

Abstract
Energy metabolism plasticity enables stemness programs during the reprogramming of somatic cells to an induced pluripotent stem cell (iPSC) state. This relationship may introduce a new era in the understanding of Warburg’s theory on the metabolic origin of cancer at the level of cancer stem cells (CSCs). Here, we used Yamanaka’s stem cell technology in an attempt to create stable CSC research lines in which to dissect the transcriptional control of mTOR—the master switch of cellular catabolism and anabolism—in CSC-like states. The rare colonies with iPSC-like morphology, obtained following the viral transduction of the Oct4, Sox2, Klf4, and c-Myc (OSKM) stemness factors into MCF-7 luminal-like breast cancer cells (MCF-7/Rep), demonstrated an intermediate state between cancer cells and bona fide iPSCs. MCF-7/Rep cells notably overexpressed SOX2 and stage-specific embryonic antigen (SSEA)-4 proteins; however, other stemness-related markers (OCT4, NANOG, SSEA-1, TRA-1–60, and TRA-1–81) were found at low to moderate levels. The transcriptional analyses of OSKM factors confirmed the strong but unique reactivation of the endogenous Sox2 stemness gene accompanied by the silencing of the exogenous Sox2 transgene in MCF-7/Rep cells. Some but not all MCF-7/Rep cells acquired strong alkaline phosphatase (AP) activity compared with MCF-7 parental cells. SOX2-overexpressing MCF-7/Rep cells contained drastically higher percentages of CD44+ and ALDEFLUOR-stained ALDHbright cells than MCF-7 parental cells. The overlap between differentially expressed mTOR signaling-related genes in 3 different SOX2-overexpressing CSC-like cell lines revealed a notable downregulation of 3 genes, PRKAA1 (which codes for the catalytic α 1 subunit of AMPK), DDIT4/REDD1 (a stress response gene that operates as a negative regulator of mTOR), and DEPTOR (a naturally occurring endogenous inhibitor of mTOR activity). The insulin-receptor gene (INSR) was differentially upregulated in MCF-7/Rep cells. Consistent with the downregulation of AMPK expression, immunoblotting procedures confirmed upregulation of p70S6K and increased phosphorylation of mTOR in Sox2-overexpressing CSC-like cell populations. Using an in vitro model of the de novo generation of CSC-like states through the nuclear reprogramming of an established breast cancer cell line, we reveal that the transcriptional suppression of mTOR repressors is an intrinsic process occurring during the acquisition of CSC-like properties by differentiated populations of luminal-like breast cancer cells. This approach may provide a new path for obtaining information about preventing the appearance of CSCs through the modulation of the AMPK/mTOR pathway.
Keywords: mTOR, AMPK, reprogramming, breast cancer, cancer stem cells, SOX2
Introduction
Cancer stem cells (CSCs), which are multipotent and have self-renewal capabilities, play a critical role in the origin and propagation of human carcinomas.1-5 Individual tumors have recently been shown to harbor multiple phenotypically or genetically distinct CSCs, because differentiated, normal, and non-CSC tumor cells can acquire CSC states via the activation of partially understood paths to stemness, i.e., CSCs are made and not just born.6-9 The mechanisms of CSC formation are not fully amenable to analysis using patient samples, because these tumors can only be studied after the transformation events have already occurred. Therefore, a unique system for modeling the molecular processes that are causally involved in generating CSC states may involve reprogramming the heterogeneous populations of human cancer cells to pluripotency.10-12
Remarkable similarities exist between the processes leading to the acquisition of CSC states and the reprogramming of somatic cells to pluripotency, i.e., induced pluripotent stem cells (iPSCs).13-19 Yamanaka’s reprogramming method involves the transient expression of 4 transcription factors (Oct4, Sox2, Klf4, and c-Myc [OSKM]) that have been demonstrated to be critical for stemness and cell differentiation in normal differentiated somatic cells. Several of these reprogramming factors, which can reset the epigenetic status of the cells and allow them to adopt a plethora of new possible fates, were previously known for their oncogenic activity. Indeed, after Yamanaka factors induce differentiated cells to trans-differentiate into iPSCs with self-renewal and differentiation capabilities, iPSCs spontaneously form teratocarcinomas upon transplantation into nude mice. The malignant behavior of iPSCs is due to the presence of a subpopulation of undifferentiated CSC-like teratoma-initiating pluripotent stem cells that are responsible for the growth of the teratocarcinomas and are intermixed with the desired, non-tumorigenic iPSC derivatives that terminally differentiate into multiple lineages.20,21 Consequently, CSCs have recently been proposed to arise through a reprogramming-like mechanism.
Because the most challenging problem facing this field is the variability of CSCs based on their origin, we hypothesized that the ability of certain cancer cells to be reprogrammed to pluripotency might allow the in vitro generation of pluripotent cancer stem cell lines from human tumors, which would provide a unique opportunity for discovering the intrinsic processes of CSCs. The subsequent determination of the mechanisms that are over-represented in “breast cancer iPSCs” may provide crucial insights into the self-renewing tumor-initiating mechanisms that regulate both the number and aberrant functionality of CSCs. The recently discovered parallels between the metabolic changes that occur during oncogenesis and the induction of pluripotency strongly suggest that cell reprogramming is a naturally occurring phenomenon.22-29 Because energy metabolism plasticity enables stemness programs during the reprogramming of somatic cells to an iPSC state, we recently reasoned that this could introduce a new era in the understanding of Warburg’s theory on the metabolic origin of cancer, at the level of the so-called CSCs, i.e., similar to the observations in iPSCs, cell bioenergetics can operate as a/the pivotal decision-making parameter during the reprogramming and acquisition of stem cell properties in non-CSC tumor cells.30-35
Here, we used Yamanaka stem-cell technology in an attempt to create stable de novo breast CSC lines in which to dissect the transcriptional control of mTOR, the master switch of cellular catabolism and anabolism in CSC-like states. We first performed reprogramming experiments with MCF-7 human breast cancer cells using the OSKM Yamanaka factors. After assessing the iPSC-like nature of the MCF-7/Rep derivatives using morphological criteria and a battery of stem cell-associated pluripotency markers, we explored whether the transcriptional control of cellular metabolism by the AMP-activated protein kinase (AMPK) and mammalian target of rapamycin (mTOR) pathways (which serve as a signaling nexus for regulating cellular metabolism, energy homeostasis, and cell growth and are frequently deregulated in genetic tumor syndromes and cancers) is poised to be a major driver of the metabolic conversion of differentiated tumor cells to CSC-like states. Using an in vitro model of the de novo generation of CSC-like states through the nuclear reprogramming of an established breast cancer cell line, we now reveal that the transcriptional suppression of mTOR repressors is an intrinsic process occurring during the acquisition of CSC-like properties by differentiated populations of breast cancer cells.
Results
Nuclear reprogramming of MCF-7 human breast cancer cells generates intermediate cellular states between cancer cells and bona fide iPSCs
MCF-7 cells were first transduced with individual retroviruses containing Oct-4, Sox2, Klf4, and c-Myc at a 1:1:1:1 ratio on day 0. On days 10–15, we began to observe human embryonic stem (hES) cell-like morphological changes, including the emergence of pronounced individual cell borders and a cobblestone appearance, with a remarkably high nucleus-to-cytoplasm ratio in individual cells. On days 15–20, clearly recognizable, hES-like colonies composed of very small, tightly packed cells with large nuclei and notable nucleoli appeared in the MCF-7 cell cultures (now called MCF-7/Rep cells) that usually formed round-shaped colonies with clear boundaries (Fig. 1). Upon seeding onto mouse embryonic fibroblast (MEF) feeder layers, MCF-7/Rep cells formed flat and round-edged hES-like colonies, and 6 single clones (designated MCF-7/Rep clone #1 to clone #6) were chosen at random and successfully expanded for further analyses; for simplicity in this study, we only show 3 representative clones (#1, #3, and #5) for each set of data.

Figure 1. Nuclear reprogramming of human MCF-7 breast cancer cells. Left. Experimental scheme for the reprogramming of MCF-7 cells. Right. Phase-contrast images of iPSC-like colonies from MCF-7 cells (top). Representative phase-contrast images of either MCF-7/Rep clones or parental MCF-7 cells during EB-mediated differentiation (bottom). As for normal iPSCs, the MCF-7/Rep clones but not the MCF-7 parental cells formed EB-like spherical aggregates in suspension culture containing differentiation-promoting medium.
To prepare MCF-7/Rep cells to assess their immature status, we manually isolated colonies from the feeder layer-supported culture. The MCF-7/Rep colonies were gently broken into small clumps containing approximately 10–50 cells and seeded onto Matrigel-coated plastic slides. The cells were cultured in the MEF-conditioned medium for 36–48 h to allow attachment to slides as small groups. This procedure allowed us to analyze a desired MCF-7/Rep clone with minimal contamination from MEFs. We first evaluated changes in the expression of several glycan surface antigens, specifically the stage-specific embryonic antigens SSEA-1 and SSEA-4 and the tumor rejection antigens TRA-1–60 and TRA-1–81, which are specific and useful markers of pluripotent stem cells. Undifferentiated ES cells from human, Rhesus macaque, and Callithrix jacchus (the common marmoset monkey) express SSEA-3 and SSEA-4 but not SSEA-1 (Lewis X-CD15); human iPSCs display the same pattern of expression of these markers. Figure 2 shows that virtually all of the cells in the re-seeded clumps of MCF-7/Rep cells were strongly positive for SSEA-4 compared with parental MCF-7 cells, indicating that the MCF-7/Rep cells from this “pick up and re-seed” procedure maintained well-recognized, undifferentiated iPSC-like features within the 48 h experimental timeframe. To further corroborate these findings, clonally expanded MCF-7/Rep cells were harvested and subjected to flow cytometry to evaluate SSEA-4 expression. Our results confirmed the drastic increase in SSEA-4+ cells after the nuclear reprogramming of MCF-7 cells and subsequent expansion of MCF-7/Rep clones (Fig. 2). The baseline SSEA-1 positivity in MCF-7 cells was slightly increased in all the MCF-7/Rep clones (Fig. 3). Regarding TRA-1–60 and TRA-1–81, human and non-human primate ES cells, human iPSCs, and immortal embryonic germ cells (EGCs) express TRA-1–60 and TRA-1–81. In MCF-7/Rep cells, only weak TRA-1–60 signals were detected when compared with SSEA-4. TRA-1–81 was similarly expressed by MCF-7/Rep cells, but not by its MCF-7 parental counterparts; TRA-1–81 labeling was much stronger than that of TRA-1–60. Immunofluorescence analyses confirmed that MCF-7/Rep cells strongly expressed the pluripotency marker SOX2; however, the other stemness markers, i.e., OCT4 and NANOG, were found at low to moderate levels in most of the MCF-7/Rep cells (Fig. 3).

