

Information on genomic variation provided by novel sequencing technologies offers new and powerful approaches to identify regulatory elements and characterize gene promoter sequences.
Abstract
Identifying regulatory elements and revealing their role in gene expression regulation remains a central goal of plant genome research. We exploited the detailed genomic sequencing information of a large number of Arabidopsis (Arabidopsis thaliana) accessions to characterize known and to identify novel cis-regulatory elements in gene promoter regions of Arabidopsis by relying on conservation as the hallmark signal of functional relevance. Based on the genomic layout and the obtained density profiles of single-nucleotide polymorphisms (SNPs) in sequence regions upstream of transcription start sites, the average length of promoter regions in Arabidopsis could be established at 500 bp. Genes associated with high degrees of variability of their respective upstream regions are preferentially involved in environmental response and signaling processes, while low levels of promoter SNP density are common among housekeeping genes. Known cis-elements were found to exhibit a decreased SNP density than sequence regions not associated with known motifs. For 15 known cis-element motifs, strong positional preferences relative to the transcription start site were detected based on their promoter SNP density profiles. Five novel candidate cis-element motifs were identified as consensus motifs of 17 sequence hexamers exhibiting increased sequence conservation combined with evidence of positional preferences, annotation information, and functional relevance for inducing correlated gene expression. Our study demonstrates that the currently available resolution of SNP data offers novel ways for the identification of functional genomic elements and the characterization of gene promoter sequences.
Despite the recent discoveries of alternative mechanisms of gene expression regulation such as microRNA-mediated phenomena (Filipowicz et al., 2008; Chekulaeva and Filipowicz, 2009), epigenetic effects (Razin and Kantor, 2005; Karlić et al., 2010; Bronner et al., 2011), as well as the influence of global genome structural properties (Hatfield and Benham, 2002; Mellor, 2006), the control of gene transcription via gene-upstream cis-elements, most prominently those that act as specific binding sites for transcription factors, referred to as transcription factor-binding sites (TFBSs), remains a pivotal mode of gene expression regulation. Massively parallel joint sequencing and gene expression profiling platforms, such as ChIP-Chip and ChIP-seq, enabled the discovery of many novel cis-elements across all kingdoms and species (Smith et al., 2005; Kharchenko et al., 2008; Valouev et al., 2008). In parallel with the increased set of experimentally determined sequence motifs, a range of different bioinformatic approaches have been conceived to detect functional motifs in gene promoters in silico (Stormo, 2000; Wasserman and Sandelin, 2004; Das and Dai, 2007). As cis-elements are relatively short (6–15 nucleotides [nt]), the de novo motif discovery by bioinformatic approaches alone is challenging. Furthermore, regulatory elements often function in combination to increase specificity, rendering their identification even more complex (Kato et al., 2004; Zhu et al., 2005; Waleev et al., 2006). Conceptually, most established bioinformatic methods aim to identify sequence motifs that are specifically enriched in a set of gene promoter sequences relative to a control set (Sinha and Tompa, 2002; D’Haeseleer, 2006). The grouping of genes is typically based on observed coexpression patterns as a hallmark of coordinated expression regulation mediated by the presence of a particular motif or motif combination in the respective promoter sequences (Bussemaker et al., 2001; Pavesi et al., 2004; Vandepoele et al., 2009). A number of convenient and Web-accessible bioinformatic tools to identify candidate motifs in sets of sequences have been developed and are being used routinely by molecular biologists, such as MEME (Bailey et al., 2009) and Amadeus (Linhart et al., 2008). The information about discovered TFBSs and other cis-elements are collected in central repositories (Wingender et al., 1996; Sandelin et al., 2004; O’Connor et al., 2005; Yamamoto and Obokata, 2008; Ling et al., 2010).
