

Abstract
CDKN1A (p21) and CDKN2A (p16) inhibit CDK4/6, initiating senescence. According to our view on senescence, the role of p21 and p16 is to cause cell cycle arrest, whereas MTOR (mechanistic target of rapamycin) drives geroconversion to senescence. Recently we demonstrated that one of the markers of p21- and p16-initiated senescence is MEK-dependent hyper-elevation of cyclin D1. We noticed that a synthetic inhibitor of CDK 4/6 (PD0332991) also induced cyclin D1-positive senescence. We demonstrated that PD0332991 and p21 caused almost identical senescence phenotypes. p21, p16, and PD0332991 do not inhibit MTOR, and rapamycin decelerates geroconversion caused by all 3 molecules. Like p21, PD0332991 initiated senescence at any concentration that inhibited cell proliferation. This confirms the notion that a mere arrest in the presence of active MTOR may lead to senescence.
Keywords: aging, cancer, rapalogs, CDKN1A, CDKN2A
Introduction
In cell culture, cellular senescence is defined as an irreversible state. To reach this state, cells first need to be arrested. At first, this arrest is reversible.1 However, over time, active MTOR and MEK/MAPK drive geroconversion (conversion to irreversible senescence), leading to a large morphology (hypertrophy), hyperfunctional and hyper-secretory phenotypes, hyper-elevated cyclin D1, and loss of replicative and regenerative potential (RP).2-7 For example, induction of ectopic p21 and p16 in HT1080 cells (HT-p21 and HT-p16 cells) causes cell cycle arrest, which is reversible during 2 days.8-10 If p21 and p16 switched off, then most cells can recover and resume proliferation.8-10 It is most important that, during p21- and p16-induced arrest, MTOR and MEK are still active3,7 and drive cellular growth in size (hypertrophy) and loss of regenerative/replicative potential (RP). In the presence of IPTG, which induces ectopic p21 and p16, cells can remain senescent for a seemingly unlimited period of time. When IPTG is removed, senescent cells either cannot resume proliferation or die. This system allows one to observe reversibility vs. irreversibility by switching on and off p21 by simple removal of IPTG. However, this model is restricted to one cell line. To study cellular aging in any cell line, including normal and primary cells, one needs a removable drug, which acts on non-transfected cells (note: HT-p21 and HT-p16 have IPTG- inducible exogenous gene). Majority of agents that induce arrest, such as doxorubicin, are not easily removable, and they are toxic or DNA damaging. Given that p21 and p16 inhibit CDK 4/6, we chose a small-molecule CDK 4/6 inhibitor, PD033299111,12 However, it is believed that p21 and p16 cause senescence not by simply inducing arrest, but by running a putative senescence program including interaction with cytoplasmic proteins and trans-regulation of numerous genes. In our view on senescence, the role of p21 and p16 is mere inhibition of CDK 4/6, while MTOR causes geroconversion to senescence.
Results
PD0332991 and p21 cause cyclin-D1-positive senescence
First we compared the effects of PD0332991 and IPTG-induced p21 in HT-p21 cells. Like IPTG, PD0332991 did not inhibit phosphorylation of the MTOR target p70 S6K (on either Thr389 or Thr421/Ser424 phosphorylation sites) and ERK1/2 (Fig. 1A). IPTG and PD0332991 equally hyper-induced cyclin D1 and E, as seen on days 1 and 3 (Fig. 1A). This confirmed that cyclin D1 hyper-induction is a common marker of geroconversion, regardless of which CDK inhibitor used (p21, p16 or the synthetic small molecule PD0332991). Thus, PD0332991 and IPTG caused identical effects, even though PD0332991 is a direct inhibitor of CDK4/6 and IPTG is acting via induction of ectopic p21 (Fig. 1A). Second, we compared the effects of rapamycin and U0126 on cyclin D1 induction. U0126 was more potent in inhibiting cyclin D1 in senescent cells. As expected, unlike rapamycin, U0126 inhibited phosphorylation of the mostly “MEK-dependent site” (Thr421/Ser424) on p70S6K and predominantly on day 1 (Fig. 1A), while rapamycin completely inhibited phosphorylation of “MTOR site” (Thr389) and partially “MEK site” (Thr421/Ser 424) (Fig. 1A). In contrast, only U0126 inhibited ERK phosphorylation, a target of MEK (Fig. 1A). We conclude that hyper-induction of cyclin D1 is mostly regulated by MEK, and just partially via MTOR. Like IPTG, PD0332991 caused senescent morphology in HT-p21 cells (Fig. 1B). This senescent morphology was partially suppressed by co-treatment with rapamycin (Fig. 1B). Next, we investigated replicative potential (RP) of PD0332991-arrested cells after the drug was washed out, allowing quiescent cells to proliferate and form colonies, whereas senescent cells could not resume proliferation13 (Fig. 1C and D). Yet, PD0332991 (at non-toxic concentrations) did not arrest every single cell and therefore senescent/quiescent cells coexisted with proliferating cells, which rapidly overgrew in the culture (Fig. 1D, right panel, no Noco). To avoid overgrowth of non-arrested cells during treatment with PD0332991, we treated cells for the last 2 days with nocodazole to kill off proliferating cells, thus leaving intact only PD0332991-arrested cells (Fig. 1C–D), as described previously.13 This method revealed that replicative potential of PD0332991-arrested cells was low and comparable with that of IPTG-treated cells (Fig. 1D left panel, + Noco). Co-treatment with rapamycin increased RP of both IPTG- and PD0332991-treated cells (Fig. 1D), confirming that PD0332991-induced senescence was MTOR-dependent. Furthermore, we investigated if the duration of treatment with PD0332991 affects the extent of geroconversion using a lower (0.5 µM) concentration of the drug. In agreement with data shown in Figure 1D, most cells were able to resume proliferation after 3.5 days of treatment with 0.5 µM PD0332991 (Fig. S1A). By day 7, however, most cells (~90%) acquired senescent morphology (Fig. S1B), and cells lost their RP (Fig. S1A).