Figure 2. Nuclear reprogramming of human MCF-7 breast cancer cells. Representative immunofluorescence images of parental MCF-7 cells and MCF-7/Rep clones stained with an antibody against SSEA-4. MCF-7 and MCF-7/Rep cell populations were also examined by flow cytometry for the stem cell marker SSEA-4 (green histograms) vs. isotype controls (purple histograms).
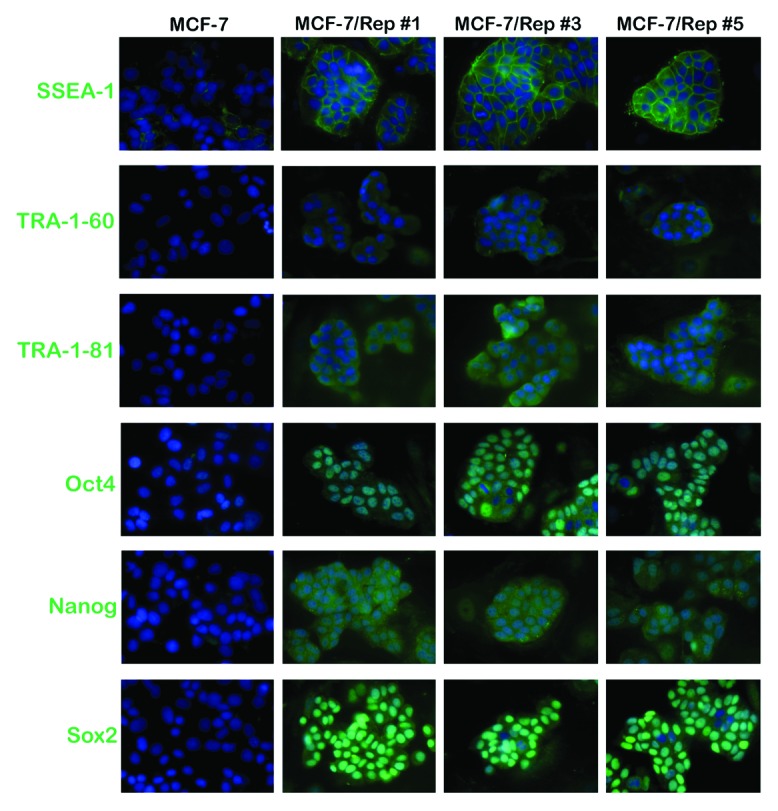
Figure 3. Nuclear reprogramming of human MCF-7 breast cancer cells. Representative immunofluorescence images of iPSC-like colonies from MCF-7/Rep clones and parental MCF-7 cells stained with antibodies against SSEA-1, TRA-1–60, TRA-1–80, OCT4, NANOG, and SOX2.
While we acknowledge that the fluorescence imaging data are qualitative, these data provide a good reference for the quantitative analyses of stem cell-associated pluripotency markers at the single cell level. We therefore concluded that the nuclear reprogramming of MCF-7 human breast cancer cells appears to generate intermediate states between differentiated cancer cells and bona fide pluripotent iPSCs. To unambiguously corroborate this suggestion, we took advantage of the Human OSKM factors Expression qBiomarker iPSC PCR array, which has been designed as an iPSC induction validation tool for analyzing the endogenous and total expression levels for the 4 reprogramming transcription factors used in the Yamanaka cocktail. The endogenous gene expression levels are measured by employing primer sets that are located outside the coding sequence of the mRNA, whereas the total gene expression levels are measured by employing primer sets that anneal within the coding sequence of the mRNA for each stemness transcription factor. A complete reprogramming, therefore, is indicated by an equal amount of endogenous and total expression of the transcription factors used in the cocktail. As observed in Figure 4, endogenous Sox2 was strongly reactivated (>5-fold in the MCF-7/Rep clone#1), whereas exogenous transgenic Sox2 was fully silenced in MCF-7/Rep cells. Endogenous Oct4, Klf-4, and c-Myc, however, were not expressed in any of the MCF-7/Rep clones. Therefore, Sox2 was the sole transgene that was overexpressed in all the MCF-7/Rep clones, indicating that none of the clones were reprogrammed successfully.

Figure 4. Nuclear reprogramming of human MCF-7 breast cancer cells. Left: RT-PCR analyses to detect the expression of exogenous (exo) and endogenous (endo) Oct4, Sox2, Klf-4, and c-Myc transcripts in MCF-7/Rep cells. Figure shows the difference of each transcript as fold-change mean values ± SD vs. MCF-7 parental cells; n = 3). Right: Total cell lysates (750 μg) from MCF-7 parental cells and 2 representative MCF-7/Rep clones were incubated with membranes of the Human Pluripotent Stem Cell Antibody Array, including 15 pluripotent stem cell markers, as per the manufacturer’s instructions (Proteome Profiler; R&D Systems). Figure shows representative proteome analyses that were developed on X-ray film following exposure to chemiluminescent reagents. Of the 15 pluripotent stem cell markers analyzed on the Human Pluripotent Stem Cell Array, SOX2 underwent the highest induction in protein expression when comparing MCF-7/Rep clones to MCF-7 parental cells.
Because morphological traits and SSEA-4 expression may not be sufficient to identify the successfully reprogrammed cancer iPSCs that are expected to have acquired pluripotent characteristics, we then used a commercial antibody array that allows the assessment of the expression of OCT4, SOX2, NANOG, and other pluripotency-associated proteins in a single test to compare protein extracts obtained from MCF-7 parental cells and their MCF-7/Rep counterparts. We did not observe significant expression of OCT4, NANOG, or SOX2 proteins in the MCF-7 control population; however, the induction of SOX2 protein, but not OCT4 or NANOG, was clearly observable in MCF-7/Rep cells (Fig. 4A). These findings supported the immunofluorescence analyses showing that the expression of several reprogramming-associated proteins was generally unevenly distributed in the colonies of the MCF-7/Rep clones, in striking contrast to the homogeneous and prominent nuclear expression of SOX2 observed in most cells of the MCF-7/Rep colonies.
Sox2-overexpressing MCF-7/Rep cells are enriched with CSC-like attributes
We assessed whether the sole reactivation of endogenous Sox2 in the MCF-7/Rep cells was sufficient to promote the acquisition of some degree of pluripotency properties. Using a cell-permeable fluorescent substrate for alkaline phosphatase (AP) that differentially stains pluripotent stem cells, we concluded that the overexpression of Sox2 appeared to be correlated with AP expression, because MCF-7/Rep clones expressing Sox2 exhibited more intense AP staining compared with AP-negative MCF-7 parental cells (Fig. 5A). Nevertheless, the AP distribution showed mosaic expression patterns in MCF-7/Rep clones, suggesting that Sox2-overexpressing MCF-7/Rep cells may be enriched with pluripotent-like subpopulations with CSC attributes.

Figure 5. Nuclear reprogramming of human MCF-7 breast cancer cells. (A) AP Live Stain was applied to MCF-7 parental cells and Sox2-overexpressing MCF-7/Rep cells growing in Matrigel. Figure shows representative microphotographs of immunofluorescence staining for AP Live Stain. (B). MCF-7 parental cells and Sox2-overexpressing MCF-7/Rep cells were subjected to ALDEFLUOR® assay to identify cells with high ALDH activity (ALDHbright). The ALDH inhibitor DEAB was used as a negative control. The cells without inhibitor shifted to the right and were considered ALDHbright cells. Figure shows fold changes in the expansion of the ALDHbright subpopulation calculated by dividing the number of ALDHbright cells counted in MCF-7/Rep cells by those counted in MCF-7 parental cells. (C) Representative immunofluorescence images of MCF-7 parental cells and MCF-7/Rep clones stained with antibodies against CD44 and E-cadherin.
Because MCF-7/Rep cells were not fully pluripotent but exhibited distinct molecular characteristics that were compatible with the acquisition of a breast CSC-like state, we investigated whether well-accepted breast CSC hallmarks were overrepresented in Sox2-overexpressing MCF-7/Rep cells. In several types of tumors including breast cancer, cell subpopulations enriched for cancer-initiating activity have been readily identified by flow cytometry analysis using the ALDEFLUOR® reagent on the basis of the high levels of aldehyde dehydrogenase (ALDH) activity; these cells are considered ALDHbright cells. The ALDEFLUOR® assay quantifies ALDH activity by measuring the conversion of the ALDH substrate, BODIPY aminoacetaldehyde, to the fluorescent product, BODIPY aminoacetate. The addition of the ALDH inhibitor DEAB specifically reduces ALDH-dependent fluorescence, thus confirming that ALDHbright cells are being correctly identified. When we used flow cytometry and the ALDEFLUOR® reagent to measure the proportion of MCF-7/Rep cells exhibiting high levels of ALDH activity, we confirmed that Sox2-overexpressing MCF-7/Rep cancer cell populations contained drastically higher percentages of ALDHbright cells than MCF-7 parental cells, a fact that is likely attributable to the overexpression of the ALDH1/3 isoforms in MCF-7/Rep cells (Fig. 5B). To identify ALDHbright cells, a control aliquot (+DEAB) was analyzed by flow cytometry and set for the detection of only the brightest ALDH-positive cells. Using this cutoff, the test (-DEAB) aliquot was analyzed to identify its ALDHbright cell content. In MCF-7 parental cells, ~1% of the cells were expressing high ALDH activity. In Sox2-overexpressing MCF-7/Rep clone#1 and #5, however, 25 and 30% of the cells were ALDHbright; therefore, the ALDHbright cell population in MCF7/Rep cells impressively increased by >20 times compared with MCF-7 parental cells. Because CD44 is another commonly used biomarker to identify the breast CSC phenotype, we performed immunofluorescence analysis to confirm that, in striking contrast to CD44-negative MCF-7 parental cells, MCF-7/Rep cells notably gained a significant membrane-associated expression of the CSC-related marker CD44 (Fig. 5C). Notably, there was no evidence of epithelial-to-mesenchymal (EMT) activation in Sox2-overexpressing CSC-like MCF-7/Rep cells, as they maintained a strong E-cadherin-mediated cell–cell adhesion, accompanied by a typical epithelial cobblestone appearance (Fig. 5C).
