

Transcriptomics of N-deprived Chlamydomonas sta6, CC-4349 (a wild-type strain), and three complemented STA6 strains showed upregulation of glyoxylate and gluconeogenesis pathways, validated by enzyme and metabolite analyses. Resequencing of all strains revealed that sta6 and CC-4349 are distantly related, highlighting the importance of using complemented strains for relating phenotype to genotype.
Abstract
To understand the molecular basis underlying increased triacylglycerol (TAG) accumulation in starchless (sta) Chlamydomonas reinhardtii mutants, we undertook comparative time-course transcriptomics of strains CC-4348 (sta6 mutant), CC-4349, a cell wall–deficient (cw) strain purported to represent the parental STA6 strain, and three independent STA6 strains generated by complementation of sta6 (CC-4565/STA6-C2, CC-4566/STA6-C4, and CC-4567/STA6-C6) in the context of N deprivation. Despite N starvation–induced dramatic remodeling of the transcriptome, there were relatively few differences (5 × 102) observed between sta6 and STA6, the most dramatic of which were increased abundance of transcripts encoding key regulated or rate-limiting steps in central carbon metabolism, specifically isocitrate lyase, malate synthase, transaldolase, fructose bisphosphatase and phosphoenolpyruvate carboxykinase (encoded by ICL1, MAS1, TAL1, FBP1, and PCK1 respectively), suggestive of increased carbon movement toward hexose-phosphate in sta6 by upregulation of the glyoxylate pathway and gluconeogenesis. Enzyme assays validated the increase in isocitrate lyase and malate synthase activities. Targeted metabolite analysis indicated increased succinate, malate, and Glc-6-P and decreased Fru-1,6-bisphosphate, illustrating the effect of these changes. Comparisons of independent data sets in multiple strains allowed the delineation of a sequence of events in the global N starvation response in C. reinhardtii, starting within minutes with the upregulation of alternative N assimilation routes and carbohydrate synthesis and subsequently a more gradual upregulation of genes encoding enzymes of TAG synthesis. Finally, genome resequencing analysis indicated that (1) the deletion in sta6 extends into the neighboring gene encoding respiratory burst oxidase, and (2) a commonly used STA6 strain (CC-4349) as well as the sequenced reference (CC-503) are not congenic with respect to sta6 (CC-4348), underscoring the importance of using complemented strains for more rigorous assignment of phenotype to genotype.
INTRODUCTION
Starch is the predominant energy storage molecule of photosynthetic organisms in the Viridiplantae, and its synthesis and degradation are well characterized in many organisms, including Chlamydomonas reinhardtii (Ball and Deschamps 2009; Zeeman et al., 2010). Starch is formed in the plastid by the sequential activities of ADP-Glc pyrophosphorylase, converting Glc-1-P to ADP-Glc, and starch synthases and starch branching enzymes, which catalyze the formation of α-1,4- and α-1,6-glycosidic bonds, respectively, to generate amylose and amylopectin (Smith, 1999). The genes encoding these enzymes in C. reinhardtii have been identified through classical genetics or homology-based approaches. Mutations in either starchless6 (sta6), encoding the small subunit of ADP-Glc pyrophosphorylase, or sta7, encoding isoamylase, abolish starch synthesis (Mouille et al., 1996; Zabawinski et al., 2001). The biosynthesis of storage carbohydrate polymers (starch or chrysolaminaran, previously known as leucosin) as well as triacylglycerols (TAGs) is promoted in green algae and diatoms during N starvation as a strategy for storing reduced carbon when there is insufficient N nutrition for growth and division (Varum and Myklestad, 1984; Granum et al., 2002; Guschina and Harwood, 2006; Hu et al., 2008; Rodolfi et al., 2009; Wang et al., 2009; Work et al., 2010; Siaut et al., 2011) .
C. reinhardtii sta mutants and equivalent mutants in other photosynthetic organisms accumulate higher amounts of TAG. It is assumed that this occurs because carbon flow to storage carbohydrate is blocked (Ramazanov and Ramazanov, 2006; Wang et al., 2009; Li et al., 2010a, 2010b; Work et al., 2010; Fan et al., 2011, 2012; Sanjaya et al., 2011). The increased yield of TAG in acetate-supplemented N-starved sta6 is not inconsistent with this model (Goodson et al., 2011; Ramanan et al., 2013). Because of the potential for using algae to generate biodiesel precursors, there is interest in understanding carbon metabolism and its regulation in the context of TAG accumulation in algae (reviewed in Johnson and Alric, 2013). In addition to triggering starch and TAG synthesis, N starvation also induces a number of other responses, including ribosome turnover, gametogenesis, and chlorosis (Siersma and Chiang, 1971; Martin and Goodenough, 1975; Plumley and Schmidt, 1989; Bulté and Wollman, 1992).
C. reinhardtii, a unicellular chlorophyte alga containing a pyrenoid within its single chloroplast representing the site of starch accumulation and ribulose-1,5-bis-phosphate carboxylase/oxygenase function, is an excellent reference system for addressing this issue (Harris, 2001). A draft genome sequence (http://www.phytozome.net/search.php for the version 5 assembly), resources for transcriptomics, proteomics, metabolomics (Bölling and Fiehn, 2005; May et al., 2008; Atteia et al., 2009; Rolland et al., 2009; Mühlhaus et al., 2011; Urzica et al., 2012; Wang et al., 2012), and well-characterized mutants in starch metabolism are available (Ball et al., 1991; Mouille et al., 1996; Wattebled et al., 2002, 2003). In addition, the growth medium and nutrient requirements are well defined, and there is considerable ongoing effort to discover and understand the operation of carbon metabolism pathways under different trophic situations, including anaerobiosis, respiratory, and phototrophic conditions (Kropat et al., 2011; Brueggeman et al., 2012; Catalanotti et al., 2012; Fang et al., 2012; Johnson and Alric, 2012). Recent investigations of TAG accumulation have documented two sites for lipid droplet accumulation in C. reinhardtii, one extraplastidic, likely in endoplasmic reticulum–derived compartments, and the other within the chloroplast (Li-Beisson et al., 2010; Fan et al., 2011; Goodson et al., 2011). The plastid lipid bodies are more evident in sta6, again consistent with the redirection of reduced carbon from starch (synthesized in the plastid) to TAG accumulation (Goodson et al., 2011).
Although N starvation is the best trigger for TAG accumulation in C. reinhardtii under both photoautotrophic or photoheterotrophic growth conditions, other nutrient deficiencies also promote TAG biosynthesis, including P, S, Fe, and Zn starvation (Matthew et al., 2009; Kropat et al., 2011; Breuer et al., 2012; Cakmak et al., 2012; Msanne et al., 2012). Recent transcriptome experiments have sought to identify the key acyltransferases and other proteins involved in algal TAG accumulation and lipid body biosynthesis (Miller et al., 2010; Boyle et al., 2012). These studies have resulted in the identification of both type I and type II acyltransferases, DGAT1 and DGTT1 through DGTT5, respectively, for de novo TAG biosynthesis, where TAG is synthesized from diacylglycerol and acyl-coenzyme A (acyl-CoA), as well as a phospholipid diacylglyceroltransferase, PDAT1, in which TAG is synthesized by transesterification using a membrane lipid as a substrate (Miller et al., 2010; Merchant et al., 2012; Boyle et al., 2012; Msanne et al., 2012; Yoon et al., 2012).
With a view to understanding the effect of redirecting carbon from starch biosynthesis toward TAG accumulation, we used comparative transcriptomics of the sta6 mutant and its presumed parental strain in a time course of N starvation to distinguish sta6-dependent differences. We validated the discoveries by documentation of changes in enzyme activity and selected metabolites to conclude that gluconeogenesis is counterintuitively stimulated in the sta6 mutant. In the course of the work, we also discovered that strain CC-4349, originally listed in the culture collection as the parent of sta6, is mislabeled. Genome resequencing identified strain CC-4568 as the true parent. Therefore, we compared the sta6 mutant to three independent complemented strains to understand the contribution of the STA6 locus to the phenotype. Genome resequencing and transcriptomics give an indication of the variability between individual transformants, which may be useful in other work.
RESULTS
Comparative Transcriptomics of CC-4349, sta6, and Three Independent STA6 Strains under N Deprivation
The sta6 mutant, which is incapable of starch synthesis because of a mutation in the small subunit of ADP-Glc pyrophosphorylase, can accumulate only TAG when it is starved for N (Zabawinski et al., 2001). Several studies indicate that this and other sta mutants accumulate more TAG than do the corresponding wild-type or complemented strains (Wang et al., 2009; Li et al., 2010b; Work et al., 2010). When we transferred the sta6 mutant (CC-4348), a wild-type presumed parental strain (but see below), CC-4349, and three previously characterized complemented strains STA6-C2, STA6-C4, and STA6-C6 (CC-4565, CC-4566, and CC-4567, respectively) to Tris-acetate-phosphate (TAP) medium lacking ammonium (the usual N source), we recapitulated this phenotype (Figure 1). None of the strains grows appreciably in N-free medium, experiencing at most one doubling during the course of the experiment (48 h). Chlorophyll content decreased in all strains, and consistent with a previous report, the decrease of chlorophyll is slightly accelerated in the sta6 mutant (Work et al., 2010). As expected, the starch content increased rapidly in STA6 strains as a function of time after transfer to N-free medium but was near zero in the sta6 mutant strain (Zabawinski et al., 2001). Although we did not measure a significant difference in TAG accumulation in CC-4349 versus sta6 in the first 48 h after N deprivation, we noted that by 96 h, the TAG content of sta6 exceeded that of CC-4349 (52 versus 24 fmol cell−1, respectively) (Figure 1D), as documented by others (Li et al., 2010a; Work et al., 2010). sta6 accumulated more major lipid droplet protein (MLDP1), a protein associated with the algal lipid droplet, than did STA6 (Figure 1E).
Figure 1.

Increased TAG but Not Starch in the sta6 Mutant.
In all panels, CC-4349 is shown in red, sta6 (CC-4348) in black, and three strains complemented for STA6 (STA6-C2, STA6-C4, and STA6-C6) in blue. Chlorophyll content (A), cell density (B), starch content per cell (C), and TAG per cell (D). Error bars represent one sd calculated from three separate cultures (biological replicates). (E) shows an immunoblot of MLDP1 using protein extracts obtained from N-deprived cultures of sta6 and STA6-C2, STA6-C4, and STA6-C6, sampled at 0, 24, 48, and 96 h as indicated.
In previous work, we identified several genes, DGAT1, DGTT1, and PDAT1, encoding acyltransferases whose expression was increased in C. reinhardtii in response to N removal from the medium, and in a time course experiment established that changes in transcript abundance preceded changes in TAG accumulation (Boyle et al., 2012). Based on that work, we analyzed the transcriptomes of sta6, strain CC-4349, and the three complemented strains in response to N deprivation in a time-course experiment that would allow us to distinguish primary responses as well as longer term ones (Figure 2). Therefore, we collected cells from triplicate cultures of sta6 and CC-4349 prior to washing in N-free medium (0') and at 0, 0.5, 4, 8, 12, 24, and 48 h after transfer to N-free medium for preparation of total RNA. Additionally, in a second time-course experiment, cells were collected from cultures of sta6, CC-4349, STA6-C2, STA6-C4, and STA6-C6 at 0.5, 4, and 48 h after transfer to N-free medium. For strains sta6 and CC-4349 in the first time-course experiment, two of the RNA preparations were analyzed by RNA sequencing (RNA-Seq) on the Illumina platform, and the third sample was retained for independent validation by quantitative RT-PCR (qRT-PCR), while single samples were taken for cultures of sta6 and the STA6 strains collected at 0.5, 4, and 48 h. The reads (totaling 146,992,656 and 447,140,526 for sta6 and CC4349 in the first experiment and 87,214,220, 91,346,488, 123,682,836, and 131,687,452 for sta6, STA6-C2, STA6-C4, and STA6-C6, respectively, in the second experiment; see Supplemental Data Set 1 online) were aligned to the genome and to the 17,301 gene models in the version 4 assembly of the C. reinhardtii genome, normalized, and quantified for gene expression estimates (see Methods).
Figure 2.
