

Abstract
Triple-negative breast cancer (TNBC) represents an aggressive subtype, for which radiation and chemotherapy are the only options. Here we describe the identification of disulfiram, an FDA-approved drug used to treat alcoholism, as well as the related compound thiram, as the most potent growth inhibitors following high-throughput screens of 3185 compounds against multiple TNBC cell lines. The average IC50 for disulfiram was ~300 nM. Drug affinity responsive target stability (DARTS) analysis identified IQ motif-containing factors IQGAP1 and MYH9 as direct binding targets of disulfiram. Indeed, knockdown of these factors reduced, though did not completely abolish, cell growth. Combination treatment with 4 different drugs commonly used to treat TNBC revealed that disulfiram synergizes most effectively with doxorubicin to inhibit cell growth of TNBC cells. Disulfiram and doxorubicin cooperated to induce cell death as well as cellular senescence, and targeted the ESA+/CD24-/low/CD44+ cancer stem cell population. Our results suggest that disulfiram may be repurposed to treat TNBC in combination with doxorubicin.
Keywords: triple-negative breast cancer, high-throughput screens, disulfiram, cancer stem cells, IQGAP1, MYH9
Introduction
Breast cancer (BC) is a highly heterogeneous disease that includes ERα+, HER2+, and triple-negative forms (TNBC; ERα −, progesterone receptor [PR]−, and HER2−).1 TNBC can be further divided into several different subtypes, including basal-like and claudin-low/mesenchymal-like BC.2 Cell lines established from these tumors are referred to as Basal-A and Basal-B, respectively. These cell lines resemble the BCs from which they were derived and can be used as surrogates for primary tumors.3
Patients affected by TNBC are currently treated with single agent or combination therapies of doxorubicin, paclitaxel, 5-fluorouracil, epirubicin, methotrexate, cyclophosphamide, cisplatin, and gemcitabine.4-12 Although effective, most combinations have adverse side effects, including neutropenia, neuropathy, and cardiotoxicity, and many tumors still progress to metastasis.4,9 Regimes with a more tolerable toxicity tend to yield a much lower overall response rate.10,11 There is, therefore, a great need to improve efficacy of existing combination therapies by mitigating toxic side effects and improving complete response rates. One way of achieving this is through discovery of new drugs, either as single-agent treatments or in combination with existing regimes. Here we performed a high-throughput drug screen against human TNBC cells to identify novel therapeutics and identified disulfiram, an FDA-approved drug used to treat alcoholism, as the most potent growth inhibitor.
Results
High-throughput drug screening identifies disulfiram as an effective growth inhibitor of TNBC cells
To discover novel therapeutics for TNBC, we performed a robotic-assisted high-throughput screen of 4 different TNBC cell lines with 3185 small molecules, including 2000 and 1185 compounds from the Spectrum and Prestwick libraries, respectively. These partially overlapping libraries consist of FDA-approved drugs and additional agents with known biological activity. The 4 cell lines used in our screens (HCC70, MDA-MB-231, MDA-MB-436, and Bt549) represent a wide range of TNBCs with respect to pRb and p53 tumor suppressors status as well as subtype (basal-like and claudin-low). Each screen and validation of hits was performed in 384-well format, using alamar blue viability assay readout. Figure 1A–D depicts the average response of TNBC lines against both libraries and the top 5 most potent drugs in each (top 50 most potent drugs listed in Tables S1 and S2). Among the most potent compounds were known antineoplastic agents such as doxorubicin. In addition, a small number of compounds not previously known to target TNBC were identified, including disulfiram (DSF) and its structurally related analog thiram (Fig. 1C and D).

Figure 1. High-throughput screen of 3185 compounds with known biological activities against 4 human-derived TNBC cell lines (MDA-MB-231, MDA-MB-436, HCC70, Bt549). Shown are the average responses by the 4 lines to (A) Spectrum library (1 µM, 2000 drugs), (B) Prestwick library (0.8 µM, 1185 drugs). (C and D) Top 5 hits from the Spectrum and Prestwick libraries; disulfiram and thiram are highlighted in red. Values represent the average cell viability of all 4 lines expressed as a percentage of vehicle treated control. (E–J) Validation and dose-response curves for select hits using alamar blue viability assay, performed in triplicate.
Dose-response curves for DSF and a number of top hits were performed on all 4 TNBC lines. DSF was more effective against each cell line than doxorubicin, daunorubicin, mitoxantrone, colchicine, or paclitaxel (Fig. 1E–J). Notably, MDA-MB-436 cells were resistant to the mitotic inhibitors colchicine and paclitaxel but highly susceptible to DSF. To further test for efficacy of DSF against TNBC, we performed MTT viability assays on a panel of 13 human-derived TNBC lines (Fig. 2A and B). Both DSF and thiram effectively suppressed growth of TNBC cells, with an average IC50 across all lines of 300 nM and 360 nM, respectively. The effect of these drugs was similar for both Basal-A and Basal-B TNBC cell lines (Fig. 2C and D).

Figure 2. Dose-response curves for a panel of 13 human-derived TNBC cell lines treated with disulfiram or thiram. (A) Response to disulfiram for each individual line by MTT viability assay. Average IC50 = 300 nM. n = 3–5, each performed in triplicate. (B) Response to thiram for each individual line. Average IC50 = 360 nM. n = 3–5, each performed in triplicate. (C) Average response to disulfiram based on TNBC subtype. (D) Average response to thiram based on TNBC subtype. Basal-like (BaA): HCC1954, HCC1569, HCC3153, HCC70, HCC1937, and MDA-MB-468. Claudin-low (BaB): MDA-MB-436, MDA-MB-231, MDA-MB-157, Bt549, SUM149, Hs578t, and HCC38.
Identification of IQGAP1 scaffold protein and myosin heavy chain 9 as binding targets for disulfiram
Thiram, a sulfur fungicide applied directly to the skin for treatment of scabies, is moderately toxic by ingestion and highly toxic when inhaled.13 Disulfiram (DSF) is used to treat alcoholism based on its inhibitory effect on acetaldehyde dehydrogenases, which are encoded by 3 of the 19 different aldehyde dehydrogenase (ALDH) genes, ALDH1A1, ALDH2, and ALDH1B1/ALDH5. It has mild side effects in humans that include headache, metallic taste, and drowsiness due to increased production of tryptophol in the liver.14,15 Beyond treatment of alcoholism, disulfiram was found in non-biased drug screens to show anti-neoplastic activity against prostate cancer and glioblastoma16-18 and is the subject of ongoing clinical trials for lung and liver cancer.19,20 DSF has been implicated in multi-drug resistance, NFκB-mediated apoptosis, phosphoinositide 3-kinase signaling, and induction of p53.21-24 ALDH1A3 is commonly elevated in cancer stem cells (CSC).25 However, this isoform is not expressed in MDA-MB-231 cells,26 in which we showed DSF to have a potent inhibitory effect (Figs. 1 and 2). Thus, it is unlikely that DSF exerts its effects through ALDH1A3.
To identify targets of DSF, we performed Drug Affinity Responsive Target Stability (DARTS) analysis27-29 using MDA-MB-231 cells treated with 100 nM DSF. Cell lysates were incubated with varying concentrations of DSF before digestion with pronase followed by SDS-PAGE and silver staining (Fig. 3A). DSF-protected bands were identified, extracted, and subject to mass spectrometry. Remarkably, this analysis identified 2 IQ motif-containing factors as candidate targets for DSF: IQ motif containing GTPase activating protein 1 (IQGAP1) and myosin heavy chain 9 (MYH9) (Fig. 3B–C).