Transcriptional control of mTOR signaling regulators is significantly altered in Sox2-overexpressing CSC-like MCF-7/Rep cells
We investigated whether the transcriptional control of cellular metabolism by mTOR signaling was significantly altered in our in vitro model of the de novo generation of CSC-like states. Eighty-four key genes involved in the mTOR-signaling pathway were evaluated by qRT-PCR using total RNA from MCF-7 parental cells and MCF-7/Rep clones #1, #3, and #5. This commercially available gene array (Human mTOR Signaling PCR Array, SABiosciences) includes members of the mTORC1 and mTORC2 complexes, upstream regulators of many mTOR responses, and genes that are downstream of the many cellular processes regulated by mTOR complex activation. Imposing a 3-fold change in the mRNA expression level as the cut-off to determine significant regulatory effects on mTOR-related genes demonstrated that the acquisition of a Sox2-overexpressing CSC-like state involved significant alterations in 12, 13, and 28 mTOR-related genes in MCF-7/Rep clones #1, #3, and #5, respectively (Fig. 6, right). We identified 4 genes that were commonly regulated by all 3 MCF-7/Rep clones, PRKAA1 (−3.7-fold), DDIT4 (−5.1-fold), DEPTOR (−7.5-fold), and INSR (+3.0-fold). The PRKAA1 gene codes for the catalytic α 1 subunit of the AMP-activated protein kinase (AMPK).36-38 DDIT4 is a stress response gene, also known as REDD1 (regulated in development and DNA damage responses), which operates as a negative regulator of mTOR.39-43 DEPTOR has been identified as a naturally occurring endogenous inhibitor of mTOR that can suppress mTOR activity in vivo.44-46 INSR is a receptor tyrosine kinase that mediates the pleiotropic actions of insulin (e.g., insulin binding to the insulin receptor stimulates glucose uptake).47-49 The downregulation of AMPK expression was verified and confirmed using western blot analysis as a marker of end-point protein expression (Fig. 6A). Nuclear reprogramming of MCF-7 cells significantly decreased AMPKα1/α2 protein content, which was drastically suppressed in the MCF-7/Rep clone #5. We then analyzed the activation status of mTOR signaling pathway in MCF-7/Rep cells. mTOR activity was monitored by analyzing the phosphorylation level of its main downstream target, p70S6K1. Of note, immunoblotting analysis revealed that total levels of p70S6K1 in MCF-7/Rep cells were increased prominently as compared with MCF-7 parental cells (Fig. 6B). Consistent with the stimulation of p70S6K1, we detected a clear increase in the phosphorylation of mTOR at Ser2448 in MCF-7/Rep cells, thus confirming that mTOR pathway activity is notably augmented in Sox2-overexpressing CSC-like cell lines.

Figure 6. Activation of the mTOR pathway in Sox2-overexpressing CSC-like cellular states. (A) Overlaps between differentially expressed mTOR signaling-related genes in three MCF-7/Rep clonal groups. Venn diagram showing common changes in gene expression between three clonal groups (groups #1, #3, and #5) of MCF-7/Rep cells vs. parental MCF-7 cells. These Venn diagrams illustrate the overlaps between genes that were scored as being either up- or downregulated (n = 3 replicates for each clone). To be included as an overlap, a gene must be differentially expressed in at least one time point in both time courses (>3.0-fold and P value < / = 0.05). The 4 genes commonly regulated in the 3 MCF-7/Rep clonal groups are depicted on the left. Genes that are positively regulated over time (red), and genes that are negatively regulated over time (green) are indicated. The P value for the 3 clonal groups was calculated using a hypergeometric test and indicates that the probability of identifying the overlapping genes by random chance is very low. (B) Representative immunoblotting analyses of total AMPK, total p70S6K1, total mTOR, and phospho-mTORSer2448 in MCF-7 parental cells and MCF-7/Rep clones. The protein extracted from the harvested cells was resolved by SDS-PAGE, transferred to nitrocellulose membranes, and probed with the indicated antibodies. (C) Activation of the lipogenic phenotype in MCF-7/Rep cells. Robust expression of the lipogenic enzyme fatty acid synthase (FASN) in MCF-7/Rep cells as measured by immunofluorescence microscopy. Note that MCF-7/Rep cells express drastically higher levels of FASN than parental MCF-7 cells.
Sox2-overexpressing CSC-like MCF-7/Rep cells exhibit a fatty acid synthase (FASN)-overexpressing lipogenic phenotype
iPSCs generated from MEFs strongly enhance lipogenesis by triggering regulatory circuits that activate and provide substrates for lipogenic enzymes, such as FASN.33 Compared with non-CSCs, CSC-like cells express significantly higher levels of all lipogenic enzymes, which improve cell survival capacities,50-52 and studies published over the last few years have shown that mTOR signaling controls lipid biosynthesis through various mechanisms.53-56 Therefore, we hypothesized that, because reprogramming breast cancer cells to a Sox2-overexpressing CSC-like state involves the transcriptional suppression of mTOR repressors, MCF-7/Rep cells might overexpress FASN. A comparison of the starting population of MCF-7 cells and their CSC-like cell progeny using immunofluorescence microscopy clearly revealed that reprogrammed breast cancer cells display a dramatic upregulation of the lipogenic phenotype. Thus, the cytoplasmic accumulation of FASN was highly prominent in all of the MCF-7/Rep clones (#1, #3, and #5), whereas control cultures of parental MCF-7 cells expressed dramatically lower levels of FASN (Fig. 6C).
Discussion
The properties and molecular hallmarks of the rare population of undifferentiated tumor cells that retain the ability to self-renew, proliferate, and develop into more differentiated tumor cells (i.e., CSCs) are not well understood. Therefore, the development of novel methods for the molecular characterization of CSCs is urgently needed, because these approaches may ultimately facilitate the potential discovery of new targets that are specifically involved in tumor initiation. Master regulators of self-renewal and pluripotency (such as the transcription factors Oct4 and Sox2) have been shown to be expressed in a subpopulation of breast and ovarian cancer cells that possesses self-renewal abilities, and some cancers appear to hijack the underlying self-renewal transcription factor machinery to support aberrant proliferation and tumor initiation (i.e., some self-renewal transcription factors are overexpressed in early breast cancer lesions and in poorly differentiated, high-grade tumors).11,57-61 Therefore, we hypothesized that the nuclear reprogramming of differentiated cancer cells to a pluripotent-like state by introducing the Yamanaka stemness factors Oct4, Sox2, Klf4, and c-Myc might generate novel in vitro model systems to identify new mechanisms that are causally involved in breast cancer self-renewal and tumor initiation.62 Moreover, because key events governing the acquisition of CSC-like states are primarily regulated by reversible and transient modifications, rather than the accumulation of irreversible and stable modifications, studying the intrinsic ability of cancer cells to generate iPSCs in response to pluripotency-stimulating transcription factors may provide us with pivotal information on the molecular links between malignant transformation, pluripotency, and tumorigenesis. Particularly, by decoding the biological barriers that prevent the cancer (differentiated) phenotype from being reversed to a pluripotent (stem) status, we could directly explore the mechanisms governing how differentiated cancer cells can dynamically enter the CSC state.
Here, we described the molecular consequences observed upon the nuclear reprogramming of the well-characterized human breast cancer cell line MCF-7, an estrogen receptor-positive (ER+) breast cancer cell line that was isolated in 1973 from a 69-y-old Caucasian woman suffering from an invasive ductal carcinoma. The rare colonies with iPSC-like morphology that appeared following the viral transduction of OSKM stemness factors into MCF-7 cells appeared to demonstrate an intermediate state between cancer cells and the expected bona fide breast cancer-iPSCs. Although the reprogrammed MCF-7/Rep cells isolated based on their hES-like morphology strongly expressed some of the standard human pluripotent markers (for example SSEA-4) and exhibited stronger AP activity, albeit in a mosaic pattern, than MCF-7 parental cells, none of the examined clones homogenously exhibited other commonly accepted criteria by which to determine the status of successful reprogramming. The expression of OCT4, NANOG, SSEA-1, TRA-1–60, and TRA-1–81 proteins, if present, mostly showed an uneven expression pattern among the MCF7/Rep clones. Taken together, these data suggest that morphological traits, SSEA-4 and AP may not be sufficient to identify the successfully reprogrammed human cancer cells after transduction with the OSKM stemness factors. Because the expected derivation of breast cancer iPSCs required multiple individual viral vectors to deliver all the transcription factors needed to induce reprogramming, it could be argued that many cancer cells will receive only 1, 2, 3, or 4 factors, making it difficult to study the biochemistry of reprogramming on a highly heterogeneous population of breast cancer cells. However, we should acknowledge that our reprogramming efficiency was not significantly higher than those reported for somatic cell reprogramming, as might be expected due to pre-existing tumor suppressor loss and/or the endogenous expression of reprogramming factors. Additionally, MCF-7 cells infected with a single lentiviral vector expressing the 4 transcription factors from a single multicistronic transcript (data not shown) similarly failed to be reprogrammed into a bona fide pluripotent state. Although MCF-7/Rep cells, but not MCF-7 parental cells, were able to form aggregates of nonadherent spheroids known as embryoid bodies, which is the principal step in the differentiation of pluripotent stem cells, they failed to differentiate into all 3 primary germ layers and form benign mature tissue elements (as would have been expected for reprogrammed somatic cells) when we assessed teratoma formation in vivo. Indeed, when profiling their in vivo tumorigenicity, MCF-7/Rep cells engrafted with significantly higher efficacy and formed tumors at a faster rate than their MCF-7 parental counterparts in nude mice implanted subcutaneously with estrogen pellets, and the pathology of the reprogrammed breast cancer cells still best resembled highly aggressive, undifferentiated invasive breast carcinomas (unpublished observations). Despite all this evidence, it remains unclear whether human solid tumor cells can be reprogrammed into cells capable of dedifferentiating and forming benign teratomas. Whereas teratoma formation from solid human cancer cells has been reported previously,63 the authors reported only xenograft formation with no histology or immunohistochemistry. Zhang et al.,64 who recently reported the terminal differentiation and loss of tumorigenicity of human sarcomas via pluripotency-based reprogramming, failed to observe benign mature tissue elements in their teratoma assays.