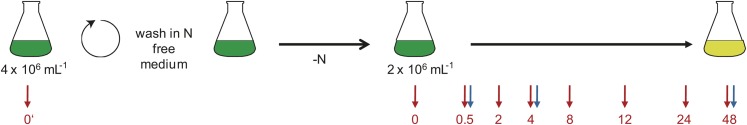
Experimental Design for Sampling the Transcriptome.
Cultures were grown to a density of 4 × 106 cells mL−1 in +N medium and washed in N-free medium before resuspending cells to a final density of 2 × 106 cells mL−1 in N-free medium. For the time-course experiment comparing CC-4349 and sta6, samples were taken in triplicate at 0’ (i.e., from +N medium before washing), 0, 0.5, 2, 4, 8, 12, 24, and 48 h after N deprivation as indicated with red arrows. In a separate experiment, sta6, STA6-C2, STA6-C4, and STA6-C6 were sampled at 0.5, 4, and 48 h after N deprivation, as indicated with blue arrows. The light-green color of an N-free culture at 48 h indicates the cells are chlorotic.
N-Deprivation Responses Unique to CC-4349 and sta6
A key objective of this investigation was to identify the effect of the mutation in STA6 on the N-deficiency transcriptome. Therefore, we used a hierarchical clustering method to retrieve all genes that were expressed differently in the two strains (see Supplemental Figure 1 online).
The analysis revealed a small number of genes encoding enzymes in central carbon metabolism as being differently expressed in sta6 versus CC-4349 and STA6. ACS3 and PAT2, encoding acetyl-CoA synthetase and phosphate acetyl-transferase, which catalyze acetate assimilation, are upregulated severalfold at 24 h in sta6 compared with CC-4349, reaching 543 versus 149 reads per kilobase of mappable length per million aligned reads (RPKM) and 61 versus 37 RPKM, respectively, and approximately twofold higher at 48 h in sta6 versus STA6 (Figure 3; see Supplemental Figure 2 online). Transcripts encoding two enzymes of the glyoxylate cycle, which enables the conversion of the activated acetyl group to gluconeogenic precursors, are more abundant in sta6 compared with CC-4349. ICL1 and MAS1, encoding isocitrate lyase and malate synthase (both of which are unique to the glyoxylate pathway), are upregulated 12- and 14-fold, respectively, at 48 h, relative to CC-4349, peaking at 700 and 224 RPKM. PCK1 and FBP1, encoding phosphoenolpyruvate carboxykinase and Fru-1,6-bisphosphatase, respectively, catalyzing two steps unique to gluconeogenesis, and TAL1, encoding transaldolase, cocluster with ICL1 and MAS1 with respect to expression pattern and are also ∼10-fold upregulated in sta6 compared with CC-4349 (Figure 3; see Supplemental Data Set 2 online). The transcript abundances of each of these genes are severalfold higher in sta6 than in STA6 at 48 h (Figure 3; see Supplemental Figure 2 and Supplemental Data Set 3 online). Four of the five enzymes encoded by these upregulated transcripts are predicted to localize to the chloroplast (see Supplemental Data Set 3 online). All five genes display strikingly similar patterns of expression, consistent with a pathway connection, and suggesting the potential for increased movement of carbon in the direction of gluconeogenesis (Figures 3 and 4). Additionally, PGH1, encoding enolase, is upregulated approximately twofold in sta6 versus CC-4349 (peaking at 1168 and 543 RPKM in the two strains, respectively, at 4 h). With respect to other transcripts encoding enzymes of glycolysis/gluconeogenesis and the pentose phosphate pathway, all are found at similar levels in both strains (see Supplemental Data Set 3 online).
Figure 3.

Greater Upregulation of Genes Encoding Enzymes of Acetate Metabolism, Gluconeogenesis, and the Oxidative Pentose Phosphate Pathway in sta6 versus CC-4349 and STA6.
Heat map showing fold change of RPKM (relative to 0 h in CC-4349), ranging from 0.18 (yellow) to 18 (violet), of genes involved in acetate assimilation, the glyoxylate cycle, glycolysis/gluconeogenesis, and the oxidative pentose phosphate pathway as indicated. Heat map was generated using Matrix2png (Pavlidis and Noble, 2003). RPKM values for complete pathways are shown in Supplemental Data Set 3 and Supplemental Figure 2 online.
Figure 4.

Upregulated Genes Encode Key Enzymes in Central Carbon Metabolism.
Steps in the glyoxylate cycle (A), gluconeogenesis (B), and oxidative pentose phosphate pathway (C) are shown. The complete oxidative pentose phosphate pathway is shown in Supplemental Figure 11 online. For clarity, only relevant enzymes are shown.
The coordinated increase in transcripts encoding these enzymes in sta6 versus STA6 suggests an increase in acetate utilization in the glyoxylate cycle followed by gluconeogenesis to generate hexose-phosphates in sta6 (Figures 3 and 4). This is counterintuitive given that sta6 is unable to utilize hexose-phosphate for starch synthesis. These results contrast with the findings from previous studies on STA6 lines in which transcripts encoding enzymes of the glyoxylate cycle were decreased in -N compared with +N (Miller et al., 2010), indicating that the phenomenon is sta6 specific.
There is no difference in the abundance of transcripts encoding enzymes of the citric acid cycle, suggesting that acetate oxidation is similar in sta6 and CC-4349 (see Supplemental Data Set 3 online). Transcripts encoding enzymes of the Calvin Benson cycle are typically decreased in abundance across the time course in both strains, consistent with the rapid downregulation of the genes encoding components of the light reactions (see Supplemental Data Set 4 online).
Surprisingly, the abundance of transcripts encoding enzymes in fatty acid and lipid metabolism were similar in sta6 versus CC-4349 despite the TAG overaccumulation phenotype of sta6 (see Supplemental Data Set 5 online). The expression profiles of the four acetyl-CoA carboxylase subunits, and most diacylglycerol acyltransferases (DAGATs) responsible for the final step of TAG synthesis, are highly comparable. Three exceptions to this were DGTT2, Cre06.g310200, and MLDP1 for which the transcript numbers are higher in sta6 versus CC-4349 and STA6 (Figure 5; see Supplemental Data Set 5 online). DGTT2 was originally predicted to encode a type-2 diacylglycerol transferase (DGAT) on the basis of high sequence similarity to a protein exhibiting acyltransferase activity in Morterialla ramanniana and has since been functionally validated (Lardizabal et al., 2001; Sanjaya et al., 2013) Cre06.g310200 possesses high similarity to a type-3 DGAT. Acyltransferase activity has been demonstrated for type-3 DGATs in several organisms, although not yet in an alga (Saha et al., 2006; Rani et al., 2010; Hernández et al., 2012). MLDP1 has been suggested to play a structural role in the algal lipid body analogous to that of oleosins in land plants (Moellering and Benning, 2010). Immunoblot analysis validated that the increased MLDP1 mRNA abundance is correlated with increased polypeptide accumulation in sta6 versus STA6 (Figure 1E), which is consistent with its proposed function as a structural protein of the lipid body. Additionally, the mRNA predicted to encode biotin synthase (Cre01.g261150) accumulates to a threefold higher level in sta6 than in CC-4349 at several time points. As acetyl-CoA carboxylase is biotin dependent, this suggests the possibility that the cofactor could be rate-limiting.
Figure 5.

Expression Profiles of Genes Related to TAG Accumulation.
DGTT2, encoding a type-2 diacylglycerolacyltransferase (A); DGAT3, encoding a putative soluble diacylglycerolacyltransferase (B); and MLDP1 encoding major lipid droplet protein (C). Expression level is expressed as RPKM for each mRNA at 0.5, 4, and 48 h. CC-4349 is in red, sta6 is in black, and STA6 is in blue.
In response to N starvation, vegetative C. reinhardtii cells differentiate into gametes whose mating type is determined by one of two mating loci (called plus and minus) (Sager and Granick, 1954; Beck and Haring, 1996). Gametes subsequently fuse to form zygotes (reviewed in Pan and Snell, 2000). We noted that SAD1, GSM1, and FUSM, all specific to the minus mating type, are expressed in CC-4349 (Ferris and Goodenough, 1994; Ferris et al., 1996; Lin and Goodenough, 2007). Curiously, these genes were not expressed in either sta6 or in strains complemented for STA6. Instead, MTA3A, located within the mt+ locus (Ferris et al., 2002), is expressed in sta6 and the complemented strains but not in CC-4349 (see Supplemental Data Set 6 online). Indeed, qRT-PCR analysis confirmed that CC-4349 is mt−, while sta6 and the STA6 strains are mt+ (see Supplemental Figure 3 online).
Genome Sequences Reveal Mutations besides STA6 Deletion
In addition to the mating type discrepancy, we noted that sta6 cells are smaller than CC-4349. A small cell size was observed already by others for sta6 and sta7 (another starchless mutant), although CC-124 rather than the parental was used as a wild-type comparison (Work et al., 2010). The small cell size phenotype was not rescued by the STA6 gene (see Supplemental Figure 4 online). CC-4349 is also not an Arg auxotroph, as described in the original publication, although this was assumed to be the result of a reversion in the laboratory (U. Goodenough, personal communication). Therefore, we resequenced the genomes of sta6, the purported wild-type (CC-4349) strain and one of the three complemented STA6 strains (STA6-C6; CC-4567). The original parental cw strain from which sta6 is derived was acquired independently from the Dauvillée laboratory (CC-4568) and was also resequenced (see Methods and Supplemental Table 1 online).
The sta6 strain was generated by insertion of pARG7.8 into the genome of CC-4568 (Zabawinski et al., 2001). The read coverage validates the insertion of this plasmid in the sta6 genome and furthermore identified the position of integration, which is in the fourth exon of STA6 (Figure 6; see Supplemental Table 2 online). The read coverage dropped from >50× in the flanking upstream region to 0 at this locus (genome v5.3 coordinates: chromosome 3, 5742415 to 5748751). However, the drop in coverage extended beyond the STA6 gene into the neighboring RBO1 gene. Reads could be aligned throughout this region in all other strains, indicating the reduction in read alignment was not due to difficulty in sequencing the area or attributable to problematic alignment to the reference genome (CC-503). The loss of these two genes from the genome of sta6 is supported by very low abundance of the corresponding transcripts in the sta6 transcriptome (see Supplemental Figure 5 online). In addition, transcript abundance of the paralogous RBO2 gene downstream of RBO1 was also reduced (peaking at 19 RPKM in CC-4349 but only 1 RPKM in sta6), perhaps revealing regulation of RBO2 by RBO1. Besides this issue, we noted a large number of insertions and deletions (InDels) and single-nucleotide variations between sta6 and the reference strain as well as between sta6 and CC-4349, the purported parental wild-type strain used also in other studies of TAG accumulation (Wang et al., 2009; Goodson et al., 2011) (see Supplemental Table 2 online). The genome of CC-4568 revealed 15,095 single-nucleotide variants (SNVs) relative to the reference genome (Table 1). This compared with 14510, 15,049, and 15,878 SNVs for sta6, STA6-C6, and CC-4349, respectively, versus the reference. A pairwise analysis of all genomes indicated 1408 SNVs between CC-4568 and sta6, and 2086 between STA6-C6 and CC-4568, strongly suggesting that CC-4568 is more closely related to sta6 and that CC-4349 is more distantly so (Table 1; see Supplemental Figure 6 online).
Figure 6.

Resequencing of the sta6 Genome Reveals Extent of Gene Disruption.
The reads were aligned to the C. reinhardtii reference strain (CC-503) using the Burrows-Wheeler Aligner and displayed via the Integrative Genomics Viewer. The depth of coverage, indicated in the figure by vertical gray bars on a scale from 0 to 100, drops off precipitously at the boundaries of the disrupted genes. The breakout tracks above and below display the left and right boundaries, respectively, magnified to the nucleotide level. Gene models for STA6 and RBO1 are shown with exons represented by thick bars and introns represented by thin bars. The nucleotide track indicates the reference sequence at these loci.
Table 1. Pairwise Analysis of SNVs in Each Strain Relative to CC-503.
| Strain | CC-503a | CC-4567 (sta6-C6) | CC-4568 | CC-4349 |
|---|---|---|---|---|
| CC-503a | ||||
| CC-4567 (sta6-C6) | 15,049 | |||
| CC-4568 | 15,095 | 2,086 | ||
| CC-4349 | 15,878 | 21,759 | 21,750 | |
| CC-4348 (sta6) | 14,510 | 1,434 | 1,408 | 21,145 |
CC-503 is the sequenced reference strain (Merchant et al., 2007).