One approach to bioinformatically detect functional sequence motifs relies on the assumption that they are likely conserved in orthologous gene promoter sequences across diverging species (Wasserman et al., 2000; Blanchette and Tompa, 2002, 2003). These so-called phylogenetic footprinting approaches search for conserved sequence segments across a set of orthologous gene promoter sequences. This strategy has been applied successfully to four different yeast strains: Saccharomyces cerevisiae, S. paradoxus, S. mikatae and S. bayanus (Kellis et al., 2003). As the alignment depth was shallow (only four strains), the density of mutations in the promoter of a single gene was correspondingly low. Thus, rather than searching only across a single site, the authors searched for the presence of conservation of sequence motifs across all their instances in the genome, thereby substantially enlarging the sequence set, resulting in an increased motif detection sensitivity leading to the identification of 72 elements, including most known, but also 42 novel cis-element motifs (Kellis et al., 2003).
A combination of evolutionary conservation and coexpression-based approaches was performed for the plant Arabidopsis (Arabidopsis thaliana) combined with Brassica oleracea (Haberer et al., 2006) and poplar (Populus spp.; Vandepoele et al., 2009). Both studies led to the confirmation of known motifs and the identification many novel motifs.
Evidently, the larger the set of promoter sequences to be compared (i.e. the more genomes included in the cross-genome comparison), the larger the set and density of detected mutations, and thus, the finer the resolution at which conserved sequence elements can be detected. However, with more included species, the sequences will become increasingly divergent, making deciding on the correct orthology relationships between genes and their promoters ever more challenging. By contrast, if the considered genomes are very similar, as in the case of different strains of a species or even allelic variants in a population of a single species, the density of accumulated mutations may be too low to differentiate conserved from variable regions. Thus, a large number of genomes is necessary to effectively detect short conserved and potentially functional regulatory motifs from sets of genomes of genetically close organisms. While this is a tall requirement, the correct detection of equivalent sites across different genomes is much facilitated.
Several initiatives are currently being pursued to fully sequence a large number of individuals from a particular species and to detect all sequence variants. Most prominently, the 1,000-genomes project aims to comprehensively identify genetic differences in the human population (Siva, 2008). With the rapid increase of sequence information depth, inspecting regulatory regions and individual cis-elements with regard to their variability has become possible (Spivakov et al., 2012). Similarly, for the model plant Arabidopsis, the 1,001-genomes initiative has been launched to sequence a large set of Arabidopsis accessions (Cao et al., 2011). As a result, a comprehensive set of frequent genetic variants, including single-nucleotide polymorphisms (SNPs), has already become available (Schneeberger et al., 2011) and is enabling systemic screens for phenotype-genotype associations in Arabidopsis (Horton et al., 2012). Thus, the above-mentioned requirement for a deep coverage of genomic regions in order to obtain high resolution for an effective detection of conserved functional sequence elements may be met already.
In this study, we exploited the available SNP set in Arabidopsis resulting from the 1,001-genomes project with the aim to characterize known and identify novel cis-regulatory elements. Unlike previous studies that detected polymorphisms using hybridization microarrays (Childs et al., 2010), the 1,001-genomes data yield SNP information at the resolution of single base positions, as they were obtained using resequencing technologies. Currently, there are about 150 described cis-motifs known in Arabidopsis that have been determined and validated in a number of different experiments and by bioinformatic approaches, and various plant-specific cis-element motif databases are available (Higo et al., 1999; Davuluri et al., 2003; O’Connor et al., 2005).
The large SNP set from the 1,001-genomes project allowed us to characterize the known motifs with regard to SNP density, not only globally but also in a location-specific manner. Even though constraints on the positions of cis-elements relative to the transcription start site (TSS) have been observed to vary between different motifs (Wray et al., 2003), it can be assumed that for many cis-elements, in order to be functional, they should reside in a relatively narrow positional range relative to the TSS to ensure proper binding of all proteins forming the transcription initiation complex. In human, 28% to 35% of cis-motifs exhibit distinctive positional preferences (Xie et al., 2005). Positional preferences have also been exploited for motif prediction purposes (Kiełbasa et al., 2001). Thus, elevated motif counts in a small sequence interval may be taken as a positive indicator of the functional relevance of a given motif. Consequently, and as demonstrated for yeast (Moses et al., 2003), SNP frequencies should be reduced in those intervals, as functional cis-elements should be conserved. Therefore, the two properties, positional occurrence and SNP density, can be expected to be anticorrelated, which may serve as a criterion to identify functional instances of known motifs in Arabidopsis.