MCF-7/Rep cells failed to exhibit a significant reactivation of the endogenous pluripotency-associated genes Oct4, Klf-4, and c-Myc. Although it is tempting to conclude that MCF-7/Rep cells merely represent incompletely reprogrammed clones with hES-like morphology that did not fully express the 4 exogenous transgenes, re-transduction experiments of the 3 other transgenes (i.e., Oct4, Klf-4, and c-Myc) into the selected Sox2-overexpressing MCF-7/Rep clones that lacked their expression should be performed before concluding that the failure to activate the pluripotency-related genes in the MCF-7/Rep clones was not due to technical barriers or the need for improved reprogramming technologies. It should be noted that whereas the present practices for reprogramming somatic cells to iPSCs involve the simultaneous introduction of reprogramming factors, Liu et al.65 have recently established that the simultaneous delivery of OSKM results in opposing activities among the 4 Yamanaka factors, thus diminishing their effectiveness. In their hands, the highest efficiency was observed with the OK, M, and S sequential protocol. It is noteworthy that MCF-7 cells naturally express Klf-4 and c-Myc,66,67 whereas MCF-7/Rep cells had up to 6- and 4-fold reductions in the endogenous levels of the Klf-4 and c-Myc genes, respectively. Together, these findings strongly suggest that the endogenous status of the Yamanaka factors may contribute to the reprogramming process differentially, i.e., positively or negatively, to determine the best procedure for reprogramming. The strong epithelial nature of MCF-7 cells, which is thought to be an optimal cell state for reprogramming, and the fact that E-cadherin is a competent substitute for Oct4 during somatic cell reprogramming68-70 may imply that estrogen receptor-positive MCF-7 luminal cells represent a breast cancer cellular state extremely sensitive to the dosage of Sox2;71 accordingly, the knockdown of endogenous E-cadherin notably compromised the time-sensitive generation of hES-like colonies in MCF-7 cells transduced with OSKM factors (data not shown), whereas the transcriptional analyses of the OSKM factors confirmed the strong but unique reactivation of the endogenous Sox2 oncogene accompanied by the silencing of the exogenous Sox2 transgene in MCF-7/Rep cells. Although Sox2-overexpressing MCF-7/Rep cells were not fully pluripotent, they exhibited distinct molecular characteristics that were compatible with the acquisition of a breast CSC-like state; thus, well-accepted breast cancer stem cell markers (SSEA-4, ALDEFLUOR, CD44)72-78 were overrepresented in Sox2-overexpressing MCF-7/Rep cells. If a differentiation hierarchy exists even in well-established cancer cell cultures, as predicted by the CSC theory, the most undifferentiated component in the cell culture may be the most permissive to reprogramming with Sox2 toward CSC-like states. In this regard, Sox2 transcriptional activity, as measured by GFP expression from a Sox2 reporter construct, has been detected in only a small subset of MCF-7 cells.79,80 GFP+ cells with Sox2 activity constitute a phenotypically distinct cell population, with an enhanced CSC-like phenotype and tumorigenicity in ER-positive MCF-7 luminal breast cancer cells.79 Because the measurement of Sox2 transcriptional activity depends on the binding of Sox2 to the promoter of the EMT gene driver Twist1,80 it may be relevant to establish whether the 2 distinct subsets of MCF-7 cells separated based on their differential reporter activity also display a differential responsiveness to the activation of nuclear reprogramming events.
Accumulating evidence suggests that changes in the cellular bioenergetics (i.e., the metabotype) may be a novel prerequisite for acquired stemness.35 For example, an experimental model comparing oncogenic transformation and cellular reprogramming has recently confirmed that somatic cells must first acquire changes that lead to the downregulation of cell differentiation machinery and the upregulation of glycolysis and other glycolysis-related metabolic pathways.29 Once these changes have occurred, the oncogenic transformation/induced pluripotency pathways can then diverge due to other factors, such as the activity of pluripotency genes. Given this scenario, we decided to look beyond cancer-specific genetic mutations, epigenetic remodeling, the accumulation of DNA damage, or reprogramming-induced cellular senescence and explore whether the nuclear reprogramming of breast cancer cells to Sox2-overexpressing CSC-like states involves the remodeling of bioenergetic and biosynthetic metabolism. We explored whether the evolutionarily conserved AMPK/mTOR pathway (which plays critical roles in the regulation of energy metabolism) is significantly over- or underrepresented upon the acquisition of stemness properties and a new cell identity. Importantly, we present the first characterization of the mTOR signaling-related transcriptome during the conversion of differentiated MCF-7 tumor cells to CSC-like-MCF-7 cells, and we reveal that the acquisition of stem cell-like states appears to utilize redundant molecular mechanisms aimed at ensuring the activation of mTOR; these mechanisms involve the significant transcriptional suppression of mTOR repressors (PRKAA1, DDIT4/REDD1, and DEPTOR) and the activation of mTOR enhancers (INSR) (Fig. 6A).
First, we confirmed that the downregulation of PRKAA1 may be a universal metabolic feature for the acquisition of stem cell-related properties, regardless of either normal or cancerous genetic background. PRKAA1 has been shown to be significantly and consistently downregulated upon the generation of iPSCs from normal fibroblasts. As suggested by Prigione et al.,24 iPSCs generated from fibroblasts may suppress the activation of AMPK (a master regulator of energy homeostasis that can switch off biosynthetic pathways to avoid anabolic inhibition), similar to the phenomenon observed in cancer cells. The downregulation of AMPK may account for the activation and/or maintenance of the Warburg effect,81 which fuels the induction of stemness during the reprogramming of somatic cells. Accordingly, the pharmacological activation of AMPK has been shown to establish a metabolic barrier to somatic cell reprogramming that cannot be bypassed even through p53 deficiency,30 which is a fundamental mechanism used to greatly improve the efficiency of stem cell production.82-85 In this scenario, hypothesizing that the subpopulation of MCF-7 epithelial cancer cells targeted by the stem cell factor Sox2 necessarily acquires distinctive AMPK-related bioenergetic signatures is tempting; this acquisition of AMPK-related bioenergetic signatures is a molecular strategy that has been shown to similarly induce and maintain the repression of mitochondrial oxidative phosphorylation in “normal” iPSCs.
Second, the generation of Sox2-overexpressing CSC-like states upon the nuclear reprogramming of ER-positive MCF-7 luminal breast cancer cells is accompanied by the suppression of the mTOR repressor DDIT4/REDD1. REDD1 loss has been shown to elicit tumorigenesis in a mouse model (immortalized REDD1−/− cells are highly tumorigenic compared with their wild-type counterparts), and the downregulation of REDD1 in a subset of human cancers (including breast CSCs) has been observed to promote selectively oncogenic anchorage-independent growth under hypoxic conditions.39-43 Therefore, the net result of REDD1 downregulation may be increased tumor cell adaptation to hypoxic conditions and increased cellular translation and cell growth potential; when combined, these effects may explain the dramatic induction of tumorigenesis that has been observed in the context of the genetic loss of REDD1. Our findings support the recent discovery that the elaborate regulation of mTOR activity is required for somatic cell reprogramming induced by defined transcription factors86; mechanistic studies should unambiguously elucidate whether novel regulatory pathways centered on REDD1 can actively contribute to the acquisition of breast CSC properties.87
Third, we also reveal that the inhibitory role of DEPTOR in the mTOR signaling pathway may be impacted during the Sox2-driven conversion of differentiated MCF-7 cells into CSC-like MCF-7 cells. Previous evidence has shown that DEPTOR plays a pivotal role in the development and progression of human malignances by regulating many cellular processes, such as cell growth, apoptosis, autophagy, epithelial-to-mesenchymal transition (EMT), and drug resistance; all these outcomes could be partially mediated through the inhibitory effect of DEPTOR on mTOR.44-46 Consistent with the activation of the mTORC1 and mTORC2 pathways in many human cancers, DEPTOR expression is low in most cancers. Moreover, the mTOR complexes and DEPTOR negatively regulate each other, suggesting that a feedforward loop exists in which the loss of DEPTOR leads to an increase in mTOR activity. This increased mTOR activity then further reduces DEPTOR expression at the transcriptional and post-translational levels. Our findings suggest that the upregulation of DEPTOR may be required to regulate mTOR activity in targeting stem-like breast cancer cells that express low levels of DEPTOR. In this regard, Liu et al.45 have shown that resveratrol (a naturally occurring polyphenol) promotes the interaction between DEPTOR and mTOR, leading to the inhibition of mTOR activity. Notably, resveratrol can induce apoptosis in cancer stem-like cells through the suppression of lipogenesis by modulating FASN expression,51,88 a downstream marker of AMPK deactivation/mTOR activation. We accordingly observed that FASN was overexpressed in MCF-7/OSKM cells.
Fourth, the mitogenic and anti-apoptotic effects of insulin, the principal regulator of glucose metabolism that plays a crucial role in cancer development and progression, may be directly mediated by its own receptor (INSR). Recent findings have begun to suggest that both IGF-IR and INSR may contribute to the expansion of adult progenitor/stem-like cells cultured as non-adherent spheres and the stimulation of self-renewal. Thus, stem and progenitor cells that survive in serum-free suspension to produce spherical colonies that self-renew are strongly positive not only for several putative stem cell markers (e.g., Sox2), but also for high levels of INSR.89 Moreover, the insulin axis plays a key role in the “breast stem cell burden” hypothesis,90-92 which suggests that factors that expand the normal breast stem cell pool would increase the probability that one such cell might undergo an oncogenic mutation or epigenetic change and favor the generation of mammary tissue-specific stem cells. Therefore, because breast stem/progenitor cells can serve as direct targets for transformation, the number of these cells (which is positively associated with mammary gland mass) is an important determinant of breast cancer risk. In an alternate scenario, CSCs may arise through reprogramming-like mechanisms, in which the target of transformation could be a tissue stem cell, progenitor cell, or differentiated cell that acquires self-renewal ability. INSR may be similarly involved in stimulating the insulin/mTOR pathway during the reprogramming of cancer cells to a pluripotent state. Our current findings may be relevant to the recent studies suggesting that the use of the extended activity basal insulin analog glargine increases the rate of development and subsequent detection of pre-existing undetectable malignancies rather than malignant cell transformation and new cancer formation.93
Corollary
Cancer, cellular plasticity, and cell fate reprogramming are tightly intertwined processes. Not all cancer cells possess the plasticity necessary for reprogramming to CSC-like states, and only some cancer genes harbor the required capacity to fully elicit the reprogramming process in the right cellular context. By understanding cancer as a disease of cellular reprogramming in which the formation of cancer-maintaining cells is a developmentally unfavorable process that imposes a great stress to the cells, it is conceivable that most of the driving forces in CSC generation play a permissive role in tumor progression. Not surprisingly, the elimination of the p53-regulated DNA damage control checkpoint notably enhances the efficiency of the somatic reprogramming process, similar to the effects of the loss of p53 in allowing the genesis of tumor-initiating cells and the development of cancer. We now suggest that the remodeling of AMPK/mTOR-regulated bioenergetic and biosynthetic metabolism (which occurs in the context of bona fide oncogenes and tumor suppressor genes) may be an active contributor (rather than a consequence) that defines cancer cell fate and is responsible for the acquisition of CSC-like states in breast cancer. The dysfunction of negative metabolic regulators, including defects in the AMPK-mTOR pathway, could result in permissive metabolic reprogramming that allows a differentiated cancer cell to be reprogrammed to regain stem cell characteristics in a process of tumorigenic reprogramming to pluripotency.35,94 Determining how the AMPK/mTOR-regulated bioenergetic and biosynthetic metabolism regulates stem cell fate may facilitate the development of therapies for aging-related metabolic diseases, such as obesity, diabetes, and cancer.95 Although the final results demonstrated that the rare colonies with iPSC-like morphology obtained following the viral transduction of Oct4, Sox2, Klf4, and c-Myc into MCF-7 breast cancer cells (MCF-7/Rep) demonstrated an intermediate state between cancer cells and bona fide iPSCs, they still exhibited a significant enhancement of breast CSC-like molecular characteristics. If our current findings reflect the fact that stem/progenitor cells within heterogeneous populations of MCF-7 breast cancer epithelial cells reprogram at a higher frequency than more differentiated MCF-7 cells (i.e., the target cells mediating the relatively low frequency of colonies with the Sox2-overexpressing phenotype in the transduced MCF-7 cultures were a subpopulation that was not fully differentiated ab initio), both the intrinsic flexibility and reversibility of the AMPK/mTOR-driven metabolic program may have crucial implications for the pharmacological manipulation of the self-renewal and pluripotent capabilities that fuel CSC-driven tumorigenesis (Fig. 7). Because our current findings suggest that the antagonistic nature of the AMPK and mTOR pathways may participate in the process of tumorigenic reprogramming to CSC-like states, it is reasonable to predict that when stable CSC-like research lines are used in drug screening, they may provide a new path for obtaining valuable molecular information about killing breast CSCs.