Figure 1. Comparison of p21- and PD0332991-induced senescence in HT-p21 cells. (A) Immunoblot analysis. Cells were treated with IPTG and 1 µM PD0332991 with or without 500 nM Rapamycin or 10 µM U0126. Cells were lysed on day 1 and 3 and immunoblotting was performed with the indicated antibodies.(B) Beta-Gal staining. Cells were treated with IPTG and 1 µM PD0332991 with or without 500 nM Rapamycin. (C) RP: Schema of experiment for measuring RP, presented in (D). (D) RP in IPTG- and PD0332991- treated HT-p21 cells. Cells were treated with IPTG or 1 µM PD0332991 with or without 500 nM rapamycin (R). After 1 day, 200 nM nocodazole was added in half of the wells (+Noco). After 3 days of drug treatment, drugs were washed out and cells were allowed to recover for 9 days and colonies were stained with crystal violet.
Time- and MTOR-dependent senescence caused by PD0332991 in MEL 10 cells
We next investigated the effect of PD0332991 in MEL10 cells, which are prone to senescence. MEL10 cells are easily arrested by nutlin-3a, etoposide, and U0126. These cells are senescence-prone not only because they are easily arrested, but also because they possess a resilient MTOR pathway, and none of the drugs used (nutlin, etoposide, or U0126) inhibit it.7,14 This creates a condition for geroconversion. At a wide range of concentrations, PD0332991 caused dephosphorylation of retinoblastoma protein (Rb) at Ser780, which is phosphorylated by CDK4/6, indicating that PD0332991 inhibited its target (Fig. 2). A longer exposure of the blot revealed residual phosphorylation of Rb at Ser 780 at concentrations of 0.125–0.5 µM, indicating that some cells were not arrested on day 1. Importantly, PD0332991 did not cause pS6 dephosphorylation at any concentration used, thus creating a situation for geroconversion to occur. In fact, at all concentrations, PD0332991 induced cyclin D1, an early marker of geroconversion.7 This marker was decreased by co-treatment with rapamycin, which blocked S6 phosphorylation. Importantly rapamycin did not restore but even decreased Rb phosphorylation (see overexposed blot), consistent with continuous arrest of the cell cycle. In agreement, PD0332991 inhibited proliferation at the same concentrations, and rapamycin did not restore proliferation, but in turn, it was cytostatic by itself (Fig. 3A and C). We also determined RP of the arrested cells after 3.5 and 7 days of treatment with PD0332991 (Fig. 3B and D). Loss of RP was prominent at 1–2 µM PD0332991 after 3.5 days treatment (Fig. 3B), and it was prevented by co-treatment with rapamycin. By day 7, cells treated with 0.5 µM PD0332991 completely lost RP, which was prevented by rapamycin (Fig. 3D). Thus, geroconversion was dependent on the duration of treatment. After 3.5 days, cells started to acquire senescent morphology, which was not prominent, consistent with some retention of RP (Fig. 4). By day 7, all cells were senescent (Fig. 4), consistent with loss of RP (Fig. 3D). Rapamycin prevented both senescent morphology (Fig. 4) and loss of RP (Fig. 3D) in MEL10 cells treated with PD0332991.
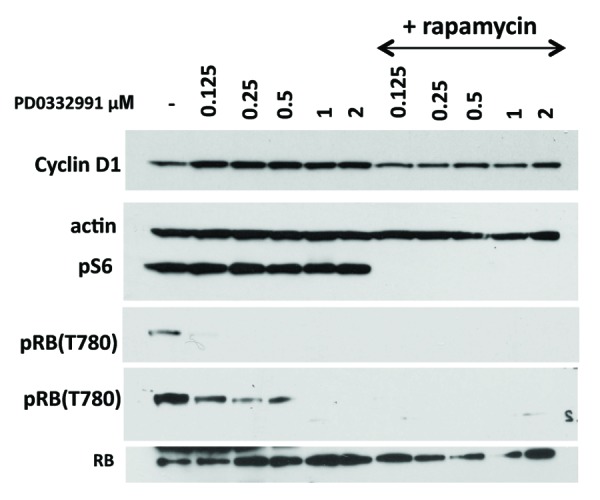
Figure 2. Immunoblot analysis of PD0332991-induced senescence in MEL10 cells. Cells were treated with indicated concentrations of PD0332991 with or without 10 nM rapamycin. After 1 day, cells were lysed and immunoblotting was performed with the indicated antibodies.
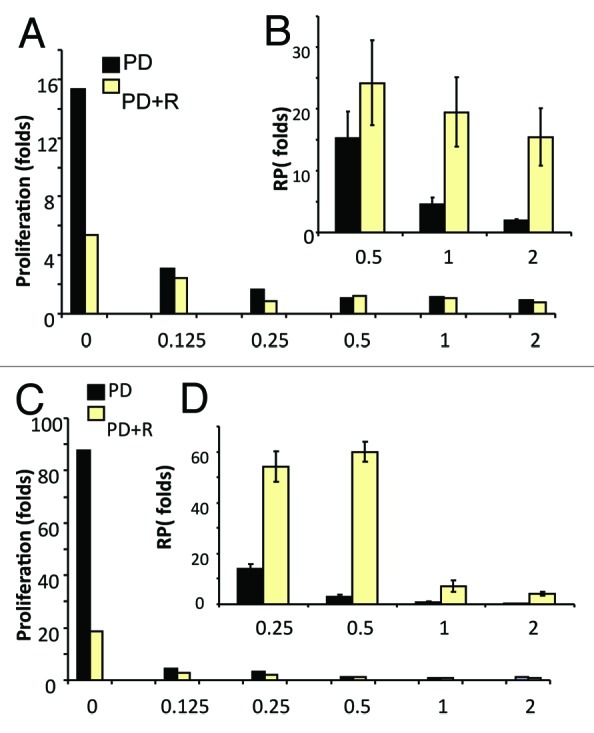
Figure 3. The effect of PD0332991 on proliferation and replicative potential (RP) in MEL10 cells. Cells were treated with the indicated concentrations of PD0332991 with or without 10 nM rapamycin. (A and B) After 3.5 days, proliferation (A) was measured by cell counting, as fold increase compared with initial (seeding) cell numbers. In parallel sets (B), drugs were washed out, and cells were allowed to proliferate, and RP was measured after 7 days of culture, as described in “Materials and Methods”. (C and D) After 7 days treatment, proliferation (A) was measured by cell counting, as fold increase compared with initial (seeding) cell numbers. In parallel sets (B), drugs were washed out, and cells were allowed to proliferate, and RP was measured after 7 days of culture, as described in “Materials and Methods”.
