Cancer Res. 1998;58:3163–72. [PubMed] [Google Scholar]
- 40.Zhang J, Harrison JS, Studzinski GP. Isoforms of p38MAPK gamma and delta contribute to differentiation of human AML cells induced by 1,25-dihydroxyvitamin D₃. Exp Cell Res. 2011;317:117–30. doi: 10.1016/j.yexcr.2010.08.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Congleton J, MacDonald R, Yen A. Src inhibitors, PP2 and dasatinib, increase retinoic acid-induced association of Lyn and c-Raf (S259) and enhance MAPK-dependent differentiation of myeloid leukemia cells. Leukemia. 2012;26:1180–8. doi: 10.1038/leu.2011.390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Gothot A, Pyatt R, McMahel J, Rice S, Srour EF. Functional heterogeneity of human CD34(+) cells isolated in subcompartments of the G0 /G1 phase of the cell cycle. Blood. 1997;90:4384–93. [PubMed] [Google Scholar]
- 43.Itahana K, Campisi J, Dimri GP. Methods to detect biomarkers of cellular senescence: the senescence-associated beta-galactosidase assay. Methods Mol Biol. 2007;371:21–31. doi: 10.1007/978-1-59745-361-5_3. [DOI] [PubMed] [Google Scholar]
- 44.Miranda MB, Redner RL, Johnson DE. Inhibition of Src family kinases enhances retinoic acid induced gene expression and myeloid differentiation. Mol Cancer Ther. 2007;6:3081–90. doi: 10.1158/1535-7163.MCT-07-0514. [DOI] [PubMed] [Google Scholar]
- 45.Noh EK, Kim H, Park MJ, Baek JH, Park JH, Cha SJ, Won JH, Min YJ. Gefitinib enhances arsenic trioxide (AS2O3)-induced differentiation of acute promyelocytic leukemia cell line. Leuk Res. 2010;34:1501–5. doi: 10.1016/j.leukres.2010.02.016. [DOI] [PubMed] [Google Scholar]
- 46.Jensen HA, Styskal LE, Tasseff R, Bunaciu RP, Congleton J, Varner JD, Yen A. The Src-family kinase inhibitor PP2 rescues inducible differentiation events in emergent retinoic acid-resistant myeloblastic leukemia cells. PLoS One. 2013;8:e58621. doi: 10.1371/journal.pone.0058621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Weber C, Schreiber TB, Daub H. Dual phosphoproteomics and chemical proteomics analysis of erlotinib and gefitinib interference in acute myeloid leukemia cells. J Proteomics. 2012;75:1343–56. doi: 10.1016/j.jprot.2011.11.004. [DOI] [PubMed] [Google Scholar]
- 48.Boehrer S, Galluzzi L, Lainey E, Bouteloup C, Tailler M, Harper F, Pierron G, Adès L, Thépot S, Sébert M, et al. Erlotinib antagonizes constitutive activation of SRC family kinases and mTOR in acute myeloid leukemia. Cell Cycle. 2011;10:3168–75. doi: 10.4161/cc.10.18.16599. [DOI] [PubMed] [Google Scholar]
- 49.Jones JE, Wang L, Kropf PL, Duan R, Johnson DE. Src family kinase gene targets during myeloid differentiation: identification of the EGR-1 gene as a direct target. Leukemia. 2009;23:1933–5. doi: 10.1038/leu.2009.118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Stabile LP, He G, Lui VW, Henry C, Gubish CT, Joyce S, Quesnelle KM, Siegfried JM, Grandis JR. c-Src activation mediates erlotinib resistance in head and neck cancer by stimulating c-Met. Clin Cancer Res. 2013;19:380–92. doi: 10.1158/1078-0432.CCR-12-1555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Huang HL, Chen YC, Huang YC, Yang KC, Pan Hy, Shih SP, Chen YJ. Lapatinib induces autophagy, apoptosis and megakaryocytic differentiation in chronic myelogenous leukemia K562 cells. PLoS One. 2011;6:e29014. doi: 10.1371/journal.pone.0029014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Quintás-Cardama A, Kantarjian H, Cortes J. Imatinib and beyond--exploring the full potential of targeted therapy for CML. Nat Rev Clin Oncol. 2009;6:535–43. doi: 10.1038/nrclinonc.2009.112. [DOI] [PubMed] [Google Scholar]