Figure 3. Disulfiram binds IQGAP1 and MYH9 to partially inhibit cell growth. (A) DARTS was performed on MDA-MB-231 cell lysates incubated with either vehicle (DMSO) or varying concentrations of disulfiram and visualized via silver staining. Mass spectrometry analysis of disulfiram-protected bands identified IQGAP1 (red arrowhead) and MYH9 (black arrowheads). (B) DARTS–western blot analysis was performed to validate IQGAP1 and MYH9 as binding targets of disulfiram. β-actin was used as a negative control. IQGAP1 and MYH9 over β-actin ratios, calculated from ImageJ analysis,91 are shown. Arrow indicates band used to calculate enrichment ratio over control. (C) DARTS–western blot analysis using BT-549 (Basal B) cell lysates. GAPDH was used as a negative control. (D and E) western blot analysis demonstrating efficient knockdown of IQGAP1 and MYH9 after treatment with RNAi relative to disulfiram (DSF), DharmaFECT 4 transfection reagent, or no treatment. (F) MTT viability assay after treatment with MYH9 or IQGAP1 siRNA, or disulfiram (DSF). Values represent % of vehicle control. n = 3, each performed in triplicate.
IQGAP1 is a ubiquitously expressed scaffold protein involved in cytoskeletal organization, cellular adhesion, and cell cycle regulation.30-34 MYH9 is a subunit of myosin IIA that plays a role in cell motility, cell shape, and cytokinesis.35-37 Both proteins contain IQ calmodulin-binding motifs, a highly basic α-helical sequence of ~20–25 amino acids.38 To investigate these targets, we used Dharmacon RNA interference (RNAi, Thermo Scientific Dharmacon) in MDA-MB-231 cells. Western blot analysis confirmed near-complete or efficient knockdown of IQGAP1 and MYH9, respectively (Fig. 3D and E). Knockdown of each factor resulted in a 25–30% inhibition of cell growth 72 h post transfection (Fig. 3F). In contrast, DSF inhibited cell growth by 75% at 250 nM, 72 h post-treatment. Combined knockdown of both proteins did not consistently enhance growth inhibition (Fig. S1). Thus, while IQGAP1 and MYH9 interact with DSF, additional factors, possibly other IQ-motif containing proteins, likely interact with and contribute to the global effect of DSF. Notably, the partial effect seen here with IQGAP1 knockdown in MDA-MB-231 tumor cells is in stark contrast to a recent paper showing that IQGAP1 inhibition suppresses tumor initiation/hyperproliferation driven by RAS signaling activation39 (see “Discussion”).
Pathway analysis reveals divergent effects of disulfiram on cellular signaling
To identify the global effects of disulfiram on cellular signaling, MDA-MB-231 cells were treated with vehicle alone, 100 nM or 250 nM DSF. After 72 h, RNA was extracted and subjected to transcriptional profiling, followed by analysis via Database for Annotation, Visualization and Integrated Discovery (DAVID), and the results were finally visualized by “functional enrichment maps”.40 This analysis revealed that DSF affected multiple signaling pathways (Fig. 4). Upregulated pathways included anti-survival genes such as PTEN and caspases.41,42 Also upregulated were sphingolipids and their receptor EDG1/S1P1 pathways, which promote apoptosis as well as autophagy and cellular senescence.43-48

Figure 4. Pathway analysis of MDA-MB-231 cells 72 h post treatment with 100 nM or 250 nM disulfiram (DSF), compared with untreated control. Expression data were analyzed by Database for Annotation, Visualization and Integrated Discovery (DAVID) and visualized by “functional enrichment maps”.40 Pathways of interest are marked with red boxes.
Disulfiram synergizes with doxorubicin to inhibit growth of TNBC cells
We next asked whether DSF could cooperate with other drugs commonly used to treat TNBC. Specifically, we tested the combined effect of DSF plus doxorubicin, gemcitabine, paclitaxel, or the CDK4/6 inhibitor PD-0332991 (Fig. 5; Fig. S2). The most effective combination in MDA-MB-231 cells was DSF plus doxorubicin, and a synergistic response of these 2 drugs was observed across a range of concentrations (Fig. 5A). Additional analysis revealed that the effect of DSF plus doxorubicin on MDA-MB-468, Hs578t, and HCC38 cells was additive, while the effect on HCC70 cells was synergistic (Fig. 5B) (See “Materials and Methods” for criteria). As doxorubicin is highly toxic, the additive/synergistic effect observed here with DSF may allow for lower doses and better clinical outcome.
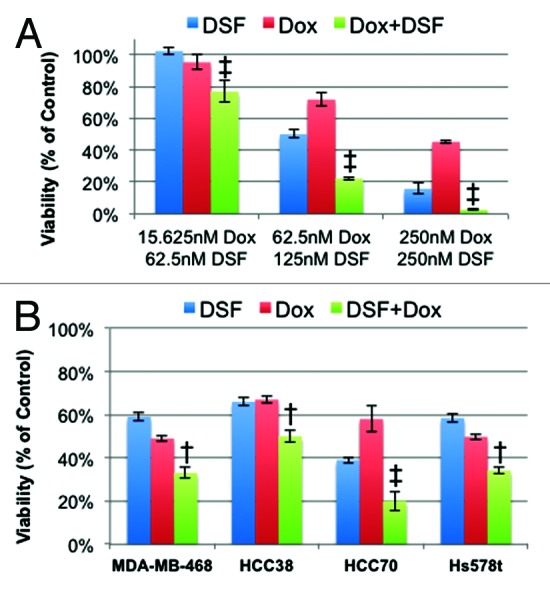
Figure 5. Effect of combination treatment with disulfiram (DSF) and doxorubicin (Dox). (A) MDA-MB-231 cells treated with DSF and Dox in combination or alone and analyzed by MTT assay. (B) MDA-MB-468, HCC38, HCC70, and Hs578t treated with their respective IC50 doses of DSF and Dox in combination or alone. All values represent % of untreated control. † denotes additive effect; ‡ denotes synergistic effect. (See “Materials and Methods”).
Disulfiram cooperates with doxorubicin to induce apoptosis and senescence in MDA-MB-231 cells and target the CSC fraction
Next, we sought to determine the mechanism by which DSF inhibits growth of TNBC cells. The pathway analysis revealed activation of pathways associated with cell death and senescence. Following doxorubicin and/or DSF treatment, many MDA-MB-231 cells died and lifted off the plate. The remaining population, including supernatant, was collected and subject to flow cytometry with Annexin-V and 7-AAD for early apoptotic (7-AAD-/Annexin-V+), late apoptotic/dead (7-AAD+/Annexin-V+), or necrotic (Annexin-V-/7-AAD+) cells. These results demonstrated cooperation between DSF and doxorubicin in inducing cell death (38% 72 h post-treatment, Fig. 6A). However, interestingly, the level of cell death and relative ratio of these populations was not altered dramatically, suggesting that while some cells undergo apoptosis, others are growth inhibited through additional mechanisms. This is consistent with the fact that MDA-MB-231 cells express a high level of mutant p53, which is stabilized by and contributes to survival signals generated by elevated phospholipase D in these cells.49

Figure 6. Apoptosis and senescence analysis of MDA-MB-231 cells treated with disulfiram and/or doxorubicin. Cells were treated with disulfiram and/or doxorubicin at 250 nM and 125 nM, respectively. (A) Flow cytometry profiles of 7-AAD and Annexin-V for each experimental condition at 24 h and 72 h post treatment. 7-AAD−/Annexin-V+ marks early stage apoptosis, 7-AAD+/Annexin-V+ late stage apoptosis, and 7-AAD+/Annexin-V− necrotic cells. (B) Percent senescence in untreated and treated MDA-MB-231 cells, and MCF7-positive control (doxorubicin 125 nM), as determined by senescence-associated β-galactosidase (X-gal/BCIG) staining.90 (C–G) Light microscope view of MDA-MB-231 cells treated with (C) vehicle control, (D) doxorubicin, (E) disulfiram, or (F) doxorubicin plus disulfiram, and (G) MCF7 positive control (doxorubicin 125 nM). Scale bars represent 200 µm.