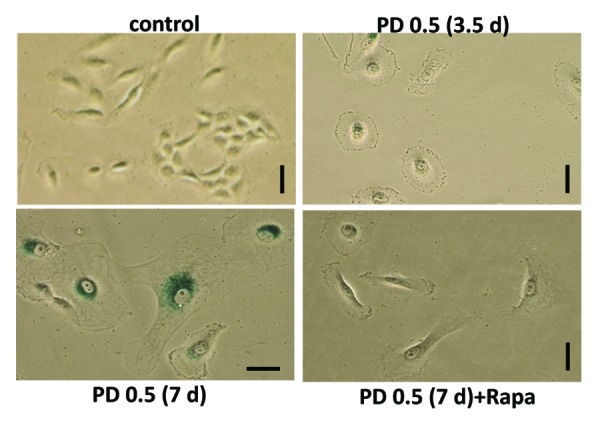
Figure 4. Senescent morphology of PD0332991-treated MEL10 cells. MEL10 cells were treated with 0.5 µM PD0332991 with or without 10 nM rapamycin. After 3.5 and 7 days, cells were stained for beta-Gal. Bar: 100 µm.
Time- and MTOR- dependent senescence caused by PD0332991 in normal RPE cells
Next we extended our observations to normal RPE cells. At a wide range of concentrations PD0332991 inhibited Rb phosphorylation at CDK4/6 site Ser780 (Fig. 5A). In agreement, at the same concentrations, PD0332991 inhibited cell proliferation (Fig. 5B), but it did not inhibit the MTOR pathway (Fig. 5A). Cyclin D1 was induced as a marker of geroconversion. In agreement, RPE cells showed decreased replicative potential (RP, Fig. 5C). Rapamycin preserved RP in PD0332991-treated RPE cells (Fig. 5D). Finally, we confirmed that U0126 prevented cyclin D1 induction during geroconversion, but it did not inhibit proliferation and Rb phosphorylation (Fig. 5A).
Figure 5.
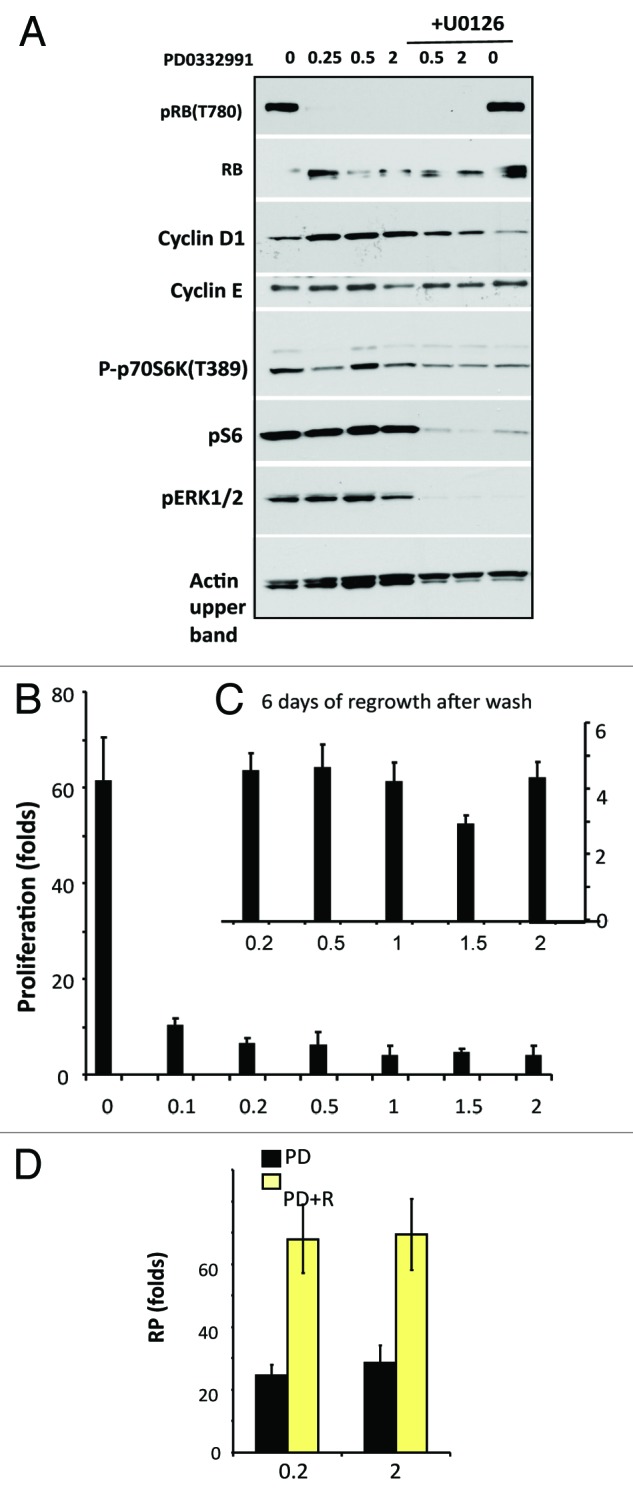
The effect of PD0332991 on RPE cells. (A). Immunoblot analysis: Cells were treated with the indicated concentrations of PD0332991 with or without 10 µM U0126. Cells were lysed after 24 h. (B and C) Proliferation (B) and RP: replicative potential (C). After 7 days treatment proliferation (B) was measured by cell counting. In parallel set (C), drugs were washed out and cells were allowed to proliferate. RP was measured by counting after 6 days of culture. (D) The effect of rapamycin on RP of PD0332991-treated RPE cells. Cells were treated with 0.2 or 2 µM PD0332991 with or without 5 nM rapamycin (R). After 5 days treatment drugs were washed out and RP was determined.