Figure 7. A yin-yang balancing act of the AMPK/mTOR link in reprogramming to Sox2-overexpressing breast cancer stem-like cellular states. We suggest that the AMPK/mTOR signaling link in reprogramming to Sox2-overexpressing breast cancer stem-like cellular states can be viewed much like a yin-yang balancing act, based on the ancient Chinese scientific thinking concerning how things work (i.e., the “universe” is governed by the balance of yin and yang, where yin represents the negative element, while yang represents the positive element). Consistent with this concept, AMPK and mTOR appear to have opposing activation patterns during abnormal differentiation in cancer. The forced activation of AMPK in cancer stem-like cells could lead to differentiation,32,33,100 whereas an elaborate regulation of the activation status of mTOR might be required to allow defined transcription factors to reprogram differentiated cancer cells toward stemness.68 The exploitation of this AMPK/mTOR signaling link with the use of the yin-yang concept may allow for the development of novel therapeutic strategies that target CSC states.
Materials and Methods
Reprogramming of human breast cancer cells
MCF-7 cells originally obtained from American Type Culture Collection (ATCC) were transduced with retroviral vectors encoding the reprogramming factors Oc4, Sox2, Klf4, and c-Myc following Yamanaka original protocol with modifications that have been previously validated in our laboratory.30-33 Briefly, the retroviral vector plasmids pMXs-hOCT4, pMXs-hSOX2, pMXs-hKlf4, pMXs-hc-Myc (Addgene), and VSV-G were packaged in 293T cells using Plus™ Reagent (Invitrogen) according to the manufacturer’s protocol. After 48 h, virus-containing supernatants were passed through a 0.45 μm filter and supplemented with 10 μg/mL polybrene. MCF-7 cells were seeded at ~5 × 105 cells per 100 mm dish 24 h before incubation with the virus/polybrene-containing supernatants. The transduction was repeated every 12 h for 2 d using the same batch of all 4 retroviruses. On day 6, after the first transduction, the cells were replated on a feeder layer of irradiated human HFF-1 fibroblasts (ATCC), and the regular media were replaced with human embryonic stem (hES) cell growth media (KNOCKOUT DMEM [Invitrogen], 20% KO-Serum Replacement [Invitrogen], 10 nmol/L β-FGF, 2 mmol/L GlutaMAX, 50 mmol/L 2-β-mercaptoethanol, 100 μmol/L non-essential amino acids [Sigma-Aldrich], 1% penicillin/streptomycin), and renewed every 2 d. Reprogramming MCF-7 cells were maintained at 37 °C in a 5% CO2 incubator for up to 30 d. The appearance of iPSC-like colonies, i.e., colonies that will form demonstrated well-defined phase-bright borders surrounded by feeder cells and comprising small cells with high nuclear/cytoplasmic ratios and prominent nucleoli, indistinguishable from standard iPSCs, was monitored every day.
Immunofluorescence staining and high-content confocal imaging
Three MCF-7/Rep clones were chosen at random and further analyzed for the expression of pluripotent stem cell markers including OCT4, SOX2, NANOG, SSEA-1, SSEA-4, TRA-1–60, TRA-1–8, CD44, and E-cadherin using immunofluorescence and/or flow cytometry procedures previously validated in our laboratory. We employed 96-well clear-bottom imaging tissue culture plates (Becton Dickinson Biosciences) optimized for automated imaging applications. Triton® X-100 permeabilization and blocking, primary antibody staining, secondary antibody staining using Alexa Fluor® 488/594 goat anti-rabbit/mouse IgGs (Invitrogen, Probes), and counterstaining (using Hoechst 33258; Invitrogen) were performed following the BD Biosciences protocols. The images were captured in different channels for Alexa Fluor® 488 (pseudocolored green) and Hoechst 33258 (pseudocolored blue) using a BD PathwayTM 855 Bioimager System (Becton Dickinson Biosciences) with 20× or 40× objectives (NA 075 Olympus). Merged images were obtained according to the recommended assay procedure using the BD Attovision™ software.
Embryoid bodies (EBs) formation
A standard and general method for stem cell differentiation in vitro is via the formation of cell aggregates in nonadherent spheroids known as embryoid bodies (EBs). MCF-7/Rep colonies (or regions of colonies) were cultured in suspension, so that they formed large aggregates as described elsewhere.97-99
Quantitative real-time polymerase chain reaction (qRT-PCR)
Total RNA was extracted from MCF-7 parental cells and MCF-7/Rep clones #1, #3, and #5 using a Qiagen RNeasy kit and Qiashredder columns according to the manufacturer’s instructions. One microgram of total RNA was reverse-transcribed into cDNA with a Reaction Ready™. First Strand cDNA Synthesis Kit (SABiosciences) and applied to either the Yamanaka reprogramming factors expression qBiomarker™ iPSC PCR Array System (Cat. No. IPHS-002, 96-well format) or the Human mTOR Signaling RT2 Profiler PCR Array (Cat. No. PAHS-098Z, 96-well format) following the SABiosciences RT-PCR manual. Plates were processed in an Applied Biosystems 7500 Fast Real-Time PCR System Applied Biosystems using automated baseline and threshold cycle detection. Data were interpreted with the SABiosciences web-based PCR array analysis tool.
Proteome profiling of pluripotent stem cell markers
Cells were rinsed with cold PBS and immediately solubilized in NP-40 lysis buffer (1% NP-40, 20 mmol/L Tris-HCl [pH 8.0], 137 mmol/L NaCl, 10% glycerol, 2 mmol/L EDTA, 1 mmol/L sodium orthovanadate, 10 μg/mL aprotinin, 10 μg/mL leupeptin) by rocking the lysates gently at 4 °C for 30 min. Following microcentrifugation at 14 000 × g for 5 min, supernatants were transferred into a clean test tube, and sample protein concentrations were determined using the Pierce Protein Assay Kit. Lysates were diluted and incubated with Human Pluripotent Stem Cell Antibody Array membranes (Cat. No. ARY010; Proteome Profiler; R&D Systems) according to the manufacturer’s instructions. In this method, capture and control antibodies have been spotted in duplicate on nitrocellulose membranes. Briefly, the membranes were blocked with 5% bovine serum album (BSA)/TBS [0.01 mol/L Tris HCl, pH 7.6] for 1 h. Membranes were then incubated overnight with 750 μg of total protein at 2–8 °C on a rocking platform shaker. After extensive washing with TBS including 0.1% v/v Tween-20 3 times for 5 min to remove unbound materials, the membranes were then incubated with reconstituted Detection Antibody Cocktail for 2 h at room temperature. Unbound HRP antibody was washed out with TBS including 0.1% v/v Tween-20. Finally, array data were developed on X-ray film using a chemiluminescence detection system (Amersham Life Sciences).
Alkaline phosphatase (AP) staining
AP activity was detected using a non-permanent, cell-viable stem cell imaging product that allows users to differentially stain pluripotent stem cells.96 The AP Live Stain is a cell-permeable fluorescent substrate for AP that is non-toxic to cells, diffusing out over the course of 2 h. Briefly, AP Love Stain (500×; Cat. No. A14353, Life Technologies Corp) was diluted in media and directly applied to adherent cell cultures of MCF-7 and MCF-7/Rep cells. All staining procedures were conducted according to product specifications.
Aldefluor activity assay
The Aldefluor kit (Stem Cell Technologies) was used to profile cells with high and low ALDH activity. Briefly, cells were suspended in ALDEFLUOR assay buffer containing the fluorescent ALDH substrate, BODIPY-aminoacetaldehyde (BAAA), and incubated for 45 min at 37 °C. The assay buffer also contains a transport inhibitor to prevent the efflux of the BAAA from the cells. BAAA passively diffuses into live cells and is then converted by intracellular ALDH into a negatively charged product, BODIPY aminoacetate, which is retained inside cells, labeling them with a bright fluorescent signal. After a washing step, the brightly fluorescent ALDH-expressing cells (ALDHbright) were detected in the green fluorescence channel (FL1; 520–540 nm) of a FACSCalibur instrument (BD Biosciences). A sample of cells was stained as above with the addition of a specific ALDH inhibitor, diethylaminobenzaldehyde (DEAB) (Sigma), to serve as a negative control for each experiment. Because only cells with an intact cellular membrane could retain the ALDH1 reaction product, only viable ALDHbright cells were identified. Cells incubated with BAAA and DEAB were used to establish the background signal and to define the ALDHbright region. The incubation of cells with the substrate in the absence of DEAB induced a shift in BAAA fluorescence defining the ALDHbright population.
Immunoblotting,
Cultures of MCF-7 parental cells and MCF-7/Rep clones were washed twice with cold PBS and then lysed as described above. Equal amounts of protein (i.e., 50 μg) were resuspended in 5× Laemmli sample buffer (10 min at 70 °C), subjected to 10% SDS-PAGE, and transferred onto nitrocellulose membranes. The nitrocellulose membranes were blocked for 1 h at RT with TBS-T buffer (25 mmol/L TRIS- HCl [pH 7.5], 150 mmol/L NaCl, 0.05% Tween 20) contain- ing 5% (w/v) nonfat dry milk to minimize non-specific binding. Subsequently, the treated membranes were washed in TBS-T and incubated with total AMPKα (#2603, Cell Signaling Technology®), total p70S6K1 (#9202, Cell Signaling Technology®), total mTOR (#2983, Cell Signaling Technology®), or phospho-mTORSer2448 (#2971, Cell Signaling Technology®), as specified, in 1× TBS-T buffer containing 5% w/v BSA and 0.1% Tween-20 at 4 °C with gentle shaking overnight. The membranes were washed with TBS-T, incubated with horseradish peroxidase-conjugated secondary anti-rabbit IgG in TBS-T for 1 h, and the immunoreactive bands were detected using a chemiluminescence reagent (Pierce). The blots were re-probed with an antibody against β-actin to control for protein loading and transfer. Densitometric values of the proteins bands were quantified using Scion Image software (Scion Corporation).