Indeed, we noticed that treatment with both drugs induces enlarged cell morphology in MDA-MB-231 cells. This prompted us to test for cellular senescence through senescence-associated β-galactosidase staining. Interestingly, we found that doxorubicin or DSF treatment cultures showed X-gal-positive cells (29.9% and 23.0%, respectively), and that doxorubicin plus DSF treatment resulted in an additive effect (42.4%) (Fig. 6B–F). We note that doxorubicin did not induce robust senescence in MDA-MB-231 as it did in MCF7 cells (Fig. 6G), the latter of which express wild-type p53 that drives cellular senescence.50,51 However, doxorubicin clearly induced large, X-gal-positive MDA-MB-231 cells compared with untreated cells (Fig. 6C and D), and combination of doxorubicin plus DSF further increased the percentage of these cells (Fig. 6F). Thus, DSF potentiates the effects of doxorubicin on both viability and proliferation.
Cancer stem cells (CSCs) sustain tumorigenesis and exhibit distinct gene expression profiles and sensitivity to drug treatment compared with non-CSCs.52-55 CSCs in TNBC cell lines were identified by flow cytometry as ESA+/CD24−/low/CD44+.56,57 To determine the effect of DSF and doxorubicin on CSCs, MDA-MB-231 cells were treated with each drug alone or in combination. After 72 h, remaining live cells were analyzed by flow cytometry for CD24, ESA, and CD44 cell surface markers. We found that DSF and doxorubicin effectively abrogated the CSC population when used in combination (65% inhibition), as compared with treatment with DSF (5% inhibition) or doxorubicin (57% inhibition) alone (Fig. 7). Taken together, these results suggest that DSF cooperates with doxorubicin to target TNBC cells and diminish the CSC population.

Figure 7. Effect of disulfiram and doxorubicin on the CSC fraction in MDA-MB-231 cells. (A and B) Gating conditions for live (7-AAD negative) and ESA+ cells. (C–E) Effect of disulfiram (250 nM) and doxorubicin (125 nM) on the CD44+/CD24-/low/ESA+ cancer stem cell (CSC) fraction (red box). (F) Average absolute values and normalized ratios of CSC fraction.
Discussion
Here, we describe identification of disulfiram (DSF) through a high-throughput screen of 3185 drugs as a potent growth inhibitor of TNBC. DSF compared favorably with several other drugs with anti-neoplastic activity, such as colchicine, paclitaxel, daunorubicin, and doxorubicin, and cooperated with the latter, which is used as mono-therapy for TNBCs, to suppress cell growth and reduce the CSC fraction. In addition, we used a non-biased target identification approach to discover IQ motif-containing proteins as novel DSF targets.
DARTS assay identified proteins containing IQ-motifs as targets of DSF
Using the DARTS method,27-29 we identified and confirmed IQGAP1 and myosin heavy-chain 9 (MYH9) as direct binding targets of DSF. IQGAP1 is a ubiquitously expressed scaffold protein involved in regulation of the actin cytoskeleton, transcription, cellular adhesion, and the cell cycle.30-34 Recent studies suggest a role for IQGAP1 in several human cancers, such as thyroid, colorectal, gastric, and breast cancer, where it acts to modulate oncogenic pathways.58-64 IQGAP1 interacts with at least 90 proteins and behaves as a control hub for mTOR, ERK-MAPK, and Wnt/b-catenin pathways.30,64-66 The IQGAP family includes IQGAP1–3, of which IQGAP2 has also been implicated in cancer.67-69 However, neither homolog is expressed ubiquitously. IQGAP2 is expressed at highest levels in liver, kidney, and platelets,70,71 while IQGAP3 is predominately expressed in brain tissue, where it promotes neurite growth.72 MYH9 is a myosin IIA heavy chain that also contains an IQ motif. MYH9 plays a role in cytokinesis, cell motility, and maintenance of cell shape.35-37 Furthermore, myosin heavy chain 9 has been implicated in cancer metastasis73-75 and regulation of EGFR-cytoskeletal interaction.76,77 Binding of DSF with these proteins is consistent with our pathway analysis, which identified divergent signaling pathways affected by DSF treatment.
Although RAS is not commonly mutated in TNBC, proto-oncogene tyrosine kinases are generally activated in TNBC, perhaps due to loss of the PTPN12 phosphatase.78 Moreover, MDA-MB-231 cells have oncogenic (G13D) mutation in K-RAS.79 Interestingly, it was recently suggested that IQGAP1 might offer a new approach to inhibit RAS-RAF-MEK-ERK signaling. Specifically, IQGAP1 knockout or knockdown inhibited initiation of tumors or hyperproliferation driven by oncogenic RAS pathway activation.80 In contrast, we found that RNAi-mediated knockdown of IQGAP1 in MDA-MB-231 cells only accounted for partial inhibition when compared with DSF-treated cells. The discrepancy between the 2 studies may be reconciled by the fact that Jameson et al.80 tested the effect of IQGAP1 on tumor initiation, whereas we tested the effect of knocking down IQGAP1 on pre-existing tumor cells.
We found that combined knockdown of IQGAP1 plus MYH9 did not lead to additive growth inhibition. Incomplete knockdown of MYH9 alone may account for the fractional decrease in cell viability observed. Nonetheless, this suggests that DSF exerts its effect through additional targets, possibly other IQGAP and MYH9 family members or other IQ motif-containing factors. Importantly, our results demonstrate that DSF has a stronger inhibitory effect than IQGAP1 (+/− MYH9) knockdown.
DSF cooperates with doxorubicin to decrease cell viability, increase cell death and cellular senescence, and target CSCs in TNBC
We found DSF to cooperate with doxorubicin, paclitaxel, gemcitabine, and PD-0332991 to suppress TNBC cell growth; the most potent combination was with doxorubicin. The latter is used in the treatment of TNBC.81-83 While highly toxic to tumor cells, doxorubicin also has strong side effects against normal cells, in particular high cardiotoxicity.84,85 Therefore, combinatorial drug treatments with doxorubicin that can further increase its efficacy or reduce the concentration of drug required for achieving complete response would be highly beneficial in the clinic. This is particularly compelling for elderly patients with pre-existing cardiac complications. We showed that a DSF/doxorubicin combination increased apoptotic cell death and cellular senescence and diminished the CSC fraction more so than each drug alone. Specifically, flow cytometry analysis on MDA-MB-231 cells treated with DSF and doxorubicin demonstrated a cooperative decrease in the CD44+/CD24-/low/ESA+ CSC population to 0.35 of the untreated fraction, compared with 0.95 and 0.43 by DSF or doxorubicin alone, respectively. CSCs contribute to tumor growth and disease relapse. Thus, DSF plus doxorubicin treatment holds promise for effective treatment of TNBC.
Materials and Methods
Cell lines and cultures
MDA-MB-231, MDA-MB-468, MDA-MB-157, Hs578T, and MCF7 were maintained in DMEM containing 10% FBS and 1% PEST. MDA-MB-436 was maintained in DMEM containing 10% FBS, 1% PEST, and 10 µg/ml insulin. HCC70, HCC1937, HCC38, HCC1954, HCC1569, HCC3153, and Bt549 were maintained in RPMI containing 10% FBS and 1% PEST. SUM149PT was maintained in Ham F-12 containing 5% FBS, 1% PEST, 5 µg/ml insulin, and 1 µg/ml hydrocortisone. All cell culture was grown at 37 °C with 5% CO2 in attachment plates. Cell lines Bt549, MDA-MB-436, MDA-MB-468, MDA-MB-231, and HCC1569 were kind gifts from Dr Mona Gauthier. HCC3153 was a gift from Dr Tak Mak lab. SUM149PT was a gift from Dr Benjamin Neel Lab (Asterand). The remaining breast cancer cell lines were purchased from the America Type Culture Collection (ATCC).