Discussion
Here we showed that the synthetic inhibitor of CDK4/6, p21, and p16 caused almost identical senescence phenotypes in HT-p21 cells. While inhibitors of CDKs cause cell cycle arrest, MTOR and MEK determine geroconversion and are responsible for the acquisition of hallmarks of senescence. p21 and p16 affect some targets besides CDKs, which were considered to be related to the senescence program. Although PD0332991 may have other off-target effects, only CDKs are the common targets for p21, p16, and PD0332991. p21, p16, and PD0332991 do not inhibit MTOR, which, in turn, drives senescent phenotype, and rapamycin decelerates geroconversion caused by all 3 molecules. This confirms the notion that a mere arrest in the presence of active MTOR may lead to geroconversion to senescence. Gerogenic conversion (geroconversion) without cell cycle arrest is associated with cancer15,16 and not coincidentally PI3K/MTOR and MEK/MAPK pathways are almost always activated in cancer.17-25 p16 is considered a marker of senescence,26-29 although it is rather a marker of cell cycle arrest, which may (or may not) be associated with senescence. A combination of cell cycle arrest (p16 or p21) with elevated cyclin D1 and active MTOR (phospho S6-positivity) may be a precise marker of gerogenic conversion (Fig. 6). Such gerogenic cells drive organismal aging,30 and rapamycin, which suppresses geroconversion,2-7,31-42 also delays age-related diseases and extends lifespan in mice.43-63 Whether inhibitors acting downstream of MTOR could suppress geroconversion is under initial investigation.64
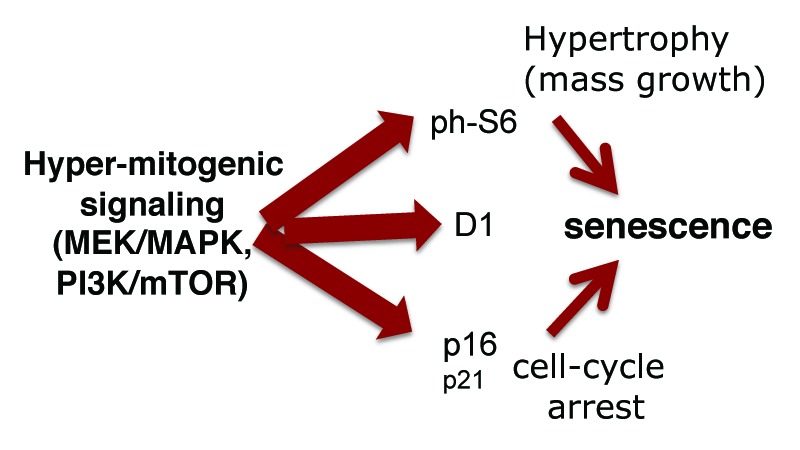
Figure 6. Hyper-mitogenic signaling and markers of senescence. When the cell cycle is arrested, active MAPK and MTOR pathways drive geroconversion from arrest to senescence. Although p16 expression is commonly considered as a marker of senescence,26-29,65 p16 merely causes cell cycle arrest. It is hyper-mitogenic signaling via MAPK and MTOR that over-stimulates arrested cells, causing senescence. (Note: p16 can be also associated with aggressive cancer66 and may have anti-aging activities67). However, p16 is induced by hyper-mitogenic signaling (Ras, Raf, MAPK, and PI-3K/mTOR),68-76 rendering p16 a proxy marker of hypermitogenic signaling and senescence (senescence = hyper-mitogenic signaling plus cell cycle arrest). The most reliable marker of senescence could be a combination of p16, cyclin D1 (Mek-dependent), and phospho-S6 (MTOR-dependent).
Material and Methods
Cell lines and reagents
HT-p21 cells, derived from HT1080 human fibrosarcoma cells (ATCC) were previously described.8-10 In HT-p21 cells, p21 expression can be turned on or off using isopropyl-thio-galactosidase (IPTG).8,9 HT-p21 cells were cultured as described previously.5,6 Melanoma MEL10 (formally, SK-MEL-147) were described previously.7,14 Normal retinal pigment epithelial RPE cell lines were obtained from (ATCC) were maintained in MEM plus 10% FBS. PD0332991 was purchased from Selleckchem. Rapamycin was obtained from LC Laboratories. IPTG (Invitrogen) was used in cell culture at final concentration 50 µg/ml. U0126 was purchased from Sigma-Aldrich.
Immunoblot analysis
Immunoblot analysis was performed as described previously.5 The following primary antibodies were used: mouse anti-phospho p70S6K (Thr389), rabbit anti-phospho Thr389 p70S6K and anti-phospho Thr421/Ser424 p70S6K, rabbit anti-phospho ERK ½ (Thr202/Tyr204), and anti-phospho Ser780 RB were purchased from Cell Signaling Biotechnology; mouse anti-cyclins D1 and E and anti-RB were obtained from Santa Cruz Biotechnology; mouse anti-p21 and rabbit anti-actin were purchased from BD Biosciences and Sigma-Aldrich, respectively. Secondary anti-rabbit and anti-mouse HRP-conjugated antibodies were purchased from Cell Signaling Biotechnology.