Analysis of the STA6-Complemented Strains
Because genome resequencing analysis indicated a large number of SNVs as well as InDels between CC-4349 and sta6, consistent with the conclusion that CC-4349 is not the parental source of the sta6 mutant, we wondered whether differences in the transcript profiles could truly be attributed to the loss of function in sta6. Therefore, we focused our analysis on the transcriptomes of N-free sta6 and the three STA6-complemented strains, STA6-C2, STA6-C4, and STA6-C6. After normalizing the raw data, we generated lists of differentially expressed genes and identified 521 that were common to all three STA6 strains, representing the true set of differentially expressed genes. Thus, only a small fraction of genes that are differentially expressed between CC-4349 and sta6 are attributable to the mutation at the STA6 locus (Figure 7; see Supplemental Data Set 7 online). In addition to the STA6 locus itself, this intersect includes many of the genes that were differentially expressed between CC-4349 and sta6 as discussed above, and especially MAS1, ICL1, TAL1, FBP1, and PCK1.
Figure 7.

Comparative Analysis of sta6 and STA6 Transcriptomes Identifies sta6-Dependent Expression Patterns.
(A) Venn diagram showing genes differentially expressed between sta6 and STA6-C2, STA6-C4, and STA6-C6 at 0.5, 4, and 48 h after N deprivation. The central intersect indicates the differentially expressed genes common to all STA6-complemented strains relative to sta6.
(B) The 521 differentially expressed genes resulting directly from STA6 were grouped according to their function in 25 categories, as assigned by MapMan ontology. For clarity, genes not assigned are not shown, but total 195.
To summarize this data set, ontology-assigned annotations of restored expression resulting from STA6 complementation indicate that the loss of the wild-type gene is responsible for the differential expression of genes relating to a number of functional groupings, including lipid metabolism and pathways of central carbon metabolism as noted above (Figure 7B).
To test the effect of the coordinate and striking increase in the expression of ICL1 and MAS1 in sta6 versus STA6, we measured the activities of the corresponding enzymes in protein extracts from N-free sta6 and the three STA6 strains. We reasoned that as the increased abundance of ICL1 and MAS1 transcripts in sta6 versus CC-4349 is evident ∼24 and 48 h after N deprivation, and since changes in transcript abundance should precede changes in protein abundance, we should test protein activities in samples at time points beyond those used for the transcriptome experiments (Figure 8). At 0 h, activity of the two enzymes remained similar in each strain. However, activity of isocitrate lyase increased approximately twofold in sta6 compared with STA6 at 48 h and approximately fourfold for malate synthase (Figure 8). A previous comparative proteomic investigation of cw15 and sta6 by another group did not identify significant increases in either of these proteins compared with cw15, although the cultures were not N starved in that work (Wang et al., 2012). These data confirm that the increase in transcript abundance translates to an increase in enzyme activity, which would suggest that in sta6, there might be increased acetate movement toward gluconeogenic precursors via the glyoxylate cycle. We note that transcripts encoding two of the regulated and unique enzymes in the gluconeogenic direction, FBP1 and PCK1, also increased in coordination with ICL1 and MAS1 (see Supplemental Data Set 3 and Supplemental Figure 2 online).
Figure 8.

Increased Isocitrate Lyase and Malate Synthase Activities in sta6 versus STA6.
Isocitrate lyase (A) and malate synthase (B) activities. C2, C4, and C6 represent the complemented strains (white), and sta6 is shown in black. Extracts were prepared from cells sampled 0, 24, 48, and 96 h after removal of N. Error bars indicate the sd of three independent biological repeats.
Metabolite Analysis Validates the Effect of Transcriptome Changes on Carbon Movement
The effect of the above-mentioned transcriptome changes was validated by liquid chromatography–tandem mass spectrometry (LC-MS/MS) analysis of specific metabolites in sta6 versus STA6 grown for 48 h in N-replete and N-free media. First, sta6 indeed contains far less ADP-Glc (3.0 μM in sta6 versus 32.6 μM in STA6 in N-free conditions) as expected from a mutation in ADP-Glc pyrophosphorylase (Figure 9). The residual ADP-Glc might be derived from a low activity of other genes encoding ADP-Glc pyrophosphorylase activities or from side reactions of UDP-Glc pyrophosphorylase in the cytosol. Second, malate and succinate, which are products of the glyoxylate pathway, also increased in sta6 compared with STA6, consistent with increased output from this pathway resulting from increased malate synthase and isocitrate lyase activity. Increases in citrate, aconitate, and isocitrate were also noted in sta6. When we looked at metabolites of glycolysis and gluconeogenesis, we noted that the Fru-1,6-bisphosphate was higher in STA6 compared with sta6 (10.8 µM compared with 3.8 µM), consistent with increased fructose-1,6-bisphosphatase activity in the mutant. Finally, we assayed cell extract prepared at 0, 24, 48, and 96 h after N deprivation for Glc-6-P levels and determined that the level in sta6 is unaltered at 0 and 48 h but is approximately threefold higher in sta6 than in the complemented strain STA6 (805 μM versus ∼300 μM) at 96 h (Figure 9H). Additions of known concentrations of Glc-6-P confirmed that the assay was quantitative in this concentration range (see Supplemental Figure 7 online). These changes in metabolite levels are N-deprivation dependent, as is also the case for changes in the transcriptome (Figure 9). Therefore, we conclude that increased mRNA and protein abundance has an effect on the operation of the cognate pathways. On this basis, we suggest that the increase in Glc-6-P observed in sta6 is a consequence of increased output from the glyoxylate and gluconeogenesis pathways, as opposed to an inability to synthesize ADP-Glc.
Figure 9.

Metabolite Profiles in sta6 and STA6-Complemented Strains Are Consistent with Predictions from Transcriptomes.
(A) to (G) sta6 (black) was grown in six independent cultures, and STA6-C (white) comprises duplicate cultures each of STA6-C2, STA6-C4, and STA6-C6. Strains were cultured in +N to a density of 4 × 106 cells mL−1 before transferring to +N and –N cultures to a final cell density of 2 × 106 cells mL−1. Samples were taken at 48 h after transfer for metabolite measurement. Significance was assessed by pairwise Student’s t test. P value correction was performed using the Bonferroni correction method (*P < 0.05, **P < 0.01, and ***P < 0.001). Error bars represent one sd.
(H) sta6 (black) and STA6-C2, STA6-C4, and STA6-C6 (white) were each grown in triplicate in +N to a density of 4 × 106 cells mL−1 before transferring to –N to a final cell density of 2 × 106 cells mL−1. Samples were taken at 0, 48, and 96 h after transfer for Glc-6-P measurement. Error bars represent one sd.
Acetate Assimilation Remains the Same in sta6 and STA6 Strains
Since genes associated with acetate assimilation are upregulated in sta6 compared with CC-4349 and STA6, we wondered whether the increased TAG accumulation in N-starved sta6 cells requires greater acetate utilization from the medium. The observation that further supplementation of the medium with acetate increases TAG accumulation is consistent with this hypothesis as well (Goodson et al., 2011). Therefore, we analyzed medium from N-free cultures of all four strains at 0, 48, and 96 h after N starvation to measure acetate utilization but found that deletion of the STA6 locus has no effect on the rate of acetate assimilation (Figure 10; see Supplemental Figure 7 online).
Figure 10.

Acetate Utilization Is Similar in All Strains.
sta6 (black) and three independent STA6 strains (STA6-C2, STA6-C4, and STA6-C6; shown in white) were cultured in +N to a density of 4 × 106 cells mL−1 before transferring to –N cultures to a final cell density of 2 × 106 cells mL−1. Acetate levels in the supernatant were assessed at 0, 48, and 96 h after transfer to –N. Error bars represent one sd.
Genome-Wide Responses to N Deprivation
Many of the changes in the transcriptome occur in all of the strains. One documented early effect of N starvation is the accumulation of starch (Figure 1C). The abundances of some transcripts encoding starch biosynthesis enzymes STA2, STA3 (encoding starch synthases), and SBE1, SBE2, and SBE3 (encoding starch branching enzymes) are increased severalfold, even in sta6 where starch is not synthesized, indicating that the increased expression of the starch synthesis pathway is a programmed response to N starvation regardless of the output from the pathway (see Supplemental Data Set 3 online). The STA6 locus is largely deleted in the sta6 mutant (see above and Supplemental Figure 5 online) and the reads therefore map only to the first three exons at the 5′ end. Therefore, while transcript abundance increases to 500 RPKM at 2 h (see Supplemental Data Set 3 online) for CC-4349, only a limited number of reads map to this locus in sta6, corresponding to ∼1 RPKM. It is likely that the truncated mRNA is degraded. On the other hand, there is no difference in the pattern of expression of STA1 (encoding the large subunit of ADP-Glc pyrophosphorylase) in sta6 compared with CC-4349, indicating that there is no mechanism for maintaining the stoichiometry of mRNAs for the two subunits. In all strains, transcript abundances for the synthases and branching enzymes increase immediately by 0.5 h, reach a peak by 0.5 to 2 h, and are reduced to the basal level by the end of the experiment. This likely reflects the switch from starch accumulation as a short-term storage molecule to TAG for long-term storage (Siaut et al., 2011) (see Supplemental Data Set 3 online). By contrast, abundances of mRNAs encoding enzymes in lipid metabolism tend to remain relatively constant across all time points in both strains. The only transcripts whose abundances increase are those encoding some of the TAG biosynthesis-specific acyltransferases and the four glycerol-3-phosphate dehydrogenase isozymes. In particular, GPD2 mRNA abundance increases >100-fold over the 48-h time course, from 6 to 916 RPKM and 15 to 895 in CC-4349 and sta6, respectively (Table 2; see Supplemental Data Set 5 online). The DGAT1, DGTT1, DGTT2, DGTT3, and PDAT1 mRNAs encoding acyltransferases specific for TAG biosynthesis are increased approximately twofold to ∼25-fold in both strains, while DGTT4 mRNA is transiently increased shortly after N deprivation in CC-4349 (but not in sta6) and then remains lowly expressed throughout the experiment as in the sta6 mutant (Table 2; see Supplemental Figure 8 online). DGTT5 was not expressed in either strain.