Western blotting
MDA-MB-231 cells were cultured in 6-well dishes with DMEM containing 10% FBS and 1% PEST and were treated 24 h after seeding at optimal density. Cells were treated with 0.25% trypsin (Sigma), washed with PBS, pelleted, and lysed with lysis buffer (0.15 M NaCl, 1% Triton X-100, 5 mM EDTA, 5 mM NaF, 0.5 mM Na3VO4, and 1:100 protease inhibitor cocktail [1 mg/mL leupeptin, 2 µg/mL aprotinin, and 100 mM PMSF]). Protein concentration was determined by a Nanodrop 1000 Spectrophotometer (Thermo Scientific). Proteins in total cell lysates were fractionated by SDS-PAGE and transferred onto nitrocellulose membranes using electrophoresis for subsequent immunoblotting. Membranes were blocked with 5% nonfat dried milk in phosphate-buffered saline containing 0.05% Tween 20 (PBST) at R.T. for 1 h. After washing the membranes 3 × 5 min with PBST, they were incubated at 4 °C overnight with rabbit anti-human IQGAP1 primary antibody (Cell Signaling), rabbit anti-human MYH9 primary antibody (Proteintech Group), or rabbit anti-tubulin primary antibody (Cell Signaling). Membranes were washed with PBST buffer 3 × 5 min each and incubated with HRP-conjugated anti-rabbit IgG secondary antibody (Cell Signaling) for 1 h. After further washing, the membranes were allowed to react with ECL (enhanced chemiluminescence substrate, Thermo Scientific); the signal was detected using autoradiography film and developed using a Konica SRX-101A developer. Primary antibodies were diluted 1:1000 in PBS (tubulin was diluted 1:2000) with 5% BSA; secondary antibody was diluted 1:2000 in PBS with 5% nonfat dried milk.
Drug screening
Screens were performed in the S.M.A.R.T. Facility of the Samuel Lunenfeld Research Institute. All libraries were prepared in 100% DMSO to facilitate drug delivery via pinning. The final concentration of DMSO in each screen was 0.4%. The breast cancer lines MDA-MB-231, MDA-MB-436, Bt549, and HCC70 were seeded with their corresponding medias in 384-well plates at a density of 900 cells/well in a total volume of 50 µL/well. The following day, plates were pinned with drug libraries to reach a final concentration of 1 µM (Spectrum library, MicroSource Discovery Systems) or 0.8 µM (Prestwick library, Prestwick Chemical). Alamar blue (Invitrogen) was added 3 d post-drug pinning at 10% of the volume (5 µL/well), and cell viability was read 4–6 h later using a Pherastar plate reader. Screen data were normalized using the B-score approach to select statistically relevant hits after correction for positional effects and general systemic errors during 72 h of incubation.86
MTT viability assays
Cells were seeded in 96-well plates at their optimal density (2–5 × 103 cells/well) and treated the following day, leaving 100 µL final volume of media. Three days (72 h) post treatment, 30 µL of 2 mg/mL MTT (3-[4,5-dimethylthiazol-2-yl]-2, 5-diphenyl tetrazolium bromide, Sigma) was added to each well and incubated for 2–4 h, depending on cell type. MTT/media solution was aspirated and replaced with 100 µL DMSO and left at R.T. for 15–20 min to dissolve the formazan dye. After gently agitation to ensure even mixture of the dye, a 96-well microplate reader (Molecular Devices) was used to determine the optical density (OD) of each well at 570 nm. Percent cell viability (CV) was determined by (treatment group OD/untreated control group OD) × 100%, using DMSO as a blank. Each assay was performed in triplicate, and repeated at least 3 times.
Drug affinity responsive target stability
DARTS was performed as reported27-29 with minor modifications. Briefly, for target identification, MDA-MB-231 cells maintained in RPMI containing 10% FBS and 1% penicillin/streptomycin were lysed with M-PER (Pierce) with phosphatase inhibitors (50 mM NaF, 10 mM β-glycerophosphate, 5 mM sodium pyrophosphate, 2 mM Na3VO4) and protease inhibitors (Roche). After lysis, cold TNC buffer (50 mM TRIS-HCl pH 8.0, 50 mM NaCl, 10 mM CaCl2) was added to the lysate, and protein concentration was measured via the BCA protein assay. Lysate was incubated with either vehicle (DMSO) or varying concentrations of disulfiram for 1 h at R.T. with shaking at 600 rpm in an Eppendorf Thermomixer. The samples were then digested with different concentrations of Pronase (Roche), which is a cocktail of proteases, for 20 min at R.T. Digestion was stopped with the addition of SDS buffer, and samples were immediately heated for 10 min at 70 °C. Proteins in each sample were separated by SDS-PAGE and visualized using the ProteoSilver staining kit (Sigma). Protected bands were excised and subjected to mass spectrometry identification (Alphalyse).
For target validation, DARTS was performed as described above. Samples were separated on a 4–12% Bis-Tris gradient gel (Invitrogen), and western blotting was performed with antibodies against IQGAP1 (Cell Signaling), MYH9 (Protein Tech Group), β-actin (Cell Signaling), and GAPDH (Ambion).
Definitions of drug interaction
We used Kaspers et al.87 model to calculate the borderlines of synergistic and additive interactions, which was developed by combining the Multiplicative88 and Maximum89 models for independently acting and mutually exclusive drugs, respectively. Combining these 2 models to define the type of drug–drug interaction, we get:
Synergistic: the observed cell viability (CV) for a drug combination is less than the product of each single agent: observed CV(A+B) < CV(A) × CV(B)
Additive: the observed CV for a drug combination is less than the max reduction in CV by either drug alone, CV(A/Bmax), but is greater than the product of each single agent: CV(A) × CV(B) < observed CV(A+B) < CV(A/Bmax)
Antagonistic: the observed CV for a drug combination is greater than CV(A/Bmax): observed CV(A+B) > CV(A/Bmax).
Flow cytometry
MDA-MB-231 cells were plated at optimal densities and treated the following day with disulfiram, doxorubicin, or a combination of both. For cancer stem cell analysis: after 72 h, supernatants from each treatment group were collected before cells were trypsinized to single cell suspension. Trypsinized cells were added to their respective supernatants, washed with PBS, pelleted, resuspended in serum-free PBS, and counted. In a volume of 100 µL serum-free PBS, 0.5–1.0 × 106 cells were incubated with 5 µL mouse anti-human ESA/Ep-CAM/CD326-PE (BioLegend), 5 µL mouse anti-human CD24-FITC (BD Biosciences) and 5 µL mouse anti-human CD44-APC (BD Biosciences) antibody on ice, in the dark, for 30–40 min with occasional pulse vortexing. Cells were then washed and strained to single cells into 5 mL polystyrene round-bottom FACS tubes (BD Falcon) to a total volume of 500 µL (1 × 106 cells/mL). Finally, 5 µL 7-AAD (BD Biosciences) was added to each tube as a viability marker, and cells were processed on a FACSCaliber (Becton Dickinson) no longer than 1 h post-staining. For apoptosis analysis: after 24 or 72 h, samples were processed as above, except resuspension occurred in 1× Annexin-V binding buffer, without subsequent washing. Positive controls were achieved with a heat-shock sample (cell suspension subjected to 55 °C for 10 min prior to processing). In a volume of 100 µL 1× Annexin-V binding buffer, 0.5–1.0 × 106 cells were incubated with 5 µL Annexin-V-PE (BD Biosciences) in the dark, at R.T. for 15–20 min with occasional pulse vortexing. Then, 5 µL 7-AAD (BD Biosciences) and an additional 400 µL 1× Annexin V binding buffer was added to each sample. Finally, cells were strained to single cells into 5 mL polysterene round-bottom FACS tubes (BD Falcon), placed on ice, and processed on a FACSCaliber (Becton Dickinson) no longer than 1 h post-staining.
Senescence-associated β-galactosidase staining
MDA-MB-231 and MCF7 cells were seeded onto 6-well plates at optimal density and treated the next day with 250 nM disulfiram, 125 nM doxorubicin, or a combination of both. After 72 h, cells were washed twice with PBS, fixed with 3% formaldehyde (Sigma), and stained with X-gal/BCIG staining solution (BD Pharminogen Senescence kit) overnight at 37 °C.90 The senescent cells (β-galactosidase-positive cells) were counted using a phase contrast light microscope and were expressed as a percentage of the total number of cells counted (minimum 500 cells counted from 4–5 plate areas).