Acknowledgments
This work was financially supported by the Ministerio de Ciencia e Innovación (SAF2012-38914, Plan Nacional de I+D+ I, MICINN, Spain). Alejandro Vazquez-Martin received a Sara Borrell post-doctoral contract (CD08/00283, Ministerio de Sanidad y Consumo, Fondo de Investigación Sanitaria -FIS-, Spain). Sílvia Cufí received a research fellowship (Formación de Personal Investigador, FPI) from the Ministerio de Ciencia e Innovación (MICINN, Spain).
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Footnotes
Previously published online: www.landesbioscience.com/journals/cc/article/26173
References
- 1.Wicha MS, Liu S, Dontu G. Cancer stem cells: an old idea--a paradigm shift. Cancer Res. 2006;66:1883–90, discussion 1895-6. doi: 10.1158/0008-5472.CAN-05-3153. [DOI] [PubMed] [Google Scholar]
- 2.Kakarala M, Wicha MS. Implications of the cancer stem-cell hypothesis for breast cancer prevention and therapy. J Clin Oncol. 2008;26:2813–20. doi: 10.1200/JCO.2008.16.3931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Liu S, Dontu G, Wicha MS. Mammary stem cells, self-renewal pathways, and carcinogenesis. Breast Cancer Res. 2005;7:86–95. doi: 10.1186/bcr1021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Michor F, Polyak K. The origins and implications of intratumor heterogeneity. Cancer Prev Res (Phila) 2010;3:1361–4. doi: 10.1158/1940-6207.CAPR-10-0234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Magee JA, Piskounova E, Morrison SJ. Cancer stem cells: impact, heterogeneity, and uncertainty. Cancer Cell. 2012;21:283–96. doi: 10.1016/j.ccr.2012.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gupta PB, Fillmore CM, Jiang G, Shapira SD, Tao K, Kuperwasser C, Lander ES. Stochastic state transitions give rise to phenotypic equilibrium in populations of cancer cells. Cell. 2011;146:633–44. doi: 10.1016/j.cell.2011.07.026. [DOI] [PubMed] [Google Scholar]
- 7.Chaffer CL, Brueckmann I, Scheel C, Kaestli AJ, Wiggins PA, Rodrigues LO, Brooks M, Reinhardt F, Su Y, Polyak K, et al. Normal and neoplastic nonstem cells can spontaneously convert to a stem-like state. Proc Natl Acad Sci U S A. 2011;108:7950–5. doi: 10.1073/pnas.1102454108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Iliopoulos D, Hirsch HA, Wang G, Struhl K. Inducible formation of breast cancer stem cells and their dynamic equilibrium with non-stem cancer cells via IL6 secretion. Proc Natl Acad Sci U S A. 2011;108:1397–402. doi: 10.1073/pnas.1018898108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Polytarchou C, Iliopoulos D, Struhl K. An integrated transcriptional regulatory circuit that reinforces the breast cancer stem cell state. Proc Natl Acad Sci U S A. 2012;109:14470–5. doi: 10.1073/pnas.1212811109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Abollo-Jiménez F, Jiménez R, Cobaleda C. Physiological cellular reprogramming and cancer. Semin Cancer Biol. 2010;20:98–106. doi: 10.1016/j.semcancer.2010.02.002. [DOI] [PubMed] [Google Scholar]
- 11.Beltran AS, Rivenbark AG, Richardson BT, Yuan X, Quian H, Hunt JP, Zimmerman E, Graves LM, Blancafort P. Generation of tumor-initiating cells by exogenous delivery of OCT4 transcription factor. Breast Cancer Res. 2011;13:R94. doi: 10.1186/bcr3019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Nishi M, Sakai Y, Akutsu H, Nagashima Y, Quinn G, Masui S, Kimura H, Perrem K, Umezawa A, Yamamoto N, et al. Induction of cells with cancer stem cell properties from nontumorigenic human mammary epithelial cells by defined reprogramming factors. Oncogene. 2013 doi: 10.1038/onc.2012.614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hanna JH, Saha K, Jaenisch R. Pluripotency and cellular reprogramming: facts, hypotheses, unresolved issues. Cell. 2010;143:508–25. doi: 10.1016/j.cell.2010.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Papp B, Plath K. Reprogramming to pluripotency: stepwise resetting of the epigenetic landscape. Cell Res. 2011;21:486–501. doi: 10.1038/cr.2011.28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Blum B, Benvenisty N. The tumorigenicity of human embryonic stem cells. Adv Cancer Res. 2008;100:133–58. doi: 10.1016/S0065-230X(08)00005-5. [DOI] [PubMed] [Google Scholar]
- 16.Knoepfler PS. Deconstructing stem cell tumorigenicity: a roadmap to safe regenerative medicine. Stem Cells. 2009;27:1050–6. doi: 10.1002/stem.37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ben-David U, Benvenisty N. The tumorigenicity of human embryonic and induced pluripotent stem cells. Nat Rev Cancer. 2011;11:268–77. doi: 10.1038/nrc3034. [DOI] [PubMed] [Google Scholar]
- 18.Castellanos A, Vicente-Dueñas C, Campos-Sánchez E, Cruz JJ, García-Criado FJ, García-Cenador MB, Lazo PA, Pérez-Losada J, Sánchez-García I. Cancer as a reprogramming-like disease: implications in tumor development and treatment. Semin Cancer Biol. 2010;20:93–7. doi: 10.1016/j.semcancer.2010.02.001. [DOI] [PubMed] [Google Scholar]
- 19.Menendez JA, Vellon L, Oliveras-Ferraros C, Cufí S, Vazquez-Martin A. mTOR-regulated senescence and autophagy during reprogramming of somatic cells to pluripotency: a roadmap from energy metabolism to stem cell renewal and aging. Cell Cycle. 2011;10:3658–77. doi: 10.4161/cc.10.21.18128. [DOI] [PubMed] [Google Scholar]
- 20.Blum B, Bar-Nur O, Golan-Lev T, Benvenisty N. The anti-apoptotic gene survivin contributes to teratoma formation by human embryonic stem cells. Nat Biotechnol. 2009;27:281–7. doi: 10.1038/nbt.1527. [DOI] [PubMed] [Google Scholar]
- 21.Menendez S, Camus S, Herreria A, Paramonov I, Morera LB, Collado M, Pekarik V, Maceda I, Edel M, Consiglio A, et al. Increased dosage of tumor suppressors limits the tumorigenicity of iPS cells without affecting their pluripotency. Aging Cell. 2012;11:41–50. doi: 10.1111/j.1474-9726.2011.00754.x. [DOI] [PubMed] [Google Scholar]
- 22.Prigione A, Fauler B, Lurz R, Lehrach H, Adjaye J. The senescence-related mitochondrial/oxidative stress pathway is repressed in human induced pluripotent stem cells. Stem Cells. 2010;28:721–33. doi: 10.1002/stem.404. [DOI] [PubMed] [Google Scholar]
- 23.Varum S, Rodrigues AS, Moura MB, Momcilovic O, Easley CA, 4th, Ramalho-Santos J, Van Houten B, Schatten G. Energy metabolism in human pluripotent stem cells and their differentiated counterparts. PLoS One. 2011;6:e20914. doi: 10.1371/journal.pone.0020914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Prigione A, Lichtner B, Kuhl H, Struys EA, Wamelink M, Lehrach H, Ralser M, Timmermann B, Adjaye J. Human induced pluripotent stem cells harbor homoplasmic and heteroplasmic mitochondrial DNA mutations while maintaining human embryonic stem cell-like metabolic reprogramming. Stem Cells. 2011;29:1338–48. doi: 10.1002/stem.683. [DOI] [PubMed] [Google Scholar]
- 25.Panopoulos AD, Yanes O, Ruiz S, Kida YS, Diep D, Tautenhahn R, Herrerías A, Batchelder EM, Plongthongkum N, Lutz M, et al. The metabolome of induced pluripotent stem cells reveals metabolic changes occurring in somatic cell reprogramming. Cell Res. 2012;22:168–77. doi: 10.1038/cr.2011.177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Folmes CD, Nelson TJ, Martinez-Fernandez A, Arrell DK, Lindor JZ, Dzeja PP, Ikeda Y, Perez-Terzic C, Terzic A. Somatic oxidative bioenergetics transitions into pluripotency-dependent glycolysis to facilitate nuclear reprogramming. Cell Metab. 2011;14:264–71. doi: 10.1016/j.cmet.2011.06.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Folmes CD, Nelson TJ, Terzic A. Energy metabolism in nuclear reprogramming. Biomark Med. 2011;5:715–29. doi: 10.2217/bmm.11.87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Folmes CD, Nelson TJ, Dzeja PP, Terzic A. Energy metabolism plasticity enables stemness programs. Ann N Y Acad Sci. 2012;1254:82–9. doi: 10.1111/j.1749-6632.2012.06487.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Riggs JW, Barrilleaux BL, Varlakhanova N, Bush KM, Chan V, Knoepfler PS. Induced pluripotency and oncogenic transformation are related processes. Stem Cells Dev. 2013;22:37–50. doi: 10.1089/scd.2012.0375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Vazquez-Martin A, Vellon L, Quirós PM, Cufí S, Ruiz de Galarreta E, Oliveras-Ferraros C, Martin AG, Martin-Castillo B, López-Otín C, Menendez JA. Activation of AMP-activated protein kinase (AMPK) provides a metabolic barrier to reprogramming somatic cells into stem cells. Cell Cycle. 2012;11:974–89. doi: 10.4161/cc.11.5.19450. [DOI] [PubMed] [Google Scholar]