SA-β-Gal staining
Beta-Gal staining was performed using Senescence-galactosidase staining kit (Cell Signaling Technology) according to manufacturer’s protocol.
RP (regenerative/replicative potential) assay
Cells were plated at low density, treated with senescence-inducing agents (IPTG or PD0332991) as indicated in figure legends. Cell numbers were determined at the end of treatment (initial cell numbers) and drugs were removed by washing. Cells were incubated in fresh drug-free medium for several days, as indicated in figure legends, and then final cell numbers were determined. RP was calculated as a ratio between final and initial cell numbers: a fold-increase in cell numbers after the drugs were washed out.
Colony formation assay
HT-p21 cells were plated at low density, treated with IPTG or PD0332991 with or without rapamycin. In some experiments, after 1 day of treatment nocodazole was added as indicated in schema in Figure 1C. Then, drugs were washed out, and cells were incubated in fresh drug-free medium for 6–7 days. Plates were fixed and stained with 1.0% crystal violet (Sigma-Aldrich).
Supplementary Material
Acknowledgments
This paper was edited by proxy editor. The authors thank Zoya Demidenko for help with experiments in Figure 1B and D. The authors declare no conflict of interests.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Supplemental Materials
Supplemental materials may be found here: www.landesbioscience.com/journals/cc/article/26130
Footnotes
Previously published online: www.landesbioscience.com/journals/cc/article/26130
References
- 1.Blagosklonny MV. Cell cycle arrest is not yet senescence, which is not just cell cycle arrest: terminology for TOR-driven aging. Aging (Albany NY) 2012;4:159–65. doi: 10.18632/aging.100443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Demidenko ZN, Blagosklonny MV. Growth stimulation leads to cellular senescence when the cell cycle is blocked. Cell Cycle. 2008;7:3355–61. doi: 10.4161/cc.7.21.6919. [DOI] [PubMed] [Google Scholar]
- 3.Demidenko ZN, Zubova SG, Bukreeva EI, Pospelov VA, Pospelova TV, Blagosklonny MV. Rapamycin decelerates cellular senescence. Cell Cycle. 2009;8:1888–95. doi: 10.4161/cc.8.12.8606. [DOI] [PubMed] [Google Scholar]
- 4.Demidenko ZN, Korotchkina LG, Gudkov AV, Blagosklonny MV. Paradoxical suppression of cellular senescence by p53. Proc Natl Acad Sci U S A. 2010;107:9660–4. doi: 10.1073/pnas.1002298107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Leontieva OV, Natarajan V, Demidenko ZN, Burdelya LG, Gudkov AV, Blagosklonny MV. Hypoxia suppresses conversion from proliferative arrest to cellular senescence. Proc Natl Acad Sci U S A. 2012;109:13314–8. doi: 10.1073/pnas.1205690109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Leontieva OV, Lenzo F, Demidenko ZN, Blagosklonny MV. Hyper-mitogenic drive coexists with mitotic incompetence in senescent cells. Cell Cycle. 2012;11:4642–9. doi: 10.4161/cc.22937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Leontieva OV, Demidenko ZN, Blagosklonny MV, Blagosklonny MV. MEK drives cyclin D1 hyperelevation during geroconversion. Cell Death Differ. 2013;20:1241–9. doi: 10.1038/cdd.2013.86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chang BD, Xuan Y, Broude EV, Zhu H, Schott B, Fang J, Roninson IB. Role of p53 and p21waf1/cip1 in senescence-like terminal proliferation arrest induced in human tumor cells by chemotherapeutic drugs. Oncogene. 1999;18:4808–18. doi: 10.1038/sj.onc.1203078. [DOI] [PubMed] [Google Scholar]
- 9.Chang BD, Broude EV, Fang J, Kalinichenko TV, Abdryashitov R, Poole JC, Roninson IB. p21Waf1/Cip1/Sdi1-induced growth arrest is associated with depletion of mitosis-control proteins and leads to abnormal mitosis and endoreduplication in recovering cells. Oncogene. 2000;19:2165–70. doi: 10.1038/sj.onc.1203573. [DOI] [PubMed] [Google Scholar]
- 10.Chang BD, Swift ME, Shen M, Fang J, Broude EV, Roninson IB. Molecular determinants of terminal growth arrest induced in tumor cells by a chemotherapeutic agent. Proc Natl Acad Sci U S A. 2002;99:389–94. doi: 10.1073/pnas.012602599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Toogood PL, Harvey PJ, Repine JT, Sheehan DJ, VanderWel SN, Zhou H, Keller PR, McNamara DJ, Sherry D, Zhu T, et al. Discovery of a potent and selective inhibitor of cyclin-dependent kinase 4/6. J Med Chem. 2005;48:2388–406. doi: 10.1021/jm049354h. [DOI] [PubMed] [Google Scholar]
- 12.Capparelli C, Chiavarina B, Whitaker-Menezes D, Pestell TG, Pestell RG, Hulit J, Andò S, Howell A, Martinez-Outschoorn UE, Sotgia F, et al. CDK inhibitors (p16/p19/p21) induce senescence and autophagy in cancer-associated fibroblasts, “fueling” tumor growth via paracrine interactions, without an increase in neo-angiogenesis. Cell Cycle. 2012;11:3599–610. doi: 10.4161/cc.21884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Leontieva OV, Demidenko ZN, Gudkov AV, Blagosklonny MV. Elimination of proliferating cells unmasks the shift from senescence to quiescence caused by rapamycin. PLoS One. 2011;6:e26126. doi: 10.1371/journal.pone.0026126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Korotchkina LG, Leontieva OV, Bukreeva EI, Demidenko ZN, Gudkov AV, Blagosklonny MV. The choice between p53-induced senescence and quiescence is determined in part by the mTOR pathway. Aging (Albany NY) 2010;2:344–52. doi: 10.18632/aging.100160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Blagosklonny MV. Molecular damage in cancer: an argument for mTOR-driven aging. Aging (Albany NY) 2011;3:1130–41. doi: 10.18632/aging.100422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Blagosklonny MV. Tumor suppression by p53 without apoptosis and senescence: conundrum or rapalog-like gerosuppression? Aging (Albany NY) 2012;4:450–5. doi: 10.18632/aging.100475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sabatini DM. mTOR and cancer: insights into a complex relationship. Nat Rev Cancer. 2006;6:729–34. doi: 10.1038/nrc1974. [DOI] [PubMed] [Google Scholar]