Supplementary Material
Acknowledgments
This study was conducted with support of the Canadian BC Foundation, Canadian BC Research Alliance, Ontario Institute for Cancer Research through funding provided by the Government of Ontario to E. Zacksenhaus, the Terry-Fox Foundation to SE. Egan and E Zacksenhaus, and the US. National Institutes of Health R01 CA124974 to JH. MYP is a trainee of the Cell and Molecular Biology (C&MB) training grant (T32 GM007185). JCL was supported in part by a fellowship from the Canadian BC Foundation.
Glossary
Abbreviations:
- 7-AAD
7-aminoactinomycin D
- ALDH
aldehyde dehydrogenase
- BaA
basal A (basal-like)
- BaB
basal B (claudin-low)
- BC
breast cancer
- BSA
bovine serum albumin
- CDK4/6
cyclin-dependant kinase 4/6
- CSC
cancer stem cell
- DARTS
drug affinity responsive target stability
- DAVID - Database for Annotation
Visualization and Integrated Discovery
- DMEM
Dulbecco modified Eagle medium
- DMSO
dimethyl sulfoxide
- Dox
doxorubicin/adriamycin
- DSF
disulfiram
- ECL
enhanced chemiluminescence
- EDG1/S1P1
endothelial differentiation gene 1/sphingosine-1-phosphate receptor 1
- EDTA
ethylenediaminetetraacetic acid
- EGFR
epidermal growth factor receptor
- ER
estrogen receptor
- ERK
extracellular signal-regulated kinase
- ESA/EpCAM
epithelial specific antigen/epithelial cell adhesion molecule
- FACS
fluorescence-activated cell sorting
- FBS
fetal bovine serum
- FDA
Food and Drug Administration
- HER2
human epidermal growth factor receptor 2
- HRP
horseradish peroxidase
- IC50
50% inhibitory concentration
- IQGAP1
IQ-motif containing GTPase activating protein 1
- MAPK
mitogen-activated protein kinase
- MEK
MAPK/ERK kinase
- mTOR
mammalian target of rapamycin
- MTT
3-(4,5-dimethylthiazol-2-yl)-2, 5-diphenyl tetrazolium bromide
- MYH9
myosin heavy chain 9
- NFkB
nuclear factor kappa-light-chain-enhancer of activated B cells
- OD
optical density
- PEST
penicillin/streptomycin
- PMSF
phenylmethylsulfonyl fluoride
- PR
progesterone receptor
- PTEN
phosphatase and tensin homolog
- R.T.
room temperature
- S.M.A.R.T.
simple modular assay & robotic technology
- TNBC
triple-negative breast cancer
- X-gal/BCIG
5-bromo-4-chloro-3-indolyl-β-D-galactopyranoside
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Supplemental Materials
Supplemental materials may be found here: www.landesbioscience.com/journals/cc/article/26063
Footnotes
Previously published online: www.landesbioscience.com/journals/cc/article/26063
References
- 1.Perou CM. Molecular stratification of triple-negative breast cancers. Oncologist. 2011;16(Suppl 1):61–70. doi: 10.1634/theoncologist.2011-S1-61. [DOI] [PubMed] [Google Scholar]
- 2.Jiang Z, Jones R, Liu JC, Deng T, Robinson T, Chung PE, Wang S, Herschkowitz JI, Egan SE, Perou CM, et al. RB1 and p53 at the crossroad of EMT and triple-negative breast cancer. Cell Cycle. 2011;10:1563–70. doi: 10.4161/cc.10.10.15703. [DOI] [PubMed] [Google Scholar]
- 3.Neve RM, Chin K, Fridlyand J, Yeh J, Baehner FL, Fevr T, Clark L, Bayani N, Coppe JP, Tong F, et al. A collection of breast cancer cell lines for the study of functionally distinct cancer subtypes. Cancer Cell. 2006;10:515–27. doi: 10.1016/j.ccr.2006.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Gehl J, Boesgaard M, Paaske T, Vittrup Jensen B, Dombernowsky P. Combined doxorubicin and paclitaxel in advanced breast cancer: effective and cardiotoxic. Ann Oncol. 1996;7:687–93. doi: 10.1093/oxfordjournals.annonc.a010717. [DOI] [PubMed] [Google Scholar]
- 5.van Kuilenburg AB, Maring JG. Evaluation of 5-fluorouracil pharmacokinetic models and therapeutic drug monitoring in cancer patients. Pharmacogenomics. 2013;14:799–811. doi: 10.2217/pgs.13.54. [DOI] [PubMed] [Google Scholar]
- 6.von Minckwitz G. Docetaxel/anthracycline combinations for breast cancer treatment. Expert Opin Pharmacother. 2007;8:485–95. doi: 10.1517/14656566.8.4.485. [DOI] [PubMed] [Google Scholar]
- 7.Kadakia A, Rajan SS, Abughosh S, Du XL, Johnson ML. CMF-regimen preferred as first-course chemotherapy for older and sicker women with breast cancer: Findings from a SEER-medicare-based population study. Am J Clin Oncol. 2013 doi: 10.1097/COC.0b013e31828f5b01. [DOI] [PubMed] [Google Scholar]
- 8.Zare N, Ghanbari S, Salehi A. Effects of two chemotherapy regimens, anthracycline-based and CMF, on breast cancer disease free survival in the Eastern Mediterranean Region and Asia: a meta-analysis approach for survival curves. Asian Pac J Cancer Prev. 2013;14:2013–7. doi: 10.7314/APJCP.2013.14.3.2013. [DOI] [PubMed] [Google Scholar]
- 9.O’Shaughnessy JA, Fisherman JS, Cowan KH. Combination paclitaxel (Taxol) and doxorubicin therapy for metastatic breast cancer. Semin Oncol. 1994;21(Suppl 8):19–23. [PubMed] [Google Scholar]
- 10.Ozkan M, Berk V, Kaplan MA, Benekli M, Coskun U, Bilici A, Gumus M, Alkis N, Dane F, Ozdemir NY, et al. Gemcitabine and cisplatin combination chemotherapy in triple negative metastatic breast cancer previously treated with a taxane/anthracycline chemotherapy; multicenter experience. Neoplasma. 2012;59:38–42. doi: 10.4149/neo_2012_005. [DOI] [PubMed] [Google Scholar]
- 11.Kohail H, Shehata S, Mansour O, Gouda Y, Gaafar R, Hamid TA, El Nowieam S, Al Khodary A, El Zawahry H, Wareth AA, et al. A phase 2 study of the combination of gemcitabine and cisplatin in patients with locally advanced or metastatic breast cancer previously treated with anthracyclines with/without taxanes. Hematol Oncol Stem Cell Ther. 2012;5:42–8. doi: 10.5144/1658-3876.2012.42. [DOI] [PubMed] [Google Scholar]
- 12.Joensuu H, Gligorov J. Adjuvant treatments for triple-negative breast cancers. Ann Oncol. 2012;23(Suppl 6):vi40–5. doi: 10.1093/annonc/mds194. [DOI] [PubMed] [Google Scholar]
- 13.Dalvi RR. Toxicology of thiram (tetramethylthiuram disulfide): a review. Vet Hum Toxicol. 1988;30:480–2. [PubMed] [Google Scholar]
- 14.Christensen JK, Møller IW, Rønsted P, Angelo HR, Johansson B. Dose-effect relationship of disulfiram in human volunteers. I: Clinical studies. Pharmacol Toxicol. 1991;68:163–5. doi: 10.1111/j.1600-0773.1991.tb01215.x. [DOI] [PubMed] [Google Scholar]
- 15.Cornford EM, Bocash WD, Braun LD, Crane PD, Oldendorf WH, MacInnis AJ. Rapid distribution of tryptophol (3-indole ethanol) to the brain and other tissues. J Clin Invest. 1979;63:1241–8. doi: 10.1172/JCI109419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Iljin K, Ketola K, Vainio P, Halonen P, Kohonen P, Fey V, Grafström RC, Perälä M, Kallioniemi O. High-throughput cell-based screening of 4910 known drugs and drug-like small molecules identifies disulfiram as an inhibitor of prostate cancer cell growth. Clin Cancer Res. 2009;15:6070–8. doi: 10.1158/1078-0432.CCR-09-1035. [DOI] [PubMed] [Google Scholar]