- 53.Kropf PL, Wang L, Zang Y, Redner RL, Johnson DE. Dasatinib promotes ATRA-induced differentiation of AML cells. Leukemia. 2010;24:663–5. doi: 10.1038/leu.2009.267. [DOI] [PubMed] [Google Scholar]
- 54.Chevalier N, Solari ML, Becker H, Pantic M, Gärtner F, Maul-Pavicic A, Hübner J, Wäsch R, Schmitt-Gräff A, Lübbert M. Robust in vivo differentiation of t(8;21)-positive acute myeloid leukemia blasts to neutrophilic granulocytes induced by treatment with dasatinib. Leukemia. 2010;24:1779–81. doi: 10.1038/leu.2010.151. [DOI] [PubMed] [Google Scholar]
- 55.Fedorov O, Müller S, Knapp S. The (un)targeted cancer kinome. Nat Chem Biol. 2010;6:166–9. doi: 10.1038/nchembio.297. [DOI] [PubMed] [Google Scholar]
- 56.ten Hacken E, Scielzo C, Bertilaccio MT, Scarfò L, Apollonio B, Barbaglio F, Stamatopoulos K, Ponzoni M, Ghia P, Caligaris-Cappio F. Targeting the LYN/HS1 signaling axis in chronic lymphocytic leukemia. Blood. 2013;121:2264–73. doi: 10.1182/blood-2012-09-457119. [DOI] [PubMed] [Google Scholar]
- 57.Lainey E, Wolfromm A, Marie N, Enot D, Scoazec M, Bouteloup C, Leroy C, Micol JB, De Botton S, Galluzzi L, et al. Azacytidine and erlotinib exert synergistic effects against acute myeloid leukemia. Oncogene. 2012 doi: 10.1038/onc.2012.469. In press. [DOI] [PubMed] [Google Scholar]
- 58.Lainey E, Sébert M, Thépot S, Scoazec M, Bouteloup C, Leroy C, De Botton S, Galluzzi L, Fenaux P, Kroemer G. Erlotinib antagonizes ABC transporters in acute myeloid leukemia. Cell Cycle. 2012;11:4079–92. doi: 10.4161/cc.22382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Collins SJ, Bodner A, Ting R, Gallo RC. Induction of morphological and functional differentiation of human promyelocytic leukemia cells (HL-60) by componuds which induce differentiation of murine leukemia cells. Int J Cancer. 1980;25:213–8. doi: 10.1002/ijc.2910250208. [DOI] [PubMed] [Google Scholar]
- 60.Jemaà M, Galluzzi L, Kepp O, Castedo M, Rello-Varona S, Vitale I, Kroemer G. Transgenerational cell fate profiling: a method for the graphical presentation of complex cell cycle alterations. Cell Cycle. 2013;12:183–90. doi: 10.4161/cc.23046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Vitale I, Galluzzi L, Vivet S, Nanty L, Dessen P, Senovilla L, Olaussen KA, Lazar V, Prudhomme M, Golsteyn RM, et al. Inhibition of Chk1 kills tetraploid tumor cells through a p53-dependent pathway. PLoS One. 2007;2:e1337. doi: 10.1371/journal.pone.0001337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Galluzzi L, Marsili S, Vitale I, Senovilla L, Michels J, Garcia P, Vacchelli E, Chatelut E, Castedo M, Kroemer G. Vitamin B6 metabolism influences the intracellular accumulation of cisplatin. Cell Cycle. 2013;12:417–21. doi: 10.4161/cc.23275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Galluzzi L, Vitale I, Senovilla L, Olaussen KA, Pinna G, Eisenberg T, Goubar A, Martins I, Michels J, Kratassiouk G, et al. Prognostic impact of vitamin B6 metabolism in lung cancer. Cell Rep. 2012;2:257–69. doi: 10.1016/j.celrep.2012.06.017. [DOI] [PubMed] [Google Scholar]
- 64.de La Motte Rouge T, Galluzzi L, Olaussen KA, Zermati Y, Tasdemir E, Robert T, Ripoche H, Lazar V, Dessen P, Harper F, et al. A novel epidermal growth factor receptor inhibitor promotes apoptosis in non-small cell lung cancer cells resistant to erlotinib. Cancer Res. 2007;67:6253–62. doi: 10.1158/0008-5472.CAN-07-0538. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.