- 18.Janku F, Wheler JJ, Naing A, Stepanek VM, Falchook GS, Fu S, Garrido-Laguna I, Tsimberidou AM, Piha-Paul SA, Moulder SL, et al. PIK3CA mutations in advanced cancers: characteristics and outcomes. Oncotarget. 2012;3:1566–75. doi: 10.18632/oncotarget.716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zhang Y, Zheng XF. mTOR-independent 4E-BP1 phosphorylation is associated with cancer resistance to mTOR kinase inhibitors. Cell Cycle. 2012;11:594–603. doi: 10.4161/cc.11.3.19096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zoncu R, Efeyan A, Sabatini DM. mTOR: from growth signal integration to cancer, diabetes and ageing. Nat Rev Mol Cell Biol. 2011;12:21–35. doi: 10.1038/nrm3025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.McCubrey JA, Steelman LS, Chappell WH, Abrams SL, Franklin RA, Montalto G, Cervello M, Libra M, Candido S, Malaponte G, et al. Ras/Raf/MEK/ERK and PI3K/PTEN/Akt/mTOR cascade inhibitors: how mutations can result in therapy resistance and how to overcome resistance. Oncotarget. 2012;3:1068–111. doi: 10.18632/oncotarget.659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Holz MK. The role of S6K1 in ER-positive breast cancer. Cell Cycle. 2012;11:3159–65. doi: 10.4161/cc.21194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hart JR, Vogt PK. Phosphorylation of AKT: a mutational analysis. Oncotarget. 2011;2:467–76. doi: 10.18632/oncotarget.293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Adams JR, Schachter NF, Liu JC, Zacksenhaus E, Egan SE. Elevated PI3K signaling drives multiple breast cancer subtypes. Oncotarget. 2011;2:435–47. doi: 10.18632/oncotarget.285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Cornu M, Albert V, Hall MN. mTOR in aging, metabolism, and cancer. Curr Opin Genet Dev. 2013;23:53–62. doi: 10.1016/j.gde.2012.12.005. [DOI] [PubMed] [Google Scholar]
- 26.Serrano M, Lin AW, McCurrach ME, Beach D, Lowe SW. Oncogenic ras provokes premature cell senescence associated with accumulation of p53 and p16INK4a. Cell. 1997;88:593–602. doi: 10.1016/S0092-8674(00)81902-9. [DOI] [PubMed] [Google Scholar]
- 27.Jacobs JJ, de Lange T. p16INK4a as a second effector of the telomere damage pathway. Cell Cycle. 2005;4:1364–8. doi: 10.4161/cc.4.10.2104. [DOI] [PubMed] [Google Scholar]
- 28.Mikheev AM, Stoll EA, Ramakrishna R, Mikheeva SA, Horner PJ, Rostomily RC. Geropotency: increased malignant potential of aging neural progenitors. Aging (Albany NY) 2012;4:854–5. doi: 10.18632/aging.100514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Baker DJ, Wijshake T, Tchkonia T, LeBrasseur NK, Childs BG, van de Sluis B, Kirkland JL, van Deursen JM. Clearance of p16Ink4a-positive senescent cells delays ageing-associated disorders. Nature. 2011;479:232–6. doi: 10.1038/nature10600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Blagosklonny MV. Answering the ultimate question “what is the proximal cause of aging?”. Aging (Albany NY) 2012;4:861–77. doi: 10.18632/aging.100525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Serrano M. Dissecting the role of mTOR complexes in cellular senescence. Cell Cycle. 2012;11:2231–2. doi: 10.4161/cc.21065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Dulic V. Be quiet and you’ll keep young: does mTOR underlie p53 action in protecting against senescence by favoring quiescence? Aging (Albany NY) 2011;3:3–4. doi: 10.18632/aging.100257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kolesnichenko M, Hong L, Liao R, Vogt PK, Sun P. Attenuation of TORC1 signaling delays replicative and oncogenic RAS-induced senescence. Cell Cycle. 2012;11:2391–401. doi: 10.4161/cc.20683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Halicka HD, Zhao H, Li J, Lee YS, Hsieh TC, Wu JM, Darzynkiewicz Z. Potential anti-aging agents suppress the level of constitutive mTOR- and DNA damage- signaling. Aging (Albany NY) 2012;4:952–65. doi: 10.18632/aging.100521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Cao K, Graziotto JJ, Blair CD, Mazzulli JR, Erdos MR, Krainc D, Collins FS. Rapamycin reverses cellular phenotypes and enhances mutant protein clearance in Hutchinson-Gilford progeria syndrome cells. Sci Transl Med. 2011;3:89ra58. doi: 10.1126/scitranslmed.3002346. [DOI] [PubMed] [Google Scholar]