- 17.Hothi P, Martins TJ, Chen L, Deleyrolle L, Yoon JG, Reynolds B, Foltz G. High-throughput chemical screens identify disulfiram as an inhibitor of human glioblastoma stem cells. Oncotarget. 2012;3:1124–36. doi: 10.18632/oncotarget.707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Triscott J, Lee C, Hu K, Fotovati A, Berns R, Pambid M, Luk M, Kast RE, Kong E, Toyota E, et al. Disulfiram, a drug widely used to control alcoholism, suppresses the self-renewal of glioblastoma and over-rides resistance to temozolomide. Oncotarget. 2012;3:1112–23. doi: 10.18632/oncotarget.604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cvek B. Targeting malignancies with disulfiram (Antabuse): multidrug resistance, angiogenesis, and proteasome. Curr Cancer Drug Targets. 2011;11:332–7. doi: 10.2174/156800911794519806. [DOI] [PubMed] [Google Scholar]
- 20.Kast RE, Boockvar JA, Brüning A, Cappello F, Chang WW, Cvek B, Dou QP, Duenas-Gonzalez A, Efferth T, Focosi D, et al. A conceptually new treatment approach for relapsed glioblastoma: coordinated undermining of survival paths with nine repurposed drugs (CUSP9) by the International Initiative for Accelerated Improvement of Glioblastoma Care. Oncotarget. 2013;4:502–30. doi: 10.18632/oncotarget.969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zhang H, Chen D, Ringler J, Chen W, Cui QC, Ethier SP, Dou QP, Wu G. Disulfiram treatment facilitates phosphoinositide 3-kinase inhibition in human breast cancer cells in vitro and in vivo. Cancer Res. 2010;70:3996–4004. doi: 10.1158/0008-5472.CAN-09-3752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Yip NC, Fombon IS, Liu P, Brown S, Kannappan V, Armesilla AL, Xu B, Cassidy J, Darling JL, Wang W. Disulfiram modulated ROS-MAPK and NFκB pathways and targeted breast cancer cells with cancer stem cell-like properties. Br J Cancer. 2011;104:1564–74. doi: 10.1038/bjc.2011.126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sauna ZE, Shukla S, Ambudkar SV. Disulfiram, an old drug with new potential therapeutic uses for human cancers and fungal infections. Mol Biosyst. 2005;1:127–34. doi: 10.1039/b504392a. [DOI] [PubMed] [Google Scholar]
- 24.Liu GY, Frank N, Bartsch H, Lin JK. Induction of apoptosis by thiuramdisulfides, the reactive metabolites of dithiocarbamates, through coordinative modulation of NFkappaB, c-fos/c-jun, and p53 proteins. Mol Carcinog. 1998;22:235–46. doi: 10.1002/(SICI)1098-2744(199808)22:4<235::AID-MC5>3.0.CO;2-I. [DOI] [PubMed] [Google Scholar]
- 25.Croker AK, Goodale D, Chu J, Postenka C, Hedley BD, Hess DA, Allan AL. High aldehyde dehydrogenase and expression of cancer stem cell markers selects for breast cancer cells with enhanced malignant and metastatic ability. J Cell Mol Med. 2009;13(8B):2236–52. doi: 10.1111/j.1582-4934.2008.00455.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Marcato P, Dean CA, Pan D, Araslanova R, Gillis M, Joshi M, Helyer L, Pan L, Leidal A, Gujar S, et al. Aldehyde dehydrogenase activity of breast cancer stem cells is primarily due to isoform ALDH1A3 and its expression is predictive of metastasis. Stem Cells. 2011;29:32–45. doi: 10.1002/stem.563. [DOI] [PubMed] [Google Scholar]
- 27.Lomenick B, Hao R, Jonai N, Chin RM, Aghajan M, Warburton S, Wang J, Wu RP, Gomez F, Loo JA, et al. Target identification using drug affinity responsive target stability (DARTS) Proc Natl Acad Sci U S A. 2009;106:21984–9. doi: 10.1073/pnas.0910040106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lomenick B, Jung G, Wohlschlegel JA, Huang J. Target identification using drug affinity responsive target stability (DARTS) Curr Protoc Chem Biol. 2011;3:163–80. doi: 10.1002/9780470559277.ch110180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lomenick B, Olsen RW, Huang J. Identification of direct protein targets of small molecules. ACS Chem Biol. 2011;6:34–46. doi: 10.1021/cb100294v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wang JB, Sonn R, Tekletsadik YK, Samorodnitsky D, Osman MA. IQGAP1 regulates cell proliferation through a novel CDC42-mTOR pathway. J Cell Sci. 2009;122:2024–33. doi: 10.1242/jcs.044644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Meyer RD, Sacks DB, Rahimi N. IQGAP1-dependent signaling pathway regulates endothelial cell proliferation and angiogenesis. PLoS One. 2008;3:e3848. doi: 10.1371/journal.pone.0003848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kozlova I, Ruusala A, Voytyuk O, Skandalis SS, Heldin P. IQGAP1 regulates hyaluronan-mediated fibroblast motility and proliferation. Cell Signal. 2012;24:1856–62. doi: 10.1016/j.cellsig.2012.05.013. [DOI] [PubMed] [Google Scholar]
- 33.Johnson MA, Henderson BR. The scaffolding protein IQGAP1 co-localizes with actin at the cytoplasmic face of the nuclear envelope: implications for cytoskeletal regulation. Bioarchitecture. 2012;2 doi: 10.4161/bioa.21182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Briggs MW, Sacks DB. IQGAP proteins are integral components of cytoskeletal regulation. EMBO Rep. 2003;4:571–4. doi: 10.1038/sj.embor.embor867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hosono Y, Usukura J, Yamaguchi T, Yanagisawa K, Suzuki M, Takahashi T. MYBPH inhibits NM IIA assembly via direct interaction with NMHC IIA and reduces cell motility. Biochem Biophys Res Commun. 2012;428:173–8. doi: 10.1016/j.bbrc.2012.10.036. [DOI] [PubMed] [Google Scholar]
- 36.Raab M, Swift J, Dingal PC, Shah P, Shin JW, Discher DE. Crawling from soft to stiff matrix polarizes the cytoskeleton and phosphoregulates myosin-II heavy chain. J Cell Biol. 2012;199:669–83. doi: 10.1083/jcb.201205056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Yang F, Wei Q, Adelstein RS, Wang PJ. Non-muscle myosin IIB is essential for cytokinesis during male meiotic cell divisions. Dev Biol. 2012;369:356–61. doi: 10.1016/j.ydbio.2012.07.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Bähler M, Rhoads A. Calmodulin signaling via the IQ motif. FEBS Lett. 2002;513:107–13. doi: 10.1016/S0014-5793(01)03239-2. [DOI] [PubMed] [Google Scholar]
- 39.Stuart DD, Sellers WR. Targeting RAF-MEK-ERK kinase-scaffold interactions in cancer. Nat Med. 2013;19:538–40. doi: 10.1038/nm.3195. [DOI] [PubMed] [Google Scholar]
- 40.Merico D, Isserlin R, Stueker O, Emili A, Bader GD. Enrichment map: a network-based method for gene-set enrichment visualization and interpretation. PLoS One. 2010;5:e13984. doi: 10.1371/journal.pone.0013984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lu Y, Lin YZ, LaPushin R, Cuevas B, Fang X, Yu SX, Davies MA, Khan H, Furui T, Mao M, et al. The PTEN/MMAC1/TEP tumor suppressor gene decreases cell growth and induces apoptosis and anoikis in breast cancer cells. Oncogene. 1999;18:7034–45. doi: 10.1038/sj.onc.1203183. [DOI] [PubMed] [Google Scholar]