Table 2. Abundance of mRNAs Involved in TAG Synthesis over a Time Course of 0 to 48 h.
| CC-4349 |
sta6 |
STA6-C2 |
STA6-C4 |
STA6-C6 |
||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Genea | Gene Model Aug10.2 | 0 | 0.5 | 2 | 4 | 8 | 12 | 24 | 48 | 0 | 0.5 | 2 | 4 | 8 | 12 | 24 | 48 | 0.5 | 4 | 48 | 0.5 | 4 | 48 | 0.5 | 4 | 48 |
| GPD1 | 12.g511150 | 5 | 15 | 11 | 13 | 15 | 16 | 19 | 16 | 8 | 7 | 6 | 8 | 10 | 10 | 9 | 8 | 8 | 7 | 14 | 9 | 6 | 12 | 7 | 7 | 14 |
| GPD2 | 01.g053000 | 6 | 77 | 113 | 219 | 518 | 599 | 729 | 916 | 15 | 11 | 312 | 478 | 407 | 469 | 609 | 895 | 69 | 85 | 811 | 89 | 107 | 772 | 51 | 84 | 625 |
| GPD3 | 01.g053150 | 0 | 0 | 0 | 1 | 3 | 5 | 7 | 8 | 0 | 0 | 1 | 2 | 2 | 2 | 3 | 5 | 1 | 1 | 1 | 0 | 1 | 1 | 0 | 0 | 1 |
| GPD4 | 10.g421700 | 8 | 18 | 59 | 66 | 99 | 134 | 128 | 127 | 14 | 8 | 99 | 172 | 228 | 219 | 191 | 177 | 35 | 73 | 224 | 31 | 101 | 228 | 37 | 93 | 221 |
| PLSB1 | 02.g143000 | 24 | 52 | 16 | 18 | 21 | 32 | 49 | 49 | 31 | 40 | 15 | 13 | 17 | 24 | 32 | 29 | 30 | 9 | 45 | 39 | 11 | 43 | 22 | 9 | 52 |
| 06.g273250 | 8 | 21 | 23 | 16 | 16 | 22 | 16 | 15 | 11 | 12 | 23 | 18 | 17 | 20 | 17 | 16 | 17 | 15 | 17 | 17 | 17 | 14 | 20 | 16 | 15 | |
| KDG1 | 07.g312400 | 2 | 8 | 9 | 7 | 7 | 8 | 9 | 10 | 2 | 2 | 6 | 6 | 6 | 6 | 6 | 6 | 6 | 4 | 6 | 5 | 5 | 5 | 7 | 5 | 7 |
| KDG2 | 05.g240000 | 37 | 13 | 17 | 21 | 20 | 23 | 17 | 19 | 38 | 35 | 13 | 13 | 18 | 16 | 14 | 16 | 13 | 16 | 12 | 17 | 14 | 12 | 14 | 17 | 13 |
| KDG3 | 07.g325550 | 3 | 10 | 11 | 13 | 12 | 13 | 12 | 13 | 4 | 2 | 8 | 10 | 12 | 11 | 11 | 9 | 10 | 12 | 9 | 8 | 12 | 10 | 9 | 10 | 10 |
| DGAT1 | 01.g045900 | 2 | 14 | 36 | 29 | 17 | 20 | 19 | 18 | 5 | 6 | 23 | 20 | 18 | 17 | 15 | 13 | 9 | 11 | 9 | 11 | 11 | 10 | 12 | 13 | 9 |
| DGTT1 | 12.g557750 | 0 | 2 | 20 | 17 | 28 | 32 | 43 | 47 | 0 | 0 | 14 | 20 | 34 | 37 | 38 | 41 | 4 | 8 | 46 | 5 | 13 | 51 | 5 | 11 | 45 |
| DGTT2 | 02.g121200 | 19 | 26 | 27 | 33 | 34 | 36 | 35 | 37 | 73 | 112 | 80 | 68 | 49 | 51 | 39 | 45 | 27 | 19 | 32 | 33 | 26 | 36 | 20 | 19 | 27 |
| DGTT3 | 06.g299050 | 12 | 32 | 27 | 31 | 31 | 34 | 39 | 38 | 18 | 18 | 37 | 46 | 45 | 47 | 41 | 41 | 48 | 41 | 46 | 44 | 41 | 48 | 38 | 35 | 47 |
| DGTT4 | 03.g205050 | 1 | 17 | 10 | 12 | 6 | 7 | 8 | 7 | 4 | 2 | 5 | 4 | 6 | 5 | 6 | 4 | 14 | 7 | 5 | 13 | 7 | 5 | 14 | 7 | 5 |
| DGTT5 | 02.g079050 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| DGAT3-like | 06.g310200 | 10 | 21 | 26 | 31 | 28 | 19 | 22 | 35 | 26 | 56 | 88 | 80 | 69 | 67 | 80 | 63 | 20 | 21 | 32 | 28 | 27 | 30 | 24 | 20 | 33 |
| PDAT1 | 02.g106400 | 6 | 9 | 22 | 17 | 13 | 16 | 15 | 14 | 9 | 4 | 24 | 20 | 15 | 14 | 12 | 9 | 15 | 13 | 11 | 14 | 15 | 11 | 15 | 13 | 11 |
| 02.g135450 | 10 | 10 | 10 | 8 | 6 | 9 | 12 | 10 | 11 | 4 | 7 | 10 | 10 | 10 | 11 | 7 | 13 | 6 | 7 | 12 | 6 | 9 | 12 | 5 | 9 | |
| 12.g506600 | 3 | 6 | 8 | 7 | 8 | 8 | 10 | 10 | 4 | 4 | 5 | 6 | 6 | 6 | 7 | 7 | 3 | 4 | 5 | 3 | 4 | 5 | 4 | 5 | 5 | |
Definitions are as follows: GPD, glycerol-3-phosphate dehydrogenase; PLSB1, glycerol-3-phosphate acyltransferase; KDG, diacylglycerol kinase; DGAT1, type 1 diacylglycerol transferase; DGTT, type-2 diacylglycerol transferase; DGAT3; type-3 diacylglycerol transferase; PDAT, phosphatidylcholine:diacylglycerol acyltransferase.
DISCUSSION
There are two major conclusions from this work. First, that there are substantial differences among common laboratory strains of C. reinhardtii, which can confound phenotypic analyses, especially when high-sensitivity assays (like RNA-Seq) are used. Therefore, it is preferable to compare mutants to complemented strains. This has its own disadvantages, including position effects that alter the expression of the complementing gene, and the possibility of deletion/rearrangement at the site of insertion of the complementing gene. However, these disadvantages can be minimized by analyzing multiple independent complementing lines. Here, we compared the sta6 mutant to three independent complemented lines, STA6-C2, STA6-C4, and STA6-C6, which reduced the number of differently expressed genes to only ∼5 × 102. In addition, this overcomes the problem of hidden additional mutations resulting from the insertional mutagenesis protocol used for generating the mutant. In this work, we found that the sta6 mutant is also deleted for the adjacent RBO1 locus and indeed some of the differences between sta6 and the purported parental STA6 strain can perhaps be attributed to the loss of RBO1 function. Sequence analysis of each complemented strain often readily identifies the site of insertion even with low coverage. This information can be used to assess whether the additional mutation affects the pathway of interest. Second, the comparative approach suggested increased routing of acetate toward hexose-phosphate in sta6 compared with STA6 under N deprivation, which we validated with measurement of enzyme activities and metabolite levels.
Mechanism of TAG Overaccumulation in sta6
A major objective of this study was to understand how/why sta6 accumulates more TAG under N deprivation than does its parent. In response to N starvation stress, C. reinhardtii cells store carbon first as starch and then as TAG (Siaut et al., 2011). In a time-course experiment, starch accumulation was observed 1 d after N starvation but TAG accumulation did not peak until day 5. Resupply of N (coupled with a switch to darkness) resulted in rapid degradation of starch within the first few hours, whereas significant TAG degradation did not occur until more than 20 h later (Siaut et al., 2011). The pathways of carbohydrate versus lipid accumulation are thus temporally resolved. In this work, we noted that the temporal separation was not affected by the sta6 mutation. Both strains upregulated starch synthesis genes early in response to N starvation (within 0.5 h). Transcript abundances peaked by 2 h after which they decreased rapidly (see Supplemental Data Set 3 online). Likewise, mRNAs encoding enzymes involved in starch degradation accumulated similarly in both strains, despite the absence of starch in sta6. Genes encoding enzymes involved in TAG synthesis showed more graduate upregulation, with mRNA abundances increasing throughout the 48-h time course (Table 2). These transcriptome level measurements are consistent with the observed rapid increase in starch accumulation compared with a later increase in TAG levels (Figures 1C and 1D).
While most of the transcriptome, including genes involved in starch and TAG metabolism, is unaffected by the function of the STA6 gene (Figure 7), mRNAs encoding enzymes that function in acetate metabolism increased dramatically in sta6 versus STA6, in particular MAS1 and ICL1, which encode enzymes unique to the glyoxylate pathway. This pathway generates the 4C precursor for gluconeogenesis; indeed, FBP1 and PCK1, also encoding enzymes unique to the gluconeogenesis direction, are regulated in parallel with MAS1 and ICL1 in N-starved sta6 (Figure 4). Increased operation of the glyoxylate pathway and gluconeogenesis is supported by analysis of a subset of enzymes and metabolites in this work (Figures 8 and 9). Nevertheless, this result was unexpected because the movement of carbon in this direction would not be expected to stimulate fatty acid or lipid synthesis. How does this change in metabolism support increased TAG accumulation in sta6? Apart from the most prominent increases in ICL1, MAS1, PCK1, TAL1, and FBP1 transcripts levels, the expression pattern of PEPC1 and PGM3 (and a further set of genes involved in starch synthesis like PGI1, SBE2, SBE3, SSS4, and STA3) is also altered in sta6 compared with CC-4349 (Figure 3). These transcripts (like STA6) increase rapidly to a peak at 0.5 to 1 h and then show a partial decline in CC-4349. The initial rise is slightly delayed in sta6. However, it is unclear if these small differences are due to the mutation in sta6 or to the differing genetic background, as these transcripts also showed small differences between CC-4349 and the complemented STA6 lines.
In these experiments, C. reinhardtii cells were grown mixotrophically on acetate. One explanation is that sta6 consumes more acetate than STA6 via increased operation of acetyl-CoA synthetase and the glyoxylate pathway. Since starch synthesis is blocked, carbon is directed toward TAG. This model is consistent with the studies of others who have shown that acetate supplementation causes an increase in TAG content and the amount of acetate provided is proportional to lipid body droplet size (Goodson et al., 2011; Ramanan et al., 2013). However, when we measured acetate utilization, we found that acetate consumption was identical in both strains (Figure 10). Therefore, increased acetate utilization is not causal for increased TAG accumulation in sta6. It is possible that the previously reported increase in TAG resulting from provision of acetate enables cell survival and, hence, enables continuing TAG synthesis as a consequence of continued viability. Another possibility is that in the absence of ADP-Glc pyrophosphorylase activity, Glc-6-P is metabolized to trehalose, a signaling metabolite that has been identified in C. reinhardtii (Bölling and Fiehn, 2005). Whether trehalose levels are altered in sta6 and, if so, whether this affects TAG synthesis remain to be tested. A third possibility is raised by the observation that TAL1 mRNA abundance increases coordinately with that of FBP1 and PCK1 in N-starved sta6 (Figure 3; see Supplemental Data Set 3 online), specifically that Glc-6-P is further metabolized by the oxidative pentose phosphate pathway. This would allow maintenance of a pool of reduced nucleotide for de novo fatty acid biosynthesis. Given that carbon availability is not the bottleneck for TAG synthesis (at least in this genetic situation), the possibility of increased reductant availability in sta6 being causal for TAG accumulation is intriguing. We cannot determine whether the threefold higher Glc-6-P content of sta6 results from increased gluconeogenesis versus reduced conversion to ADP-Glc. However, the former is possibly more likely given that we observe more hexose-phosphate only in N-deprived conditions (i.e., where the glyoxylate pathway and gluconeogenesis are upregulated). If the higher Glc-6-P content of sta6 resulted from the block in ADP-Glc synthesis, it would also be higher in N-replete conditions. On the basis of these results, we propose that future metabolic engineering projects may benefit from attempts to increase the reductant pools.
Many laboratories have monitored the expression of genes encoding TAG biosynthesis enzymes of C. reinhardtii under N starvation conditions (Miller et al., 2010; Boyle et al., 2012; Msanne et al., 2012; Ramanan et al., 2013). The functions of a few of these, DGTT1, PDAT1, and DGTT2, have been validated experimentally (Boyle et al., 2012; Sanjaya et al., 2013). Of seven putative acyltransferases in the C. reinhardtii genome, only two are differentially expressed in sta6 versus STA6: DGTT2 and Cre06.g310200, an unvalidated homolog of a type-3 acyltransferase. Acyltransferase activity has been postulated as the rate-limiting step for TAG synthesis, assuming the acyl-CoA supply is not limiting (Ichikara and Noda, 1980; Perry and Harwood, 1993; Bao and Ohlrogge, 1999). Unlike many of the mRNAs that increase in sta6 versus STA6, the mRNA abundances of both DGTT2 and Cre06.g310200 increase comparatively early (peak for DGTT2 at 0.5 h and for Cre06.g310200 at 2 h; Table 2) after initiation of the N starvation response. This indicates that these genes are upregulated independently of other genes that are differentially expressed in the two strains. Their induction might result from an unknown signaling pathway in response to a block in starch synthesis. In particular, we note that DGTT2 is one of the 521 genes specific to the sta6 response (Figure 5).
Finally, we observe that the increase in MLDP1 mRNA correlates well with TAG accumulation, supporting its use as a marker for TAG content (Figure 1E), as suggested previously (Moellering and Benning, 2010). The availability of an antibody will facilitate the development of screening assays for strains with altered lipid content.