- 36.Mercier I, Camacho J, Titchen K, Gonzales DM, Quann K, Bryant KG, Molchansky A, Milliman JN, Whitaker-Menezes D, Sotgia F, et al. Caveolin-1 and accelerated host aging in the breast tumor microenvironment: chemoprevention with rapamycin, an mTOR inhibitor and anti-aging drug. Am J Pathol. 2012;181:278–93. doi: 10.1016/j.ajpath.2012.03.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kofman AE, McGraw MR, Payne CJ. Rapamycin increases oxidative stress response gene expression in adult stem cells. Aging (Albany NY) 2012;4:279–89. doi: 10.18632/aging.100451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Pani G. From growing to secreting: new roles for mTOR in aging cells. Cell Cycle. 2011;10:2450–3. doi: 10.4161/cc.10.15.16886. [DOI] [PubMed] [Google Scholar]
- 39.Driscoll MK, Albanese JL, Xiong ZM, Mailman M, Losert W, Cao K. Automated image analysis of nuclear shape: what can we learn from a prematurely aged cell? Aging (Albany NY) 2012;4:119–32. doi: 10.18632/aging.100434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Castilho RM, Squarize CH, Chodosh LA, Williams BO, Gutkind JS. mTOR mediates Wnt-induced epidermal stem cell exhaustion and aging. Cell Stem Cell. 2009;5:279–89. doi: 10.1016/j.stem.2009.06.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Iglesias-Bartolome R, Gutkind SJ. Exploiting the mTOR paradox for disease prevention. Oncotarget. 2012;3:1061–3. doi: 10.18632/oncotarget.712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Iglesias-Bartolome R, Patel V, Cotrim A, Leelahavanichkul K, Molinolo AA, Mitchell JB, Gutkind JS. mTOR inhibition prevents epithelial stem cell senescence and protects from radiation-induced mucositis. Cell Stem Cell. 2012;11:401–14. doi: 10.1016/j.stem.2012.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Selman C, Tullet JM, Wieser D, Irvine E, Lingard SJ, Choudhury AI, Claret M, Al-Qassab H, Carmignac D, Ramadani F, et al. Ribosomal protein S6 kinase 1 signaling regulates mammalian life span. Science. 2009;326:140–4. doi: 10.1126/science.1177221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Harrison DE, Strong R, Sharp ZD, Nelson JF, Astle CM, Flurkey K, Nadon NL, Wilkinson JE, Frenkel K, Carter CS, et al. Rapamycin fed late in life extends lifespan in genetically heterogeneous mice. Nature. 2009;460:392–5. doi: 10.1038/nature08221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Miller RA, Harrison DE, Astle CM, Baur JA, Boyd AR, de Cabo R, Fernandez E, Flurkey K, Javors MA, Nelson JF, et al. Rapamycin, but not resveratrol or simvastatin, extends life span of genetically heterogeneous mice. J Gerontol A Biol Sci Med Sci. 2011;66:191–201. doi: 10.1093/gerona/glq178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Anisimov VN, Zabezhinski MA, Popovich IG, Piskunova TS, Semenchenko AV, Tyndyk ML, Yurova MN, Antoch MP, Blagosklonny MV. Rapamycin extends maximal lifespan in cancer-prone mice. Am J Pathol. 2010;176:2092–7. doi: 10.2353/ajpath.2010.091050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Anisimov VN, Zabezhinski MA, Popovich IG, Piskunova TS, Semenchenko AV, Tyndyk ML, Yurova MN, Rosenfeld SV, Blagosklonny MV. Rapamycin increases lifespan and inhibits spontaneous tumorigenesis in inbred female mice. Cell Cycle. 2011;10:4230–6. doi: 10.4161/cc.10.24.18486. [DOI] [PubMed] [Google Scholar]
- 48.Spong A, Bartke A. Rapamycin slows aging in mice. Cell Cycle. 2012;11:845. doi: 10.4161/cc.11.5.19607. [DOI] [PubMed] [Google Scholar]
- 49.Wilkinson JE, Burmeister L, Brooks SV, Chan CC, Friedline S, Harrison DE, Hejtmancik JF, Nadon N, Strong R, Wood LK, et al. Rapamycin slows aging in mice. Aging Cell. 2012;11:675–82. doi: 10.1111/j.1474-9726.2012.00832.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Zhao C, Vollrath D. mTOR pathway activation in age-related retinal disease. Aging (Albany NY) 2011;3:346–7. doi: 10.18632/aging.100303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Khanna A, Kapahi P. Rapamycin: killing two birds with one stone. Aging (Albany NY). 2011;3:1043-1044.52. Longo VD, Fontana L. Intermittent supplementation with rapamycin as a dietary restriction mimetic. Aging (Albany, NY Online) 2011;3:1039–40. doi: 10.18632/aging.100401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Selman C, Partridge L. A double whammy for aging? Rapamycin extends lifespan and inhibits cancer in inbred female mice. Cell Cycle. 2012;11:17–8. doi: 10.4161/cc.11.1.18736. [DOI] [PubMed] [Google Scholar]
- 54.Zheng XF. Chemoprevention of age-related macular regeneration (AMD) with rapamycin. Aging (Albany NY) 2012;4:375–6. doi: 10.18632/aging.100469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Komarova EA, Antoch MP, Novototskaya LR, Chernova OB, Paszkiewicz G, Leontieva OV, Blagosklonny MV, Gudkov AV. Rapamycin extends lifespan and delays tumorigenesis in heterozygous p53+/- mice. Aging (Albany NY) 2012;4:709–14. doi: 10.18632/aging.100498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Comas M, Toshkov I, Kuropatwinski KK, Chernova OB, Polinsky A, Blagosklonny MV, Gudkov AV, Antoch MP. New nanoformulation of rapamycin Rapatar extends lifespan in homozygous p53-/- mice by delaying carcinogenesis. Aging (Albany NY) 2012;4:715–22. doi: 10.18632/aging.100496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Donehower LA. Rapamycin as longevity enhancer and cancer preventative agent in the context of p53 deficiency. Aging (Albany NY) 2012;4:660–1. doi: 10.18632/aging.100494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Flynn JM, O’Leary MN, Zambataro CA, Academia EC, Presley MP, Garrett BJ, Zykovich A, Mooney SD, Strong R, Rosen CJ, et al. Late-life rapamycin treatment reverses age-related heart dysfunction. Aging Cell. 2013 doi: 10.1111/acel.12109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Leontieva OV, Paszkiewicz G, Demidenko ZN, Blagosklonny MV. Resveratrol potentiates rapamycin to prevent hyperinsulinemia and obesity in male mice on high fat diet. Cell Death Dis. 2013;4:e472. doi: 10.1038/cddis.2012.202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Nair S, Ren J. Autophagy and cardiovascular aging: lesson learned from rapamycin. Cell Cycle. 