- 42.Weng L, Brown J, Eng C. PTEN induces apoptosis and cell cycle arrest through phosphoinositol-3-kinase/Akt-dependent and -independent pathways. Hum Mol Genet. 2001;10:237–42. doi: 10.1093/hmg/10.3.237. [DOI] [PubMed] [Google Scholar]
- 43.Hannun YA, Obeid LM. The Ceramide-centric universe of lipid-mediated cell regulation: stress encounters of the lipid kind. J Biol Chem. 2002;277:25847–50. doi: 10.1074/jbc.R200008200. [DOI] [PubMed] [Google Scholar]
- 44.Kartal Yandım M, Apohan E, Baran Y. Therapeutic potential of targeting ceramide/glucosylceramide pathway in cancer. Cancer Chemother Pharmacol. 2013;71:13–20. doi: 10.1007/s00280-012-1984-x. [DOI] [PubMed] [Google Scholar]
- 45.Lavieu G, Scarlatti F, Sala G, Carpentier S, Levade T, Ghidoni R, Botti J, Codogno P. Regulation of autophagy by sphingosine kinase 1 and its role in cell survival during nutrient starvation. J Biol Chem. 2006;281:8518–27. doi: 10.1074/jbc.M506182200. [DOI] [PubMed] [Google Scholar]
- 46.Okamoto H, Takuwa N, Gonda K, Okazaki H, Chang K, Yatomi Y, Shigematsu H, Takuwa Y. EDG1 is a functional sphingosine-1-phosphate receptor that is linked via a Gi/o to multiple signaling pathways, including phospholipase C activation, Ca2+ mobilization, Ras-mitogen-activated protein kinase activation, and adenylate cyclase inhibition. J Biol Chem. 1998;273:27104–10. doi: 10.1074/jbc.273.42.27104. [DOI] [PubMed] [Google Scholar]
- 47.Venable ME, Lee JY, Smyth MJ, Bielawska A, Obeid LM. Role of ceramide in cellular senescence. J Biol Chem. 1995;270:30701–8. doi: 10.1074/jbc.270.51.30701. [DOI] [PubMed] [Google Scholar]
- 48.Sultan A, Ling B, Zhang H, Ma B, Michel D, Alcorn J, Yang J. Synergistic effect between sphingosine-1-phosphate and chemotherapy drugs against human brain-metastasized breast cancer MDA-MB-361 cells. J Cancer. 2013;4:315–9. doi: 10.7150/jca.5956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Hui L, Zheng Y, Yan Y, Bargonetti J, Foster DA. Mutant p53 in MDA-MB-231 breast cancer cells is stabilized by elevated phospholipase D activity and contributes to survival signals generated by phospholipase D. Oncogene. 2006;25:7305–10. doi: 10.1038/sj.onc.1209735. [DOI] [PubMed] [Google Scholar]
- 50.Elmore LW, Rehder CW, Di X, McChesney PA, Jackson-Cook CK, Gewirtz DA, Holt SE. Adriamycin-induced senescence in breast tumor cells involves functional p53 and telomere dysfunction. J Biol Chem. 2002;277:35509–15. doi: 10.1074/jbc.M205477200. [DOI] [PubMed] [Google Scholar]
- 51.Jackson JG, Pant V, Li Q, Chang LL, Quintás-Cardama A, Garza D, Tavana O, Yang P, Manshouri T, Li Y, et al. p53-mediated senescence impairs the apoptotic response to chemotherapy and clinical outcome in breast cancer. Cancer Cell. 2012;21:793–806. doi: 10.1016/j.ccr.2012.04.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Dick JE. Breast cancer stem cells revealed. Proc Natl Acad Sci U S A. 2003;100:3547–9. doi: 10.1073/pnas.0830967100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Bonnet D, Dick JE. Human acute myeloid leukemia is organized as a hierarchy that originates from a primitive hematopoietic cell. Nat Med. 1997;3:730–7. doi: 10.1038/nm0797-730. [DOI] [PubMed] [Google Scholar]
- 54.Rosen JM, Jordan CT. The increasing complexity of the cancer stem cell paradigm. Science. 2009;324:1670–3. doi: 10.1126/science.1171837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Nicolini A, Ferrari P, Fini M, Borsari V, Fallahi P, Antonelli A, Berti P, Carpi A, Miccoli P. Stem cells: their role in breast cancer development and resistance to treatment. Curr Pharm Biotechnol. 2011;12:196–205. doi: 10.2174/138920111794295657. [DOI] [PubMed] [Google Scholar]
- 56.Al-Hajj M, Wicha MS, Benito-Hernandez A, Morrison SJ, Clarke MF. Prospective identification of tumorigenic breast cancer cells. Proc Natl Acad Sci U S A. 2003;100:3983–8. doi: 10.1073/pnas.0530291100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Fillmore CM, Kuperwasser C. Human breast cancer cell lines contain stem-like cells that self-renew, give rise to phenotypically diverse progeny and survive chemotherapy. Breast Cancer Res. 2008;10:R25. doi: 10.1186/bcr1982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Casteel DE, Turner S, Schwappacher R, Rangaswami H, Su-Yuo J, Zhuang S, Boss GR, Pilz RB. Rho isoform-specific interaction with IQGAP1 promotes breast cancer cell proliferation and migration. J Biol Chem. 2012;287:38367–78. doi: 10.1074/jbc.M112.377499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Hayashi H, Nabeshima K, Aoki M, Hamasaki M, Enatsu S, Yamauchi Y, Yamashita Y, Iwasaki H. Overexpression of IQGAP1 in advanced colorectal cancer correlates with poor prognosis-critical role in tumor invasion. Int J Cancer. 2010;126:2563–74. doi: 10.1002/ijc.24987. [DOI] [PubMed] [Google Scholar]
- 60.Jadeski L, Mataraza JM, Jeong HW, Li Z, Sacks DB. IQGAP1 stimulates proliferation and enhances tumorigenesis of human breast epithelial cells. J Biol Chem. 2008;283:1008–17. doi: 10.1074/jbc.M708466200. [DOI] [PubMed] [Google Scholar]
- 61.Johnson M, Sharma M, Henderson BR. IQGAP1 regulation and roles in cancer. Cell Signal. 2009;21:1471–8. doi: 10.1016/j.cellsig.2009.02.023. [DOI] [PubMed] [Google Scholar]
- 62.Liu Z, Liu D, Bojdani E, El-Naggar AK, Vasko V, Xing M. IQGAP1 plays an important role in the invasiveness of thyroid cancer. Clin Cancer Res. 2010;16:6009–18. doi: 10.1158/1078-0432.CCR-10-1627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Walch A, Seidl S, Hermannstädter C, Rauser S, Deplazes J, Langer R, von Weyhern CH, Sarbia M, Busch R, Feith M, et al. Combined analysis of Rac1, IQGAP1, Tiam1 and E-cadherin expression in gastric cancer. Mod Pathol. 2008;21:544–52. doi: 10.1038/modpathol.2008.3. [DOI] [PubMed] [Google Scholar]
- 64.White CD, Brown MD, Sacks DB. IQGAPs in cancer: a family of scaffold proteins underlying tumorigenesis. FEBS Lett. 2009;583:1817–24. doi: 10.1016/j.febslet.2009.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.White CD, Erdemir HH, Sacks DB. IQGAP1 and its binding proteins control diverse biological functions. Cell Signal. 2012;24:826–34. doi: 10.1016/j.cellsig.2011.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Tekletsadik YK, Sonn R, Osman MA. A conserved role of IQGAP1 in regulating TOR complex 1. J Cell Sci. 2012;125:2041–52. doi: 10.1242/jcs.098947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.White CD, Khurana H, Gnatenko DV, Li Z, Odze RD, Sacks DB, Schmidt VA. IQGAP1 and IQGAP2 are reciprocally altered in hepatocellular carcinoma. BMC Gastroenterol. 2010;10:125. doi: 10.1186/1471-230X-10-125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.White CD, Brown MD, Sacks DB. IQGAPs in cancer: a family of scaffold proteins underlying tumorigenesis. FEBS Lett. 2009;583:1817–24. doi: 10.1016/j.febslet.2009.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Xie Y, Yan J, Cutz JC, Rybak AP, He L, Wei F, Kapoor A, Schmidt VA, Tao L, Tang D. IQGAP2, A candidate tumour suppressor of prostate tumorigenesis. Biochim Biophys Acta. 2012;1822:875–84. doi: 10.1016/j.bbadis.2012.02.019. [DOI] [PubMed] [Google Scholar]