- 31.Vazquez-Martin A, Cufi S, Corominas-Faja B, Oliveras-Ferraros C, Vellon L, Menendez JA. Mitochondrial fusion by pharmacological manipulation impedes somatic cell reprogramming to pluripotency: new insight into the role of mitophagy in cell stemness. Aging (Albany NY) 2012;4:393–401. doi: 10.18632/aging.100465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Vazquez-Martin A, Cufi S, Lopez-Bonet E, Corominas-Faja B, Oliveras-Ferraros C, Martin-Castillo B, Menendez JA. Metformin limits the tumourigenicity of iPS cells without affecting their pluripotency. Sci Rep. 2012;2:964. doi: 10.1038/srep00964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Vazquez-Martin A, Corominas-Faja B, Cufi S, Vellon L, Oliveras-Ferraros C, Menendez OJ, Joven J, Lupu R, Menendez JA. The mitochondrial H(+)-ATP synthase and the lipogenic switch: new core components of metabolic reprogramming in induced pluripotent stem (iPS) cells. Cell Cycle. 2013;12:207–18. doi: 10.4161/cc.23352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Guan JL, Simon AK, Prescott M, Menendez JA, Liu F, Wang F, Wang C, Wolvetang E, Vazquez-Martin A, Zhang J. Autophagy in stem cells. Autophagy. 2013;9:830–49. doi: 10.4161/auto.24132. [Epub ahead of print] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Menendez JA, Joven J, Cufí S, Corominas-Faja B, Oliveras-Ferraros C, Cuyàs E, Martin-Castillo B, López-Bonet E, Alarcón T, Vazquez-Martin A. The Warburg effect version 2.0: metabolic reprogramming of cancer stem cells. Cell Cycle. 2013;12:1166–79. doi: 10.4161/cc.24479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hardie DG. AMP-activated protein kinase: an energy sensor that regulates all aspects of cell function. Genes Dev. 2011;25:1895–908. doi: 10.1101/gad.17420111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hardie DG. Adenosine monophosphate-activated protein kinase: a central regulator of metabolism with roles in diabetes, cancer, and viral infection. Cold Spring Harb Symp Quant Biol. 2011;76:155–64. doi: 10.1101/sqb.2011.76.010819. [DOI] [PubMed] [Google Scholar]
- 38.Hardie DG. Sensing of energy and nutrients by AMP-activated protein kinase. Am J Clin Nutr. 2011;93:891S–6. doi: 10.3945/ajcn.110.001925. [DOI] [PubMed] [Google Scholar]
- 39.Brugarolas J, Lei K, Hurley RL, Manning BD, Reiling JH, Hafen E, Witters LA, Ellisen LW, Kaelin WG., Jr. Regulation of mTOR function in response to hypoxia by REDD1 and the TSC1/TSC2 tumor suppressor complex. Genes Dev. 2004;18:2893–904. doi: 10.1101/gad.1256804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Sofer A, Lei K, Johannessen CM, Ellisen LW. Regulation of mTOR and cell growth in response to energy stress by REDD1. Mol Cell Biol. 2005;25:5834–45. doi: 10.1128/MCB.25.14.5834-5845.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kimball SR, Do AN, Kutzler L, Cavener DR, Jefferson LS. Rapid turnover of the mTOR complex 1 (mTORC1) repressor REDD1 and activation of mTORC1 signaling following inhibition of protein synthesis. J Biol Chem. 2008;283:3465–75. doi: 10.1074/jbc.M706643200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ben Sahra I, Regazzetti C, Robert G, Laurent K, Le Marchand-Brustel Y, Auberger P, Tanti JF, Giorgetti-Peraldi S, Bost F. Metformin, independent of AMPK, induces mTOR inhibition and cell-cycle arrest through REDD1. Cancer Res. 2011;71:4366–72. doi: 10.1158/0008-5472.CAN-10-1769. [DOI] [PubMed] [Google Scholar]
- 43.Kolesnichenko M, Hong L, Liao R, Vogt PK, Sun P. Attenuation of TORC1 signaling delays replicative and oncogenic RAS-induced senescence. Cell Cycle. 2012;11:2391–401. doi: 10.4161/cc.20683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Peterson TR, Laplante M, Thoreen CC, Sancak Y, Kang SA, Kuehl WM, Gray NS, Sabatini DM. DEPTOR is an mTOR inhibitor frequently overexpressed in multiple myeloma cells and required for their survival. Cell. 2009;137:873–86. doi: 10.1016/j.cell.2009.03.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Liu M, Wilk SA, Wang A, Zhou L, Wang RH, Ogawa W, Deng C, Dong LQ, Liu F. Resveratrol inhibits mTOR signaling by promoting the interaction between mTOR and DEPTOR. J Biol Chem. 2010;285:36387–94. doi: 10.1074/jbc.M110.169284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Wang Z, Zhong J, Inuzuka H, Gao D, Shaik S, Sarkar FH, Wei W. An evolving role for DEPTOR in tumor development and progression. Neoplasia. 2012;14:368–75. doi: 10.1593/neo.12542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Zhu H, Shyh-Chang N, Segrè AV, Shinoda G, Shah SP, Einhorn WS, Takeuchi A, Engreitz JM, Hagan JP, Kharas MG, et al. DIAGRAM Consortium. MAGIC Investigators The Lin28/let-7 axis regulates glucose metabolism. Cell. 2011;147:81–94. doi: 10.1016/j.cell.2011.08.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Braun S, Bitton-Worms K, LeRoith D. The link between the metabolic syndrome and cancer. Int J Biol Sci. 2011;7:1003–15. doi: 10.7150/ijbs.7.1003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Pollak M. The insulin and insulin-like growth factor receptor family in neoplasia: an update. Nat Rev Cancer. 2012;12:159–69. doi: 10.1038/nrc3215. [DOI] [PubMed] [Google Scholar]
- 50.Pandey PR, Okuda H, Watabe M, Pai SK, Liu W, Kobayashi A, Xing F, Fukuda K, Hirota S, Sugai T, et al. Resveratrol suppresses growth of cancer stem-like cells by inhibiting fatty acid synthase. Breast Cancer Res Treat. 2011;130:387–98. doi: 10.1007/s10549-010-1300-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Pandey PR, Xing F, Sharma S, Watabe M, Pai SK, Iiizumi-Gairani M, Fukuda K, Hirota S, Mo YY, Watabe K. Elevated lipogenesis in epithelial stem-like cell confers survival advantage in ductal carcinoma in situ of breast cancer. Oncogene. 2012 doi: 10.1038/onc.2012.519. [DOI] [PubMed] [Google Scholar]
- 52.Li G, Zhao F, Cui Y. Proteomics using mammospheres as a model system to identify proteins deregulated in breast cancer stem cells. Curr Mol Med. 2013;13:459–63. [PubMed] [Google Scholar]
- 53.Yecies JL, Manning BD. Transcriptional control of cellular metabolism by mTOR signaling. Cancer Res. 2011;71:2815–20. doi: 10.1158/0008-5472.CAN-10-4158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Yecies JL, Zhang HH, Menon S, Liu S, Yecies D, Lipovsky AI, Gorgun C, Kwiatkowski DJ, Hotamisligil GS, Lee CH, et al. Akt stimulates hepatic SREBP1c and lipogenesis through parallel mTORC1-dependent and independent pathways. Cell Metab. 2011;14:21–32. doi: 10.1016/j.cmet.2011.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Bakan I, Laplante M. Connecting mTORC1 signaling to SREBP-1 activation. Curr Opin Lipidol. 2012;23:226–34. doi: 10.1097/MOL.0b013e328352dd03. [DOI] [PubMed] [Google Scholar]
- 56.Peterson TR, Sengupta SS, Harris TE, Carmack AE, Kang SA, Balderas E, Guertin DA, Madden KL, Carpenter AE, Finck BN, et al. mTOR complex 1 regulates lipin 1 localization to control the SREBP pathway. Cell. 2011;146:408–20. doi: 10.1016/j.cell.2011.06.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Ben-Porath I, Thomson MW, Carey VJ, Ge R, Bell GW, Regev A, Weinberg RA. An embryonic stem cell-like gene expression signature in poorly differentiated aggressive human tumors. Nat Genet. 2008;40:499–507. doi: 10.1038/ng.127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Babaie Y, Herwig R, Greber B, Brink TC, Wruck W, Groth D, Lehrach H, Burdon T, Adjaye J. Analysis of Oct4-dependent transcriptional networks regulating self-renewal and pluripotency in human embryonic stem cells. Stem Cells. 2007;25:500–10. doi: 10.1634/stemcells.2006-0426. [DOI] [PubMed] [Google Scholar]
- 59.Hochedlinger K, Yamada Y, Beard C, Jaenisch R. Ectopic expression of Oct-4 blocks progenitor-cell differentiation and causes dysplasia in epithelial tissues. Cell. 2005;121:465–77. doi: 10.1016/j.cell.2005.02.018. [DOI] [PubMed] [Google Scholar]
- 60.Peng S, Maihle NJ, Huang Y. Pluripotency factors Lin28 and Oct4 identify a sub-population of stem cell-like cells in ovarian cancer. Oncogene. 2010;29:2153–9. doi: 10.1038/onc.2009.500. [DOI] [PubMed] [Google Scholar]
- 61.Hu T, Liu S, Breiter DR, Wang F, Tang Y, Sun S. Octamer 4 small interfering RNA results in cancer stem cell-like cell apoptosis. Cancer Res. 2008;68:6533–40. doi: 10.1158/0008-5472.CAN-07-6642. [DOI] [PubMed] [Google Scholar]
- 62.Ramos-Mejia V, Fraga MF, Menendez P. iPSCs from cancer cells: challenges and opportunities. Trends Mol Med. 2012;18:245–7. doi: 10.1016/j.molmed.2012.04.001. [DOI] [PubMed] [Google Scholar]
- 63.Miyoshi N, Ishii H, Nagai K, Hoshino H, Mimori K, Tanaka F, Nagano H, Sekimoto M, Doki Y, Mori M. Defined factors induce reprogramming of gastrointestinal cancer cells. Proc Natl Acad Sci U S A. 2010;107:40–5. doi: 10.1073/pnas.0912407107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Zhang X, Cruz FD, Terry M, Remotti F, Matushansky I. Terminal differentiation and loss of tumorigenicity of human cancers via pluripotency-based reprogramming. Oncogene. 2013;32(2260.e1-21):2249–60, e1-21. doi: 10.1038/onc.2012.237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Liu X, Sun H, Qi J, Wang L, He S, Liu J, Feng C, Chen C, Li W, Guo Y, et al. Sequential introduction of reprogramming factors reveals a time-sensitive requirement for individual factors and a sequential EMT-MET mechanism for optimal reprogramming. Nat Cell Biol. 2013;15:829–38. doi: 10.1038/ncb2765. [DOI] [PubMed] [Google Scholar]