Genotype of Laboratory Strains
Several observations led to the discovery that the cw strain used initially in this work and prior work by others as the congenic wild-type parent of sta6 is, in fact, unrelated. The strain has now been named CC-4349. First, the expression of mating-type specific genes in the RNA-Seq experiments indicated that CC-4349 is mt− rather than the expected mt+ like sta6. Second, the strain was larger than sta6 and the size difference was not affected by complementation with STA6, suggesting other genetic differences. Most importantly, genome resequencing identified not only an order of magnitude more SNVs relative to the true parent (Table 1) but also a number of InDels. These genetic differences make it difficult to assign phenotype to the sta6 mutation and necessitated the use of complemented strains for the comparative analyses. This is becoming more important as C. reinhardtii is increasingly a reference system for genetics-based studies. For the purpose of this project, we acquired the original cw strain (carrying the Arg auxotrophic marker) independently and found it to be the true parent by genome resequencing. There are still SNVs between this strain and the present-day sta6, but this is likely attributable to over a decade of laboratory propagation. The true parent is presently deposited as CC-4568. In light of these results, future work in this area should minimally compare CC-4348 and CC-4568 and ideally also CC-4565, CC-4566, and CC-4567. We also resequenced STA6-C6 (CC-4567) and demonstrated that this strain likely contains a single insert of the complementing genetic construct, which is at the Cre16.g66450 locus. In agreement with this, this gene, predicted to encode a calcium channel, lies within the intersect unique to STA6-C6 of differentially expressed genes relative to sta6.
In addition to deleting most of the STA6 coding region, the neighboring gene, RBO1, was also partially excised in the sta6 mutant. Insertions are often associated with deletions in the flanking DNA in C. reinhardtii, which may be a consequence of a missing RAD6 (Vlcek et al., 2008). The closest homolog (mutual best hit) to RBO1 in Arabidopsis thaliana is RBOHa, which encodes an NADPH oxidase responsible for reactive oxygen species (ROS) production in response to pathogen wounding (see Supplemental Figure 9 online). ROS signaling resulting from NADPH oxidases in vascular plants has been demonstrated in response to a number of stresses, including wounding, radiation, heat, and drought, where it induces a signaling cascade via ROS production (Torres et al., 2002). In C. reinhardtii, RBO1 is induced in response to N starvation, as observed here in CC-4349 (peak of 9 RPKM in CC-4349 versus 0 in sta6), among other stress-inducing conditions, including Zn deprivation (Malasarn et al., 2013). Notably, two differentially expressed genes between sta6 and CC-4349 were LHCSR1 and LHCSR2, both of which are involved in photoquenching to dissipate excessive energy from high-light absorption (Peers et al., 2009; Bonente et al., 2011). Transcripts of both genes were more highly induced in CC-4349 than sta6. Since these stress-related mRNAs are increased in abundance in CC-4349 but not in sta6, we suggest that RBO1 may have a role in the induction of these genes. In this case, the RBO1 regulon would be expected to lie within the genes that are expressed more highly in CC-4349 versus sta6 (see Supplemental Data Set 8 online). In this light, the deletion of the 5′ end of RBO1, including the first five exons, fortuitously presents the required genetic background to test this hypothesis and to define the putative RBO1 regulon.
METHODS
Chlamydomonas reinhardtii Strains, Media, and Culture Conditions
C. reinhardtii strains used were cw15 (nit1 NIT2 mt−), sta6 (cw15 nit1 NIT2 arg7-7 sta6-1::ARG7 mt+), and three complemented strains cw15 arg7-7 sta6-1::ARG7 mt+ (STA6). The cw15 and sta6 strains are available as CC-4349 and CC-4348, respectively, from the Chlamydomonas Resource Center (CRC) (http://chlamycollection.org/; Minnesota University). The complemented STA6 mutants, strains C2, C4, and C6, are as described previously (Li et al., 2010b) and were obtained from Ursula Goodenough (Washington University, MO). They are available as CC-4565, CC-4566, and CC-4567 from the CRC. Strain cw15 was also obtained independently from David Dauvillée. This strain is an Arg auxotroph and represents the true parent of sta6. It has been submitted to the CRC and is available as CC-4568.
Cells were grown in TAP medium unless otherwise indicated. Except where stated, cultures were grown in Innova incubators (New Brunswick Scientific) at 24°C, agitated at 180 rpm with continuous light (95 µmol m−2 s−1, six cool white fluorescent bulbs at 4100K and three warm white fluorescent bulbs at 3000K per incubator). N minus TAP was prepared by omitting ammonium chloride, as described previously (Boyle et al., 2012), and is described as N-free media, although the amino group on Tris may be used as a N source. Briefly, cells subjected to N deprivation were grown to 4 × 106 mL−1 and collected by centrifugation at 1006g for 5 min at room temperature. The supernatant was discarded, and the cells were washed in N-free TAP and resuspended in the same to a final cell count of 2 × 106 cells mL−1. Samples taken prior to washing were labeled as 0’, samples taken immediately upon inoculation of N-free media were labeled as 0, and subsequent samples were labeled with respect to the time of sampling in hours. Cells were counted with a hemocytometer.
Enzyme Assays
Samples were collected at 0, 24, 48, and 96 h post N deprivation, in biological triplicate, for enzyme assays and metabolite measurements. Whole-cell lysates were prepared as follows. Eight-milliliter samples were taken at each time point, and cells were collected by centrifugation at 1006g for 5 min and resuspended in 1 mL of 16 mM K2HPO4/11 mM KH2PO4 buffer. Cells were broken by sonication (one 20-s pulse, amplitude 69%, using a Branson Digital Sonifier with probe model 102C), and particulate matter was removed by centrifugation at 16,100g for 15 min at 4°C.
Malate synthase and isocitrate lyase were assayed by modifying previously described methods (Chell and Sundaram, 1975). For malate synthase, the final reaction conditions were 30 mM imidazole, pH 8.0, 10 mM MgCl2, 0.25 mM acetyl-CoA, 1 mM glyoxylic acid, 0.2 mM 5,5′-dithio-bis(2-nitrobenzoic acid), and 100 μL soluble cell extract prepared as described above per milliliter of reaction volume. Reactions were performed at 30°C and monitored at 412 nm for 5 min. For isocitrate lyase, the final reaction concentrations used were 30 mM imidazole, pH 6.8, 5 mM MgCl2, 1 mM EDTA, 4 mM phenylhydrazine, and 1 mM dl-isocitric acid plus 100 μL clarified extract per milliliter of reaction. Reactions were performed at 30°C and monitored at 324 nm for 5 min. For both assays, continuous product formation was monitored for at least 5 min, and the linear portion of the product formation curve was used as a measurement of initial velocity to calculate the enzymatic activity of the clarified lysate. Reagents for all assays were from Sigma-Aldrich. All assays incorporated relevant controls to subtract background activity in which each reagent was sequentially subtracted from the reaction mixture. Assay linearity with respect to protein concentration was verified by measuring serial dilutions of the protein sample. Enzyme activities were determined relative to total protein content by the BCA assay (Thermo Scientific). Starch assays were performed using the starch assay kit (Sigma-Aldrich) according to the manufacturer’s directions. Chlorophyll content of cells was measured as previously described (Moseley et al., 2002). Glc-6-P was measured using a Glc-6-P assay kit (BioVision), and concentrations were determined by comparison to a standard curve as directed by the manufacturer. The concentration of acetate in culture supernatants was determined using the EnzyChrom acetate assay kit (BioTrend Chemicals) according to the manufacturer’s directions. Glc-6-P and acetate assays were performed in 96-well format and measured using a Tecan M1000 plate reader. Glc-6-P and acetate assays were validated by addition of spike-in standards (see Supplemental Figure 7 online).
Immunoblot Analysis
Soluble cell extract was prepared as described above (see Enzyme Assays). Ten micrograms of protein per sample, calculated using the BCA assay, was diluted to equal volume. Protein samples were denatured by addition of 4× Laemmli buffer (final concentration of 31.25 mM Tris, 1.525% [w/v] SDS, 5% [v/v] glycerol, 0.01% [w/v] bromophenol blue, and 5% [w/v] β-mercaptoethanol) and boiled for 10 min before separation by denaturing PAGE (12% monomer). Proteins were transferred to nitrocellulose membrane for 90 min at room temperature under constant current (1.5 mA/cm2 membrane) in 25 mM Tris, 192 mM Gly, 0.01% (w/v) SDS, and 20% (v/v) methanol using a semidry blotter. The membranes were blocked with 1% (w/v) dried nonfat milk in phosphate buffered saline-tween (PBS-T; 137 mM NaCl, 2.7 mM KCl, 10 mM Na2HPO4, 1.7 mM KH2PO4, and 0.2% [v/v] Tween-20, adjusted to pH 7.2) for 1 h with shaking at room temperature and washed three times with 1% milk in PBS-T. Incubation of the membranes with anti-MLDP antibodies was performed at a 1:2000 dilution in 1% milk in PBS-T with shaking at room temperature for 1 h, static overnight (12 to 16 h) at 4°C, followed by shaking at room temperature for 1 h (Huang et al., 2013). After three washes with 1% milk in PBS-T and incubation with alkaline phosphatase coupled goat anti-rabbit antibody (Southern Biotechnology Associates) at a dilution of 1:2000 in 1% milk in PBS-T, antibody bound to the membrane was detected using the alkaline phosphatase color reaction. After 15 min of incubation with the color reagent, the membranes were transferred to 1× TBS, pH 7.5, and left for 14 to 16 h before imaging. ColorPlus prestained Protein Marker (Broad Range) (New England Biolabs) was used as a size standard protein.
Lipid Analysis
Lipids were analyzed by gas chromatography as described previously (Boyle et al., 2012).
Nucleic Acid Preparation and Analysis
Genomic DNA was prepared as follows. Total cellular DNA was prepared from stationary phase cultures (∼1 × 107 cells mL−1) of strains CC-4349, sta6 (CC-4348), and STA6-C6 (CC-4567) using standard procedures. Specifically, cells from 30 mL of each culture were collected by centrifugation (3440g, 5 min, 22°C) and resuspended in 2 mL of milliQ-purified water. Two milliliters of the resuspended cells was transferred to a fresh tube and combined with 2 mL of 2 × lysis solution (10 mM Tris-Cl, pH 7.5, 10 mM EDTA, 10 mM NaCl, 0.5% SDS, and 200 µg mL −1 proteinase K). After incubation for 2 h at 50°C, DNA was extracted by addition of 4 mL of phenol-chloroform, followed by vigorous shaking and centrifugation to separate phases (13,800g, 10 min, 10°C). Four milliliters of the aqueous phase was transferred to a clean tube and treated with 5 μL of 5 mg mL−1 of RNaseA for 30 min at 37°C, followed by an additional phenol-chloroform extraction as before. Four milliliters of the resulting aqueous phase was transferred to a clean tube. Next, polysaccharides were selectively precipitated by the addition of 1.4 mL of room temperature 100% ethanol, incubation for 15 min on wet ice, and centrifugation (13,800g, 10 min, 10°C). Supernatant (5.4 mL) was transferred to a clean tube, and the DNA was precipitated by the addition of 5.4 mL isopropanol, incubation for 15 min at room temperature, and centrifugation (19,800g, 10 min, 22°C). After he supernatant was discarded, the DNA pellets were air-dried for 15 min at room temperature, and resuspended in 525 μL of purified water. DNA was precipitated again by the addition of 100 μL of 5 M NaCl and 625 μL of 20% polyethylene glycol-8000. The mixture was incubated for 30 min on wet ice, after which the DNA was collected by centrifugation (19,800g, 20 min, 4°C). The supernatant was removed by decanting, and the pellet was washed with 70% ethanol, and air-dried for 15 min at room temperature. The resulting DNA was resuspended in 50 μL of purified water, and the concentration determined by optical absorbance on a NanoDrop 2000 spectrophotometer (Thermo Scientific). For each strain, 1 µg of genomic DNA was sheared by the S-220 Adaptive Focused Acoustics system (Covaris) using the following settings: 10% duty cycle, 5.0 intensity, 200 bursts s−1, 120 s, and 6°C. The resulting fragments were used to make sequencing libraries using the TruSeq DNA sample preparation kit, version 1 (Illumina), following the low-throughput protocol. The concentrations of the resulting libraries were determined by Qubit double-stranded DNA Broad Range assay kit assay kit (Invitrogen). Sequencing flow cells were prepared using the TruSeq cBot PE cluster generation kit, version 3 (Illumina), and sequencing was performed as 100 + 100-nucleotide paired end reads (CC-4349, CC-4567, CC-4568) or 50 + 50-nucleotide paired-end reads (CC-4348) on a HiSeq2000 sequencer (Illumina)
The reads were aligned using Burrows-Wheeler Aligner v.0.6.2 v.0.6.2 (Li et al., 2008), with default parameters, to the version 5.0 assembly of the C. reinhardtii CC-503 genome (Merchant et al., 2007). After removing duplicates with Picard MarkDuplicates (http://picard.sourceforge.net), we applied Genome Analyzer Tool Kit (McKenna et al., 2010) base quality score recalibration, InDel realignment, and small variant discovery (DePristo et al., 2011). Larger structural variants were called using BreakDancer_max1.1 (Chen et al., 2009) and Pindel0.2.4t (Ye et al., 2009), each with default parameters, followed by extensive manual filtering of false positives using IGV (Thorvaldsdóttir et al., 2012). SNVs are available at the National Center for Biotechnology Information dbSNP Short Genetic Variations database. SNVs and read alignments are available in VCF and BAM format upon request.