- 70.Brill S, Li S, Lyman CW, Church DM, Wasmuth JJ, Weissbach L, Bernards A, Snijders AJ. The Ras GTPase-activating-protein-related human protein IQGAP2 harbors a potential actin binding domain and interacts with calmodulin and Rho family GTPases. Mol Cell Biol. 1996;16:4869–78. doi: 10.1128/mcb.16.9.4869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Schmidt VA, Scudder L, Devoe CE, Bernards A, Cupit LD, Bahou WF. IQGAP2 functions as a GTP-dependent effector protein in thrombin-induced platelet cytoskeletal reorganization. Blood. 2003;101:3021–8. doi: 10.1182/blood-2002-09-2807. [DOI] [PubMed] [Google Scholar]
- 72.Wang S, Watanabe T, Noritake J, Fukata M, Yoshimura T, Itoh N, Harada T, Nakagawa M, Matsuura Y, Arimura N, et al. IQGAP3, a novel effector of Rac1 and Cdc42, regulates neurite outgrowth. J Cell Sci. 2007;120:567–77. doi: 10.1242/jcs.03356. [DOI] [PubMed] [Google Scholar]
- 73.Derycke L, Stove C, Vercoutter-Edouart AS, De Wever O, Dollé L, Colpaert N, Depypere H, Michalski JC, Bracke M. The role of non-muscle myosin IIA in aggregation and invasion of human MCF-7 breast cancer cells. Int J Dev Biol. 2011;55:835–40. doi: 10.1387/ijdb.113336ld. [DOI] [PubMed] [Google Scholar]
- 74.Liang S, He L, Zhao X, Miao Y, Gu Y, Guo C, Xue Z, Dou W, Hu F, Wu K, et al. MicroRNA let-7f inhibits tumor invasion and metastasis by targeting MYH9 in human gastric cancer. PLoS One. 2011;6:e18409. doi: 10.1371/journal.pone.0018409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Xia ZK, Yuan YC, Yin N, Yin BL, Tan ZP, Hu YR. Nonmuscle myosin IIA is associated with poor prognosis of esophageal squamous cancer. Dis Esophagus. 2012;25:427–36. doi: 10.1111/j.1442-2050.2011.01261.x. [DOI] [PubMed] [Google Scholar]
- 76.Bedrin MS, Abolafia CM, Thompson JF. Cytoskeletal association of epidermal growth factor receptor and associated signaling proteins is regulated by cell density in IEC-6 intestinal cells. J Cell Physiol. 1997;172:126–36. doi: 10.1002/(SICI)1097-4652(199707)172:1<126::AID-JCP14>3.0.CO;2-A. [DOI] [PubMed] [Google Scholar]
- 77.Chiu HC, Chang TY, Huang CT, Chao YS, Hsu JT. EGFR and myosin II inhibitors cooperate to suppress EGFR-T790M-mutant NSCLC cells. Mol Oncol. 2012;6:299–310. doi: 10.1016/j.molonc.2012.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Sun T, Aceto N, Meerbrey KL, Kessler JD, Zhou C, Migliaccio I, Nguyen DX, Pavlova NN, Botero M, Huang J, et al. Activation of multiple proto-oncogenic tyrosine kinases in breast cancer via loss of the PTPN12 phosphatase. Cell. 2011;144:703–18. doi: 10.1016/j.cell.2011.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Kozma SC, Bogaard ME, Buser K, Saurer SM, Bos JL, Groner B, Hynes NE. The human c-Kirsten ras gene is activated by a novel mutation in codon 13 in the breast carcinoma cell line MDA-MB231. Nucleic Acids Res. 1987;15:5963–71. doi: 10.1093/nar/15.15.5963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Jameson KL, Mazur PK, Zehnder AM, Zhang J, Zarnegar B, Sage J, Khavari PA. IQGAP1 scaffold-kinase interaction blockade selectively targets RAS-MAP kinase-driven tumors. Nat Med. 2013;19:626–30. doi: 10.1038/nm.3165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Loi S, Sirtaine N, Piette F, Salgado R, Viale G, Van Eenoo F, Rouas G, Francis P, Crown JP, Hitre E, et al. Prognostic and predictive value of tumor-infiltrating lymphocytes in a phase III randomized adjuvant breast cancer trial in node-positive breast cancer comparing the addition of docetaxel to doxorubicin with doxorubicin-based chemotherapy: BIG 02-98. J Clin Oncol. 2013;31:860–7. doi: 10.1200/JCO.2011.41.0902. [DOI] [PubMed] [Google Scholar]
- 82.Mackey JR, Martin M, Pienkowski T, Rolski J, Guastalla JP, Sami A, Glaspy J, Juhos E, Wardley A, Fornander T, et al. TRIO/BCIRG 001 investigators Adjuvant docetaxel, doxorubicin, and cyclophosphamide in node-positive breast cancer: 10-year follow-up of the phase 3 randomised BCIRG 001 trial. Lancet Oncol. 2013;14:72–80. doi: 10.1016/S1470-2045(12)70525-9. [DOI] [PubMed] [Google Scholar]
- 83.Montanari M, Fabbri F, Rondini E, Frassineti GL, Mattioli R, Carloni S, Scarpi E, Zoli W, Amadori D, Cruciani G. Phase II trial of non-pegylated liposomal doxorubicin and low-dose prednisone in second-line chemotherapy for hormone-refractory prostate cancer. Tumori. 2012;98:696–701. doi: 10.1177/030089161209800604. [DOI] [PubMed] [Google Scholar]
- 84.Volkova M, Russell R., 3rd Anthracycline cardiotoxicity: prevalence, pathogenesis and treatment. Curr Cardiol Rev. 2011;7:214–20. doi: 10.2174/157340311799960645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Carvalho FS, Burgeiro A, Garcia R, Moreno AJ, Carvalho RA, Oliveira PJ. Doxorubicin-induced cardiotoxicity: From bioenergetic failure and cell death to cardiomyopathy. Med Res Rev. 2013 doi: 10.1002/med.21280. [DOI] [PubMed] [Google Scholar]
- 86.Brideau C, Gunter B, Pikounis B, Liaw A. Improved statistical methods for hit selection in high-throughput screening. J Biomol Screen. 2003;8:634–47. doi: 10.1177/1087057103258285. [DOI] [PubMed] [Google Scholar]
- 87.Kaspers GJ, Veerman AJ, Pieters R, Van Zantwijk I, Hählen K, Van Wering ER. Drug combination testing in acute lymphoblastic leukemia using the MTT assay. Leuk Res. 1995;19:175–81. doi: 10.1016/0145-2126(94)00126-U. [DOI] [PubMed] [Google Scholar]
- 88.Valeriote F, Lin Hs. Synergistic interaction of anticancer agents: a cellular perspective. Cancer Chemother Rep. 1975;59:895–900. [PubMed] [Google Scholar]
- 89.Sondak VK, Korn EL, Kern DH. In vitro testing of chemotherapeutic combinations in a rapid thymidine incorporation assay. Int J Cell Cloning. 1988;6:378–91. doi: 10.1002/stem.5530060603. [DOI] [PubMed] [Google Scholar]
- 90.Debacq-Chainiaux F, Erusalimsky JD, Campisi J, Toussaint O. Protocols to detect senescence-associated beta-galactosidase (SA-betagal) activity, a biomarker of senescent cells in culture and in vivo. Nat Protoc. 2009;4:1798–806. doi: 10.1038/nprot.2009.191. [DOI] [PubMed] [Google Scholar]
- 91.Schneider CA, Rasband WS, Eliceiri KW. NIH Image to ImageJ: 25 years of image analysis. Nat Methods. 2012;9:671–5. doi: 10.1038/nmeth.2089. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.