- 66.Yu F, Li J, Chen H, Fu J, Ray S, Huang S, Zheng H, Ai W. Kruppel-like factor 4 (KLF4) is required for maintenance of breast cancer stem cells and for cell migration and invasion. Oncogene. 2011;30:2161–72. doi: 10.1038/onc.2010.591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Wang YH, Liu S, Zhang G, Zhou CQ, Zhu HX, Zhou XB, Quan LP, Bai JF, Xu NZ. Knockdown of c-Myc expression by RNAi inhibits MCF-7 breast tumor cells growth in vitro and in vivo. Breast Cancer Res. 2005;7:R220–8. doi: 10.1186/bcr975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Li R, Liang J, Ni S, Zhou T, Qing X, Li H, He W, Chen J, Li F, Zhuang Q, et al. A mesenchymal-to-epithelial transition initiates and is required for the nuclear reprogramming of mouse fibroblasts. Cell Stem Cell. 2010;7:51–63. doi: 10.1016/j.stem.2010.04.014. [DOI] [PubMed] [Google Scholar]
- 69.Redmer T, Diecke S, Grigoryan T, Quiroga-Negreira A, Birchmeier W, Besser D. E-cadherin is crucial for embryonic stem cell pluripotency and can replace OCT4 during somatic cell reprogramming. EMBO Rep. 2011;12:720–6. doi: 10.1038/embor.2011.88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Lowry WE. E-cadherin, a new mixer in the Yamanaka cocktail. EMBO Rep. 2011;12:613–4. doi: 10.1038/embor.2011.117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Leis O, Eguiara A, Lopez-Arribillaga E, Alberdi MJ, Hernandez-Garcia S, Elorriaga K, Pandiella A, Rezola R, Martin AG. Sox2 expression in breast tumours and activation in breast cancer stem cells. Oncogene. 2012;31:1354–65. doi: 10.1038/onc.2011.338. [DOI] [PubMed] [Google Scholar]
- 72.Huang YL, Hung JT, Cheung SK, Lee HY, Chu KC, Li ST, Lin YC, Ren CT, Cheng TJ, Hsu TL, et al. Carbohydrate-based vaccines with a glycolipid adjuvant for breast cancer. Proc Natl Acad Sci U S A. 2013;110:2517–22. doi: 10.1073/pnas.1222649110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Alison MR, Guppy NJ, Lim SM, Nicholson LJ. Finding cancer stem cells: are aldehyde dehydrogenases fit for purpose? J Pathol. 2010;222:335–44. doi: 10.1002/path.2772. [DOI] [PubMed] [Google Scholar]
- 74.Marcato P, Dean CA, Giacomantonio CA, Lee PW. Aldehyde dehydrogenase: its role as a cancer stem cell marker comes down to the specific isoform. Cell Cycle. 2011;10:1378–84. doi: 10.4161/cc.10.9.15486. [DOI] [PubMed] [Google Scholar]
- 75.Visus C, Wang Y, Lozano-Leon A, Ferris RL, Silver S, Szczepanski MJ, Brand RE, Ferrone CR, Whiteside TL, Ferrone S, et al. Targeting ALDH(bright) human carcinoma-initiating cells with ALDH1A1-specific CD8⁺ T cells. Clin Cancer Res. 2011;17:6174–84. doi: 10.1158/1078-0432.CCR-11-1111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Badve S, Nakshatri H. Breast-cancer stem cells-beyond semantics. Lancet Oncol. 2012;13:e43–8. doi: 10.1016/S1470-2045(11)70191-7. [DOI] [PubMed] [Google Scholar]
- 77.Pham PV, Phan NL, Nguyen NT, Truong NH, Duong TT, Le DV, Truong KD, Phan NK. Differentiation of breast cancer stem cells by knockdown of CD44: promising differentiation therapy. J Transl Med. 2011;9:209. doi: 10.1186/1479-5876-9-209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Williams K, Motiani K, Giridhar PV, Kasper S. CD44 integrates signaling in normal stem cell, cancer stem cell and (pre)metastatic niches. Exp Biol Med (Maywood) 2013;238:324–38. doi: 10.1177/1535370213480714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Wu F, Zhang J, Wang P, Ye X, Jung K, Bone KM, Pearson JD, Ingham RJ, McMullen TP, Ma Y, et al. Identification of two novel phenotypically distinct breast cancer cell subsets based on Sox2 transcription activity. Cell Signal. 2012;24:1989–98. doi: 10.1016/j.cellsig.2012.07.008. [DOI] [PubMed] [Google Scholar]
- 80.Wu F, Ye X, Wang P, Jung K, Wu C, Douglas D, Kneteman N, Bigras G, Ma Y, Lai R. Sox2 suppresses the invasiveness of breast cancer cells via a mechanism that is dependent on Twist1 and the status of Sox2 transcription activity. BMC Cancer. 2013;13:317. doi: 10.1186/1471-2407-13-317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Faubert B, Boily G, Izreig S, Griss T, Samborska B, Dong Z, Dupuy F, Chambers C, Fuerth BJ, Viollet B, et al. AMPK is a negative regulator of the Warburg effect and suppresses tumor growth in vivo. Cell Metab. 2013;17:113–24. doi: 10.1016/j.cmet.2012.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Kawamura T, Suzuki J, Wang YV, Menendez S, Morera LB, Raya A, Wahl GM, Izpisúa Belmonte JC. Linking the p53 tumour suppressor pathway to somatic cell reprogramming. Nature. 2009;460:1140–4. doi: 10.1038/nature08311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Marión RM, Strati K, Li H, Murga M, Blanco R, Ortega S, Fernandez-Capetillo O, Serrano M, Blasco MA. A p53-mediated DNA damage response limits reprogramming to ensure iPS cell genomic integrity. Nature. 2009;460:1149–53. doi: 10.1038/nature08287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Utikal J, Polo JM, Stadtfeld M, Maherali N, Kulalert W, Walsh RM, Khalil A, Rheinwald JG, Hochedlinger K. Immortalization eliminates a roadblock during cellular reprogramming into iPS cells. Nature. 2009;460:1145–8. doi: 10.1038/nature08285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Liu Y, Hoya-Arias R, Nimer SD. The role of p53 in limiting somatic cell reprogramming. Cell Res. 2009;19:1227–8. doi: 10.1038/cr.2009.121. [DOI] [PubMed] [Google Scholar]
- 86.He J, Kang L, Wu T, Zhang J, Wang H, Gao H, Zhang Y, Huang B, Liu W, Kou Z, et al. An elaborate regulation of Mammalian target of rapamycin activity is required for somatic cell reprogramming induced by defined transcription factors. Stem Cells Dev. 2012;21:2630–41. doi: 10.1089/scd.2012.0015. [DOI] [PubMed] [Google Scholar]
- 87.Hwang-Verslues WW, Chang PH, Wei PC, Yang CY, Huang CK, Kuo WH, Shew JY, Chang KJ, Lee EY, Lee WH. miR-495 is upregulated by E12/E47 in breast cancer stem cells, and promotes oncogenesis and hypoxia resistance via downregulation of E-cadherin and REDD1. Oncogene. 2011;30:2463–74. doi: 10.1038/onc.2010.618. [DOI] [PubMed] [Google Scholar]
- 88.Pandey PR, Okuda H, Watabe M, Pai SK, Liu W, Kobayashi A, Xing F, Fukuda K, Hirota S, Sugai T, et al. Resveratrol suppresses growth of cancer stem-like cells by inhibiting fatty acid synthase. Breast Cancer Res Treat. 2011;130:387–98. doi: 10.1007/s10549-010-1300-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Malaguarnera R, Frasca F, Garozzo A, Gianì F, Pandini G, Vella V, Vigneri R, Belfiore A. Insulin receptor isoforms and insulin-like growth factor receptor in human follicular cell precursors from papillary thyroid cancer and normal thyroid. J Clin Endocrinol Metab. 2011;96:766–74. doi: 10.1210/jc.2010-1255. [DOI] [PubMed] [Google Scholar]
- 90.Savarese TM, Low HP, Baik I, Strohsnitter WC, Hsieh CC. Normal breast stem cells, malignant breast stem cells, and the perinatal origin of breast cancer. Stem Cell Rev. 2006;2:103–10. doi: 10.1007/s12015-006-0016-9. [DOI] [PubMed] [Google Scholar]
- 91.Ginestier C, Wicha MS. Mammary stem cell number as a determinate of breast cancer risk. Breast Cancer Res. 2007;9:109. doi: 10.1186/bcr1741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Trichopoulos D, Lagiou P, Adami HO. Towards an integrated model for breast cancer etiology: the crucial role of the number of mammary tissue-specific stem cells. Breast Cancer Res. 2005;7:13–7. doi: 10.1186/bcr966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Pollak M, Russell-Jones D. Insulin analogues and cancer risk: cause for concern or cause célèbre? Int J Clin Pract. 2010;64:628–36. doi: 10.1111/j.1742-1241.2010.02354.x. [DOI] [PubMed] [Google Scholar]
- 94.Zhang G, Yang P, Guo P, Miele L, Sarkar FH, Wang Z, Zhou Q. Unraveling the mystery of cancer metabolism in the genesis of tumor-initiating cells and development of cancer. Biochim Biophys Acta. 2013;1836:49–59. doi: 10.1016/j.bbcan.2013.03.001. [DOI] [PubMed] [Google Scholar]
- 95.Rafalski VA, Mancini E, Brunet A. Energy metabolism and energy-sensing pathways in mammalian embryonic and adult stem cell fate. J Cell Sci. 2012;125:5597–608. doi: 10.1242/jcs.114827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Martí M, Mulero L, Pardo C, Morera C, Carrió M, Laricchia-Robbio L, Esteban CR, Izpisua Belmonte JC. Characterization of pluripotent stem cells. Nat Protoc. 2013;8:223–53. doi: 10.1038/nprot.2012.154. [DOI] [PubMed] [Google Scholar]
- 97.Desbaillets I, Ziegler U, Groscurth P, Gassmann M. Embryoid bodies: an in vitro model of mouse embryogenesis. Exp Physiol. 2000;85:645–51. doi: 10.1017/S0958067000021047. [DOI] [PubMed] [Google Scholar]
- 98.Aasen T, Raya A, Barrero MJ, Garreta E, Consiglio A, Gonzalez F, Vassena R, Bilić J, Pekarik V, Tiscornia G, et al. Efficient and rapid generation of induced pluripotent stem cells from human keratinocytes. Nat Biotechnol. 2008;26:1276–84. doi: 10.1038/nbt.1503. [DOI] [PubMed] [Google Scholar]
- 99.Aran B, Rodríguez-Pizà I, Raya A, Consiglio A, Muñoz Y, Barri PN, Izpisúa JC, Veiga A. Derivation of human embryonic stem cells at the Center of Regenerative Medicine in Barcelona. In Vitro Cell Dev Biol Anim. 2010;46:356–66. doi: 10.1007/s11626-010-9288-0. [DOI] [PubMed] [Google Scholar]
- 100.Sato A, Sunayama J, Okada M, Watanabe E, Seino S, Shibuya K, Suzuki K, Narita Y, Shibui S, Kayama T, et al. Glioma-initiating cell elimination by metformin activation of FOXO3 via AMPK. Stem Cells Transl Med. 2012;1:811–24. doi: 10.5966/sctm.2012-0058. [DOI] [PMC free article] [PubMed] [Google Scholar]