Samples for RNA-Seq analysis of CC-4349 and sta6 were taken in triplicate. The RNA was prepared and assessed for integrity as previously described (Boyle et al., 2012). cDNA libraries were generated in duplicate from two of the three samples taken and sequenced on a GAIIx sequencer (Illumina) at Los Alamos National Laboratory as described (Boyle et al., 2012). The read lengths and sequence data obtained for each sample are shown in Supplemental Data Set 1 online, sheet 1. The transcriptomes of the complemented strains (CC-4565, CC-4566, and CC-4567) were determined, in comparison to sta6 (CC-4348), in a separate experiment. In this case, only one sample was collected from each of the three independent transformants and sta6 at each time point (see Supplemental Data Set 1 online, sheet 2), and the corresponding cDNAs sequenced on a HiSeq2000 sequencer (Illumina). Based on an estimate of library diversity and sequencing error rate, which determines the minimum score allowed in the alignment step, the sequences were mapped to the genome using alignment tools that allow for intron-like gaps (Wu and Nacu, 2010). The alignment parameters were optimized to account for the characteristic exon-intron structure of C. reinhardtii genes. The realignment of the existing genome annotation was used to select the class of allowed gapped alignments to the C. reinhardtii genome. This information was used to differentiate between unique and ambiguous alignments and to build fully mappable gene models, referred to as “mappable sets.” Only unique alignments were used to compute expression estimates. Per-sample normalized coverage vectors are built from the final set of high-quality disambiguated alignments. These vectors are displayed in the form of per-base graphs on the the University of California, Santa Cruz browser hosted locally, which is available at http://genomes.mcdb.ucla.edu/. Sequence alignments and mappable sets were used to generate matrices of counts per gene. Normalization by mappable length and sequencing depth provided the matrix of expression estimates in units of RPKMs (Mortazavi et al., 2008). The counts per gene from individual experiments are analyzed for differential expression using negative binomial statistics (Anders and Huber, 2010) with predetermined levels of statistical significance (false discovery rate < 1%) and up/downregulation (a minimum of twofold) imposed to generate sets of target genes. SNVs are available at the National Center for Biotechnology Information dbSNP Short Genetic Variations database with accession numbers 902873753 - 902923703. SNVs and read alignments are available in VCF and BAM format upon request. Given the high number of regulated genes in N-free media and the size of the analyzed data sets, we classified genes according to their temporal expression profiles across individual or combined experiments. The abundances of several mRNAs were unexpectedly high at 0 and 0.5 h. We suspected this was due to the physiological stress of centrifugation and washing, as required to achieve N deprivation. To be sure there was no physiological significance, we employed qRT-PCR using cDNA from samples collected at 0' (before washing in N-free media), 0, and 0.5 h. These data reproduce the spike in transcript copy number at T0 for both ICL1 and FBP1 (see Supplemental Figure 10 online). Since the mRNA abundance in both strains for the two genes is >10-fold higher at T0 than in the 0' and 0.5 h samples, this suggests the increase at T0 is a response to the stress conditions the cells are exposed to. qRT-PCR was performed as previously described (Allen et al., 2007) using the primers detailed in Supplemental Table 3 online.
Genes identified by the differential expression test were clustered by a model-based clustering method implemented as previously described (Fang et al., 2012). We assumed the observed counts followed a negative binomial distribution if biological replicates were available, and a Poisson distribution otherwise. Results using 100 clusters were consolidated by visual inspection to maintain the smallest number of specific clusters as possible without losing enrichment for significant biological function (see Supplemental Figure 1 online). The clusters were complemented with functional annotations for individual or consolidated gene clusters using the pathways annotation tool (Lopez et al., 2011).
Sample Collection and Preparation for Metabolite Analyses
For metabolite analysis, cells were cultured in a shaker (180 rpm, ∼75 µmol m−2 s−1 light, generated by eight cool white fluorescent bulbs). Metabolite profiles were determined according to Tohge et al. (2011). C. reinhardtii strains were grown in the presence or absence of N as described above in six flasks representing biological replicates. One mL of culture was added to 2 mL of 70% (v/v) methanol precooled to −70°C in an ethanol/dry ice bath in a 15-mL disposable screw-top plastic conical tube. Cells were sampled as rapidly as possible and under static light intensity (∼75 µmol m−2 s−1). After quenching, samples were immediately flash frozen in liquid N and stored at −80°C. For analysis, samples were thawed in an ethanol/dry ice bath at −35°C. Water-soluble metabolites were extracted using a slightly modified protocol as previously published(Tohge et al., 2011). Sample (525 μL) was added to 105 μL of prechilled chloroform (to −20°C) and incubated at −20°C for 1 h. The mixture was defrosted on ice, mixed vigorously for 1 min (3 × 20-s pulses and put back on ice in between), and 280 μL of ice-cold milliQ-grade water added. The mixture was agitated by vortexing, and following centrifugation at 16,100g in a microfuge for 5 min at 4°C, 800 μL of the upper phase was transferred to a fresh precooled 2-mL tube. Two further extractions were performed by adding 560 μL of chilled water, mixing on a vortex mixer and centrifugation to achieve a total volume of 1920 µL. This was divided into two prechilled tubes of 960 μL each. Chilled water (280 μL) was added to each aliquot, and the tubes were flash frozen and lyophilized overnight. Each sample was resuspended in 125 μL of chilled water and filtered through multiscreen filter plates Ultracel-10 (Millipore) by centrifugation for 90 min at 2300g at 12°C. Filtrates were analyzed by LC-MS/MS as described below.
Ion-Pair Chromatography–Triple Quadrupole Mass Spectrometry
LC-MS/MS was performed on a Dionex HPLC system coupled to a Finnigan TSQ Quantum Discovery MS-Q3 (Thermo Scientific) equipped with an electrospray ionization interface. It was operated as described (Arrivault et al., 2009) with slight modifications of the liquid chromatography gradient. Chromatographic separation was performed by passing aliquots through a Gemini (C18) 4 × 2.00-mm precolumn (Phenomenex), before separation on a Gemini (C18) 150 × 2.00-mm inner diameter, 5-μm 110 Å particle column (Phenomenex) at 35°C using a multistep gradient with online-degassed eluent A (10 mM tributylamine aqueous solution, adjusted to pH 5 with 15 mM acetic acid and 5% methanol) and eluent B (100% methanol): 0 to 5 min, 100% A; 5 to 15 min, 100 to 95% A; 15 to 22 min, 95 to 90% A; 22 to 37 min, 90 to 85% A; 37 to 40 min, 85 to 0% A, and maintained for 3 min; 43 to 47 min, 70 to 45% A, and maintained for 3 min; 50 min, 10% A, and maintained for 8 min; 58 min, 100% A, and maintained for 8 min. The flow rate was 0.2 mL⋅min−1 and was increased to 0.3 mL⋅min−1 between 22 and 58 min. After separation, compounds were ionized by electrospray ionization and detected by a triple quadrupole that was operated in negative ion mode with selected reaction monitoring using an ion spray voltage of 4000 V and a capillary temperature of 230°C. The Finnigan XCALIBUR 2.5 software (Thermo Scientific) was used for both instrument control and data acquisition. Prior to injection (100 µL), a mixture of 15 stable isotope reference compounds of known concentrations was added to the sample to correct for matrix effects on these analytes in the analysis. Metabolites were quantified by comparison of the integrated MS-Q3 signal peak area with a calibration curve obtained using authentic standards by the LCQuan software (Thermo Scientific). Further analysis was performed using Windows Excel and R statistics software (R version 2.9.2 and 2.14.1 provided by the Comprehensive R Archive Network (CRAN) project, http://www.R-project.org).
Accession Numbers
Sequence data from this article can be found in the GenBank/EMBL databases under the accession number GSE51642, and the dbSNP Short Genetic Variations database with accession numbers 902873753 - 902923703.
Supplemental Data
The following materials are available in the online version of this article.
Supplemental Figure 1. Differential Responses of CC-4349 and sta6 to N Starvation.
Supplemental Figure 2. Greater Upregulation of Genes Encoding Enzymes of Acetate Metabolism, Gluconeogenesis, and the Oxidative Pentose Phosphate Pathway in sta6 versus STA6.
Supplemental Figure 3. Mating-Type Genes Used to Distinguish Relationship of Strains.
Supplemental Figure 4. Size Phenotype of sta6 Is Not Rescued by the STA6 Gene.
Supplemental Figure 5. Coverage Graph Identifies the Lesion at the STA6 Locus.
Supplemental Figure 6. Evolutionary Relationships of Strains.
Supplemental Figure 7. Spike-in Controls Indicate Enzyme-Based Assays Are Quantitative.
Supplemental Figure 8. Expression Profiles of Genes Related to TAG Accumulation.
Supplemental Figure 9. Similarity between RBO1, RBO2, and RBOHA.
Supplemental Figure 10. qRT-PCR Suggests the Increase in mRNA Abundance Observed at T0 Is a Consequence of Centrifugation and Cell Resuspension Stress.
Supplemental Figure 11. Oxidative Pentose Phosphate Pathway.
Supplemental Table 1. Genome Sequence Libraries.
Supplemental Table 2. Mutations of sta6, CC-4349, and STA6-C6 Compared with the Reference Strain CC-503.
Supplemental Table 3. Primer Sequences Used for qRT-PCR.
Supplemental Data Set 1. RNA-Seq Libraries.
Supplemental Data Set 2. Transcripts with Increased Abundance in sta6 versus CC-4349.
Supplemental Data Set 3. Abundance of mRNAs Involved in Central Carbon Metabolism.
Supplemental Data Set 4. Comparison of mRNA Abundances for Genes Encoding Enzymes Relating to Photosynthesis and Chlorophyll Synthesis and Degradation.
Supplemental Data Set 5. Comparison of mRNA Abundances for Genes Encoding Enzymes Relating to Lipid Metabolism.
Supplemental Data Set 6. Comparison of mRNA Abundances for Genes Encoding Enzymes Relating to Gametogenesis.
Supplemental Data Set 7. STA6-Responsive Genes.
Supplemental Data Set 8. RNAs with Increased Abundance in CC-4349 versus sta6.
Acknowledgments
This work was supported by Department of Energy Contract DE-EE0003046 (to S.S.M., M.P., and S.J. via the National Alliance for Advance Biofuels and Bioproducts Consortium) and in part by the National Institutes of Health R24 GM092473 to S.S.M. and the U.S. Air Force Office of Scientific Research (FA9550-11-10264, to C.B.). I.K.B. is supported by a training grant from the National Institutes of Health (T32 ES015457). We thank Ursula Goodenough for forwarding us strains cw15 (CC-4349), sta6 (CC-4348), STA6-C2 (CC-4565), STA6-C4 (CC-4566), and STA6-C6 (CC-4567), David Dauvillée for an independent cw15 (CC-4568), and Anthony Huang for the MLDP1 antibody.