2012;11:2092–9. doi: 10.4161/cc.20317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Major P. Potential of mTOR inhibitors for the treatment of subependymal giant cell astrocytomas in tuberous sclerosis complex. Aging (Albany NY) 2011;3:189–91. doi: 10.18632/aging.100298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Williamson DL. Normalizing a hyperactive mTOR initiates muscle growth during obesity. Aging (Albany NY) 2011;3:83–4. doi: 10.18632/aging.100290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Van Meter M, Seluanov A, Gorbunova V. Forever young? Exploring the link between rapamycin, longevity and cancer. Cell Cycle. 2012;11:4296–7. doi: 10.4161/cc.22868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Leontieva OV, Demidenko ZN, Blagosklonny MV. S6 kinase in geroconversion. Cell Cycle. 2013;12 doi: 10.4161/cc.26248. this issue. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Burd CE, Sorrentino JA, Clark KS, Darr DB, Krishnamurthy J, Deal AM, Bardeesy N, Castrillon DH, Beach DH, Sharpless NE. Monitoring tumorigenesis and senescence in vivo with a p16(INK4a)-luciferase model. Cell. 2013;152:340–51. doi: 10.1016/j.cell.2012.12.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Witkiewicz AK, Knudsen KE, Dicker AP, Knudsen ES. The meaning of p16(ink4a) expression in tumors: functional significance, clinical associations and future developments. Cell Cycle. 2011;10:2497–503. doi: 10.4161/cc.10.15.16776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Matheu A, Maraver A, Collado M, Garcia-Cao I, Cañamero M, Borras C, Flores JM, Klatt P, Viña J, Serrano M. Anti-aging activity of the Ink4/Arf locus. Aging Cell. 2009;8:152–61. doi: 10.1111/j.1474-9726.2009.00458.x. [DOI] [PubMed] [Google Scholar]
- 68.Collado M, Gil J, Efeyan A, Guerra C, Schuhmacher AJ, Barradas M, Benguría A, Zaballos A, Flores JM, Barbacid M, et al. Tumour biology: senescence in premalignant tumours. Nature. 2005;436:642. doi: 10.1038/436642a. [DOI] [PubMed] [Google Scholar]
- 69.Michaloglou C, Vredeveld LC, Soengas MS, Denoyelle C, Kuilman T, van der Horst CM, Majoor DM, Shay JW, Mooi WJ, Peeper DS. BRAFE600-associated senescence-like cell cycle arrest of human naevi. Nature. 2005;436:720–4. doi: 10.1038/nature03890. [DOI] [PubMed] [Google Scholar]
- 70.Gagrica S, Brookes S, Anderton E, Rowe J, Peters G. Contrasting behavior of the p18INK4c and p16INK4a tumor suppressors in both replicative and oncogene-induced senescence. Cancer Res. 2012;72:165–75. doi: 10.1158/0008-5472.CAN-11-2552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Capparelli C, Chiavarina B, Whitaker-Menezes D, Pestell TG, Pestell RG, Hulit J, Andò S, Howell A, Martinez-Outschoorn UE, Sotgia F, et al. CDK inhibitors (p16/p19/p21) induce senescence and autophagy in cancer-associated fibroblasts, “fueling” tumor growth via paracrine interactions, without an increase in neo-angiogenesis. Cell Cycle. 2012;11:3599–610. doi: 10.4161/cc.21884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Qiu W, Sahin F, Iacobuzio-Donahue CA, Garcia-Carracedo D, Wang WM, Kuo CY, Chen D, Arking DE, Lowy AM, Hruban RH, et al. Disruption of p16 and activation of Kras in pancreas increase ductal adenocarcinoma formation and metastasis in vivo. Oncotarget. 2011;2:862–73. doi: 10.18632/oncotarget.357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Sarkisian CJ, Keister BA, Stairs DB, Boxer RB, Moody SE, Chodosh LA. Dose-dependent oncogene-induced senescence in vivo and its evasion during mammary tumorigenesis. Nat Cell Biol. 2007;9:493–505. doi: 10.1038/ncb1567. [DOI] [PubMed] [Google Scholar]
- 74.Bazarov AV, Lee WJ, Bazarov I, Bosire M, Hines WC, Stankovich B, Chicas A, Lowe SW, Yaswen P. The specific role of pRb in p16 (INK4A) -mediated arrest of normal and malignant human breast cells. Cell Cycle. 2012;11:1008–13. doi: 10.4161/cc.11.5.19492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Kuilman T, Michaloglou C, Vredeveld LC, Douma S, van Doorn R, Desmet CJ, Aarden LA, Mooi WJ, Peeper DS. Oncogene-induced senescence relayed by an interleukin-dependent inflammatory network. Cell. 2008;133:1019–31. doi: 10.1016/j.cell.2008.03.039. [DOI] [PubMed] [Google Scholar]
- 76.Kosar M, Bartkova J, Hubackova S, Hodny Z, Lukas J, Bartek J. Senescence-associated heterochromatin foci are dispensable for cellular senescence, occur in a cell type- and insult-dependent manner and follow expression of p16(ink4a) Cell Cycle. 2011;10:457–68. doi: 10.4161/cc.10.3.14707. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.