AUTHOR CONTRIBUTIONS
S.S.M., I.K.B., N.R.B., and D.C. designed the experiments. I.K.B., A.G.G., S.F.-G., T.M., S.D.G., B.L., N.R.B., and J.K. performed the experiments. D.C. performed the bioinformatic analysis of RNA-Seq data. S.J. generated libraries for RNA-Seq and supervised sequencing the transcriptomes. M.P. supervised all bioinformatic analysis. C.B. supervised lipid analysis. M.S. supervised the metabolite analysis. I.K.B., S.S.M., and S.F.-G. analyzed the data. S.S.M. and I.K.B. prepared and edited the article. All authors commented on and revised the article.
Glossary
- TAG
triacylglycerol
- RNA-Seq
RNA sequencing
- qRT-PCR
quantitative RT-PCR
- RPKM
reads per kilobase of mappable length per million aligned reads
- DGAT
diacylglycerol transferase
- InDel
insertions and deletion
- SNV
single-nucleotide variant
- LC-MS/MS
liquid chromatography–tandem mass spectrometry
- ROS
reactive oxygen species
- CRC
Chlamydomonas Resource Center
- TAP
Tris-acetate-phosphate
References
- Allen M.D., del Campo J.A., Kropat J., Merchant S.S. (2007). FEA1, FEA2, and FRE1, encoding two homologous secreted proteins and a candidate ferrireductase, are expressed coordinately with FOX1 and FTR1 in iron-deficient Chlamydomonas reinhardtii. Eukaryot. Cell 6: 1841–1852 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anders S., Huber W. (2010). Differential expression analysis for sequence count data. Genome Biol. 11: R106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arrivault S., Guenther M., Ivakov A., Feil R., Vosloh D., van Dongen J.T., Sulpice R., Stitt M. (2009). Use of reverse-phase liquid chromatography, linked to tandem mass spectrometry, to profile the Calvin cycle and other metabolic intermediates in Arabidopsis rosettes at different carbon dioxide concentrations. Plant J. 59: 826–839 [DOI] [PubMed] [Google Scholar]
- Atteia A., et al. (2009). A proteomic survey of Chlamydomonas reinhardtii mitochondria sheds new light on the metabolic plasticity of the organelle and on the nature of the alpha-proteobacterial mitochondrial ancestor. Mol. Biol. Evol. 26: 1533–1548 [DOI] [PubMed] [Google Scholar]
- Ball, S., and Deschamps, P. (2009). Starch metabolism. In The Chlamydomonas Sourcebook, Vol. 2. (San Diego, CA: Academic Press). [Google Scholar]
- Ball S., Marianne T., Dirick L., Fresnoy M., Delrue B., Decq A. (1991). A Chlamydomonas reinhardtii low-starch mutant is defective for 3-phosphoglycerate activation and orthophosphate inhibition of ADP-glucose pyrophosphorylase. Planta 185: 17–26 [DOI] [PubMed] [Google Scholar]
- Bao X., Ohlrogge J. (1999). Supply of fatty acid is one limiting factor in the accumulation of triacylglycerol in developing embryos. Plant Physiol. 120: 1057–1062 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beck C., Haring M. (1996). Gametic differentiation of Chlamydomonas. Int. Rev. Cytol. 168: 259–302 [Google Scholar]
- Boyle N.R., et al. (2012). Three acyltransferases and nitrogen-responsive regulator are implicated in nitrogen starvation-induced triacylglycerol accumulation in Chlamydomonas. J. Biol. Chem. 287: 15811–15825 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Breuer G., Lamers P.P., Martens D.E., Draaisma R.B., Wijffels R.H. (2012). The impact of nitrogen starvation on the dynamics of triacylglycerol accumulation in nine microalgae strains. Bioresour. Technol. 124: 217–226 [DOI] [PubMed] [Google Scholar]
- Brueggeman A.J., Gangadharaiah D.S., Cserhati M.F., Casero D., Weeks D.P., Ladunga I. (2012). Activation of the carbon concentrating mechanism by CO2 deprivation coincides with massive transcriptional restructuring in Chlamydomonas reinhardtii. Plant Cell 24: 1860–1875 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bulté L., Wollman F.A. (1992). Evidence for a selective destabilization of an integral membrane protein, the cytochrome b6/f complex, during gametogenesis in Chlamydomonas reinhardtii. Eur. J. Biochem. 204: 327–336 [DOI] [PubMed] [Google Scholar]
- Bölling C., Fiehn O. (2005). Metabolite profiling of Chlamydomonas reinhardtii under nutrient deprivation. Plant Physiol. 139: 1995–2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonente G., Ballottari M., Truong T.B., Morosinotto T., Ahn T.K., Fleming G.R., Niyogi K.K., Bassi R. (2011). Analysis of LhcSR3, a protein essential for feedback de-excitation in the green alga Chlamydomonas reinhardtii. PLoS Biology 9: e1000577–e1000577 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cakmak T., Angun P., Demiray Y.E., Ozkan A.D., Elibol Z., Tekinay T. (2012). Differential effects of nitrogen and sulfur deprivation on growth and biodiesel feedstock production of Chlamydomonas reinhardtii. Biotechnol. Bioeng. 109: 1947–1957 [DOI] [PubMed] [Google Scholar]
- Catalanotti C., Dubini A., Subramanian V., Yang W., Magneschi L., Mus F., Seibert M., Posewitz M.C., Grossman A.R. (2012). Altered fermentative metabolism in Chlamydomonas reinhardtii mutants lacking pyruvate formate lyase and both pyruvate formate lyase and alcohol dehydrogenase. Plant Cell 24: 692–707 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chell R.M., Sundaram T.K. (1975). Isolation and characterization of isocitrate lyase and malate synthase from Bacillus stearothermophilus. Biochem. Soc. Trans. 3: 303–306 [DOI] [PubMed] [Google Scholar]
- Chen K., et al. (2009). BreakDancer: An algorithm for high-resolution mapping of genomic structural variation. Nat. Methods 6: 677–681 [DOI] [PMC free article] [PubMed] [Google Scholar]
- DePristo M.A., et al. (2011). A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat. Genet. 43: 491–498 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan J., Andre C., Xu C. (2011). A chloroplast pathway for the de novo biosynthesis of triacylglycerol in Chlamydomonas reinhardtii. FEBS Lett. 585: 1985–1991 [DOI] [PubMed] [Google Scholar]
- Fan J., Yan C., Andre C., Shanklin J., Schwender J., Xu C. (2012). Oil accumulation is controlled by carbon precursor supply for fatty acid synthesis in Chlamydomonas reinhardtii. Plant Cell Physiol. 53: 1380–1390 [DOI] [PubMed] [Google Scholar]
- Fang W., Si Y., Douglass S., Casero D., Merchant S.S., Pellegrini M., Ladunga I., Liu P., Spalding M.H. (2012). Transcriptome-wide changes in Chlamydomonas reinhardtii gene expression regulated by carbon dioxide and the CO2-concentrating mechanism regulator CIA5/CCM1. Plant Cell 24: 1876–1893 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferris P.J., Armbrust E.V., Goodenough U.W. (2002). Genetic structure of the mating-type locus of Chlamydomonas reinhardtii. Genetics 160: 181–200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferris P.J., Goodenough U.W. (1994). The mating-type locus of Chlamydomonas reinhardtii contains highly rearranged DNA sequences. Cell 76: 1135–1145 [DOI] [PubMed] [Google Scholar]
- Ferris P.J., Woessner J.P., Goodenough U.W. (1996). A sex recognition glycoprotein is encoded by the plus mating-type gene fus1 of Chlamydomonas reinhardtii. Mol. Biol. Cell 7: 1235–1248 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodson C., Roth R., Wang Z.T., Goodenough U. (2011). Structural correlates of cytoplasmic and chloroplast lipid body synthesis in Chlamydomonas reinhardtii and stimulation of lipid body production with acetate boost. Eukaryot. Cell 10: 1592–1606 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Granum E., Kirkvold S., Myklestad S. (2002). Cellular and extracellular production of carbohydrates and amino acids by the marine diatom Skeletonema costatum: Diel variations and effects of N depletion. Mar. Ecol. Prog. Ser. 242: 83–94 [Google Scholar]
- Guschina I.A., Harwood J.L. (2006). Lipids and lipid metabolism in eukaryotic algae. Prog. Lipid Res. 45: 160–186 [DOI] [PubMed] [Google Scholar]
- Harris E.H. (2001). Chlamydomonas as a model organism. Annu. Rev. Plant Physiol. Plant Mol. Biol. 52: 363–406 [DOI] [PubMed] [Google Scholar]
- Hernández M.L., Whitehead L., He Z., Gazda V., Gilday A., Kozhevnikova E., Vaistij F.E., Larson T.R., Graham I.A. (2012). A cytosolic acyltransferase contributes to triacylglycerol synthesis in sucrose-rescued Arabidopsis seed oil catabolism mutants. Plant Physiol. 160: 215–225 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu Q., Sommerfeld M., Jarvis E., Ghirardi M., Posewitz M., Seibert M., Darzins A. (2008). Microalgal triacylglycerols as feedstocks for biofuel production: Perspectives and advances. Plant J. 54: 621–639 [DOI] [PubMed] [Google Scholar]
- Huang N.L., Huang M.D., Chen T.L., Huang A.H. (2013). Oleosin of subcellular lipid droplets evolved in green algae. Plant Physiol. 161: 1862–1874 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ichikara K.I., Noda M. (1980). Fatty acid composition and lipid synthesis in developing safflower seed. Phytochemistry 19: 49–54 [Google Scholar]
- Johnson X., Alric J. (2012). Interaction between starch breakdown, acetate assimilation, and photosynthetic cyclic electron flow in Chlamydomonas reinhardtii. J. Biol. Chem. 287: 26445–26452 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson X., Alric J. (2013). Central carbon metabolism and electron transport in Chlamydomonas reinhardtii: Metabolic constraints for carbon partitioning between oil and starch. Eukaryot. Cell 12: 776–793 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kropat J., Hong-Hermesdorf A., Casero D., Ent P., Castruita M., Pellegrini M., Merchant S.S., Malasarn D. (2011). A revised mineral nutrient supplement increases biomass and growth rate in Chlamydomonas reinhardtii. Plant J. 66: 770–780 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lardizabal K.D., Mai J.T., Wagner N.W., Wyrick A., Voelker T., Hawkins D.J. (2001). DGAT2 is a new diacylglycerol acyltransferase gene family: Purification, cloning, and expression in insect cells of two polypeptides from Mortierella ramanniana with diacylglycerol acyltransferase activity. J. Biol. Chem. 276: 38862–38869 [DOI] [PubMed] [Google Scholar]
- Li Y., Han D., Hu G., Dauvillee D., Sommerfeld M., Ball S., Hu Q. (2010b). Chlamydomonas starchless mutant defective in ADP-glucose pyrophosphorylase hyper-accumulates triacylglycerol. Metab. Eng. 12: 387–391 [DOI] [PubMed] [Google Scholar]
- Li Y., Han D., Hu G., Sommerfeld M., Hu Q. (2010a). Inhibition of starch synthesis results in overproduction of lipids in Chlamydomonas reinhardtii. Biotechnol. Bioeng. 107: 258–268 [DOI] [PubMed] [Google Scholar]
- Li H., Ruan J., Durbin R.D.A.N. (2008). Mapping short DNA sequencing reads and calling variants using mapping quality scores. Genome Res 18: 1851–1858 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li-Beisson, Y., et al. (2010). Acyl-lipid metabolism. The Arabidopsis Book, 8:e0133 10.1199/tab.0133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin H., Goodenough U.W. (2007). Gametogenesis in the Chlamydomonas reinhardtii minus mating type is controlled by two genes, MID and MTD1. Genetics 176: 913–925 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopez D., Casero D., Cokus S.J., Merchant S.S., Pellegrini M. (2011). Algal Functional Annotation Tool: A web-based analysis suite to functionally interpret large gene lists using integrated annotation and expression data. BMC Bioinformatics 12: 282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malasarn D., Kropat J., Hsieh S.I., Finazzi G., Casero D., Loo J.A., Pellegrini M., Wollman F.-A., Merchant S.S. (2013). Zinc deficiency impacts CO2 assimilation and disrupts copper homeostasis in Chlamydomonas reinhardtii. J. Biol. Chem. 288: 10672–10683 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin N.C., Goodenough U.W. (1975). Gametic differentiation in Chlamydomonas reinhardtii. I. Production of gametes and their fine structure. J. Cell Biol. 67: 587–605 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matthew T., Zhou W., Rupprecht J., Lim L., Thomas-Hall S.R., Doebbe A., Kruse O., Hankamer B., Marx U.C., Smith S.M., Schenk P.M. (2009). The metabolome of Chlamydomonas reinhardtii following induction of anaerobic H2 production by sulfur depletion. J. Biol. Chem. 284: 23415–23425 [DOI] [PMC free article] [PubMed] [Google Scholar]
- May P., Wienkoop S., Kempa S., Usadel B., Christian N., Rupprecht J., Weiss J., Recuenco-Munoz L., Ebenhöh O., Weckwerth W., Walther D. (2008). Metabolomics- and proteomics-assisted genome annotation and analysis of the draft metabolic network of Chlamydomonas reinhardtii. Genetics 179: 157–166 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKenna A., Hanna M., Banks E., Sivachenko A., Cibulskis K., Kernytsky A., Garimella K., Altshuler D., Gabriel S., Daly M., DePristo M.A. (2010). The Genome Analysis Toolkit: A MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 20: 1297–1303 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merchant S.S., Kropat J., Liu B., Shaw J., Warakanont J. (2012). TAG, you’re it! Chlamydomonas as a reference organism for understanding algal triacylglycerol accumulation. Curr. Opin. Biotechnol. 23: 352–363 [DOI] [PubMed] [Google Scholar]
- Merchant S.S., et al. (2007). The Chlamydomonas genome reveals the evolution of key animal and plant functions. Science 318: 245–250 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller R., et al. (2010). Changes in transcript abundance in Chlamydomonas reinhardtii following nitrogen deprivation predict diversion of metabolism. Plant Physiol. 154: 1737–1752 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moellering E.R., Benning C. (2010). RNA interference silencing of a major lipid droplet protein affects lipid droplet size in Chlamydomonas reinhardtii. Eukaryot. Cell 9: 97–106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mortazavi A., Williams B.A., McCue K., Schaeffer L., Wold B. (2008). Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nat. Methods 5: 621–628 [DOI] [PubMed] [Google Scholar]
- Moseley J.L., Page M.D., Alder N.P., Eriksson M., Quinn J., Soto F., Theg S.M., Hippler M., Merchant S. (2002). Reciprocal expression of two candidate di-iron enzymes affecting photosystem I and light-harvesting complex accumulation. Plant Cell 14: 673–688 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mouille G., Maddelein M.L., Libessart N., Talaga P., Decq A., Delrue B., Ball S. (1996). Preamylopectin processing: A mandatory step for starch biosynthesis in plants. Plant Cell 8: 1353–1366 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Msanne J., Xu D., Konda A.R., Casas-Mollano J.A., Awada T., Cahoon E.B., Cerutti H. (2012). Metabolic and gene expression changes triggered by nitrogen deprivation in the photoautotrophically grown microalgae Chlamydomonas reinhardtii and Coccomyxa sp. C-169. Phytochemistry 75: 50–59 [DOI] [PubMed] [Google Scholar]
- Mühlhaus T., Weiss J., Hemme D., Sommer F., Schroda M. (2011). Quantitative shotgun proteomics using a uniform 15N-labeled standard to monitor proteome dynamics in time course experiments reveals new insights into the heat stress response of Chlamydomonas reinhardtii. Mol. Cell. Proteomics 10: 004739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan J., Snell W.J. (2000). Signal transduction during fertilization in the unicellular green alga, Chlamydomonas. Curr. Opin. Microbiol. 3: 596–602 [DOI] [PubMed] [Google Scholar]
- Pavlidis P., Noble W.S. (2003). Matrix2png: A utility for visualizing matrix data. Bioinformatics 19: 295–296 [DOI] [PubMed] [Google Scholar]
- Peers G., Truong T.B., Ostendorf E., Busch A., Elrad D., Grossman A.R., Hippler M., Niyogi K.K. (2009). An ancient light-harvesting protein is critical for the regulation of algal photosynthesis. Nature 462: 518–521 [DOI] [PubMed] [Google Scholar]
- Perry H.J., Harwood J.L. (1993). Radiolabelling studies of acyl lipids in developing seeds of Brassica napus: Use of [1-14C]acetate precursor. Phytochemistry 33: 329–333 [Google Scholar]
- Plumley F.G., Schmidt G.W. (1989). Nitrogen-dependent regulation of photosynthetic gene expression. Proc. Natl. Acad. Sci. USA 86: 2678–2682 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramanan R., Kim B.H., Cho D.H., Ko S.R., Oh H.M., Kim H.S. (2013). Lipid droplet synthesis is limited by acetate availability in starchless mutant of Chlamydomonas reinhardtii. FEBS Lett. 587: 370–377 [DOI] [PubMed] [Google Scholar]
- Ramazanov A., Ramazanov Z. (2006). Isolation and characterization of a starchless mutant of Chlorella pyrenoidosa STL-PI with a high growth rate, and high protein and polyunsaturated fatty acid content. Phycol. Res. 54: 255–259 [Google Scholar]
- Rani S.H., Krishna T.H.A., Saha S., Negi A.S., Rajasekharan R. (2010). Defective in cuticular ridges (DCR) of Arabidopsis thaliana, a gene associated with surface cutin formation, encodes a soluble diacylglycerol acyltransferase. J. Biol. Chem. 285: 38337–38347 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodolfi L., Chini Zittelli G., Bassi N., Padovani G., Biondi N., Bonini G., Tredici M.R. (2009). Microalgae for oil: Strain selection, induction of lipid synthesis and outdoor mass cultivation in a low-cost photobioreactor. Biotechnol. Bioeng. 102: 100–112 [DOI] [PubMed] [Google Scholar]
- Rolland N., Atteia A., Decottignies P., Garin J., Hippler M., Kreimer G., Lemaire S.D., Mittag M., Wagner V. (2009). Chlamydomonas proteomics. Curr. Opin. Microbiol. 12: 285–291 [DOI] [PubMed] [Google Scholar]
- Sager R., Granick S. (1954). Nutritional control of sexuality in Chlamydomonas reinhardi. J. Gen. Physiol. 37: 729–742 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saha S., Enugutti B., Rajakumari S., Rajasekharan R. (2006). Cytosolic triacylglycerol biosynthetic pathway in oilseeds. Molecular cloning and expression of peanut cytosolic diacylglycerol acyltransferase. Plant Physiol. 141: 1533–1543 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanjaya D., Durrett T.P., Weise S.E., Benning C. (2011). Increasing the energy density of vegetative tissues by diverting carbon from starch to oil biosynthesis in transgenic Arabidopsis. Plant Biotechnol. J. 9: 874–883 [DOI] [PubMed] [Google Scholar]
- Sanjaya M., Miller R., Durrett T.P., Kosma D.K., Lydic T.A., Muthan B., Koo A.J., Bukhman Y.V., Reid G.E., Howe G.A., Ohlrogge J., Benning C. (2013). Altered lipid composition and enhanced nutritional value of Arabidopsis leaves following introduction of an algal diacylglycerol acyltransferase 2. Plant Cell 25: 677–693 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siaut M., Cuiné S., Cagnon C., Fessler B., Nguyen M., Carrier P., Beyly A., Beisson F., Triantaphylidès C., Li-Beisson Y., Peltier G. (2011). Oil accumulation in the model green alga Chlamydomonas reinhardtii: Characterization, variability between common laboratory strains and relationship with starch reserves. BMC Biotechnol. 11: 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siersma P.W., Chiang K.S. (1971). Conservation and degradation of cytoplasmic and chloroplast ribosomes in Chlamydomonas reinhardtii. J. Mol. Biol. 58: 167–185 [DOI] [PubMed] [Google Scholar]
- Smith A.M. (1999). Making starch. Curr. Opin. Plant Biol. 2: 223–229 [DOI] [PubMed] [Google Scholar]
- Torres M.A., Dangl J.L., Jones J.D.G. (2002). Arabidopsis gp91phox homologues AtrbohD and AtrbohF are required for accumulation of reactive oxygen intermediates in the plant defense response. Proc. Natl. Acad. Sci. USA 99: 517–522 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Urzica E.I., Casero D., Yamasaki H., Hsieh S.I., Adler L.N., Karpowicz S.J., Blaby-Haas C.E., Clarke S.G., Loo J.A., Pellegrini M., Merchant S.S. (2012). Systems and trans-system level analysis identifies conserved iron deficiency responses in the plant lineage. Plant Cell 24: 3921–3948 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varum K.M., Myklestad S. (1984). Effects of light, salinity and nutrient limitation on the production of β-1,3-D-glucan and exo-d-glucanase activity in Skeletonema costatum (grev.) cleve. J. Exp. Mar. Biol. Ecol. 83: 13–25 [Google Scholar]
- Vlcek D., Sevcovicová A., Sviezená B., Gálová E., Miadoková E. (2008). Chlamydomonas reinhardtii: A convenient model system for the study of DNA repair in photoautotrophic eukaryotes. Curr. Genet. 53: 1–22 [DOI] [PubMed] [Google Scholar]
- Wang H., Alvarez S., Hicks L.M. (2012). Comprehensive comparison of iTRAQ and label-free LC-based quantitative proteomics approaches using two Chlamydomonas reinhardtii strains of interest for biofuels engineering. J. Proteome Res. 11: 487–501 [DOI] [PubMed] [Google Scholar]
- Wang Z.T., Ullrich N., Joo S., Waffenschmidt S., Goodenough U. (2009). Algal lipid bodies: Stress induction, purification, and biochemical characterization in wild-type and starchless Chlamydomonas reinhardtii. Eukaryot. Cell 8: 1856–1868 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wattebled F., Buléon A., Bouchet B., Ral J.-P., Liénard L., Delvallé D., Binderup K., Dauvillée D., Ball S., D’Hulst C. (2002). Granule-bound starch synthase I. A major enzyme involved in the biogenesis of B-crystallites in starch granules. Eur. J. Biochem. 269: 3810–3820 [DOI] [PubMed] [Google Scholar]
- Wattebled F., Ral J.P., Dauvillée D., Myers A.M., James M.G., Schlichting R., Giersch C., Ball S.G., D’Hulst C. (2003). STA11, a Chlamydomonas reinhardtii locus required for normal starch granule biogenesis, encodes disproportionating enzyme. Further evidence for a function of α-1,4 glucanotransferases during starch granule biosynthesis in green algae. Plant Physiol. 132: 137–145 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Work V.H., Radakovits R., Jinkerson R.E., Meuser J.E., Elliott L.G., Vinyard D.J., Laurens L.M.L., Dismukes G.C., Posewitz M.C. (2010). Increased lipid accumulation in the Chlamydomonas reinhardtii sta7-10 starchless isoamylase mutant and increased carbohydrate synthesis in complemented strains. Eukaryot. Cell 9: 1251–1261 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu T.D., Nacu S. (2010). Fast and SNP-tolerant detection of complex variants and splicing in short reads. Bioinformatics 26: 873–881 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye K., Schulz M.H., Long Q., Apweiler R., Ning Z. (2009). Pindel: A pattern growth approach to detect break points of large deletions and medium sized insertions from paired-end short reads. Bioinformatics 25: 2865–2871 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoon K., Han D., Li Y., Sommerfeld M., Hu Q. (2012). Phospholipid:diacylglycerol acyltransferase is a multifunctional enzyme involved in membrane lipid turnover and degradation while synthesizing triacylglycerol in the unicellular green microalga Chlamydomonas reinhardtii. Plant Cell 24: 3708–3724 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zabawinski C., Van Den Koornhuyse N., D’Hulst C., Schlichting R., Giersch C., Delrue B., Lacroix J.-M., Preiss J., Ball S. (2001). Starchless mutants of Chlamydomonas reinhardtii lack the small subunit of a heterotetrameric ADP-glucose pyrophosphorylase. J. Bacteriol. 183: 1069–1077 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeeman S.C., Kossmann J., Smith A.M. (2010). Starch: Its metabolism, evolution, and biotechnological modification in plants. Annu. Rev. Plant Biol. 61: 209–234 [DOI] [PubMed] [Google Scholar]