Furthermore, the set of available Arabidopsis genome sequences may already permit the detection of novel motifs following the phylogenetic footprinting concept as implemented similarly in the yeast study (Kellis et al., 2003): that is, searching for signs of conservation not only across a single mapping location but combining all motif mappings across all gene promoters to effectively increase the SNP density and thereby the resolution at which motifs can be detected.
Our study demonstrates that the available SNP density in Arabidopsis already enables the profiling of known motifs and the discovery of additional candidate cis-motifs in Arabidopsis.
RESULTS
Estimating the Effective Promoter Length
The type of cis-regulatory motifs that are the focus of this study are defined as genomic sequence motifs in the 5′ upstream regions of genes that function as regulatory switches on the downstream gene via motif-specific protein-binding events. The region harboring the set of all cis-elements associated with a particular gene is referred to as the gene promoter. Therefore, when it is the task to map known cis-element sequence motifs to Arabidopsis gene promoters and to identify novel motifs in them, the boundaries of the candidate promoter regions need to be defined. As there is no promoter start signal known so far, we attempted to determine the effective promoter length in Arabidopsis by taking advantage of the gene-mapping information and the associated statistic of intergenic sequence lengths. Furthermore, as promoters have likely evolved under evolutionary conservation pressure, the SNP density distribution as a function of distance from the TSS may also inform the effective average promoter sequence length. Unlike the promoter start, its end is relatively clearly, at least operationally, defined as the annotated TSS (see “Discussion”).
The length of the intergenic region that is available to function as a promoter may depend on the orientations of the two respective neighboring genes. If both genes have the same orientation (i.e. they are both on the same strand and transcribed in the same direction and are collinear), the respective intergenic region between them can potentially act as a promoter in its entirety up to the next respective upstream gene (Fig. 1, case 1, “head-to-tail”). Alternatively, the two genes can be oriented with their respective 3′ ends (“tail-to-tail”) toward each other (Fig. 1, case 2). In this case, the intergenic space between the two genes does not contain any promoter but other types of motifs associated with the specific 3′ end processing of genes (e.g. polyadenylation signals), cis/trans-acting enhancers (Kollias et al., 1987), or others, which can be assumed to require less space compared with gene promoters. Therefore, neighboring genes oriented in a tail-to-tail fashion may also, despite the presence of the mentioned motif types. Lastly, two neighboring genes may also be oriented with their respective 5′ ends (“head-to-head”) toward each other (Fig. 1, case 3), such that either the two genes can only come as close so their respective individual promoter regions do not overlap, or if they do, this region is used as a bidirectional promoter. Even though bidirectional promoters have been reported to be common phenomena in the Arabidopsis genome (Wang et al., 2009), it nonetheless creates extra evolutionary pressure, as two genes are now coupled (Li et al., 2006). Changes in the regulation of one gene will also modulate the regulation of the other, such that two functional constraints need to be met simultaneously before any change is tolerated. Coupling may also be mediated via the unwinding and opening of the DNA double helix upon transcription, rendering two genes in head-to-head orientation more likely to be coexpressed, which may not always be advantageous. Therefore, evolution may have led to a decreased frequency of such promoter overlaps. In addition to the discussed cases, overlapping genes on the opposite strand are also possible and do occur (Jen et al., 2005).
Figure 1.

Frequencies of intergene distances. Proportions of intergenic distances between neighboring genes for the three possible relative orientations of neighboring genes (right panel) and as a function of distance between them are shown. A region length interval bin size of 25 nt was used.
In agreement with the simple reasoning of the influence of gene orientation on intergene spacing, head-to-head-oriented genes (case 3) were found to be significantly farther apart (median = 1,307 nt) compared with same-orientation (case 1, median = 958 nt) and tail-to-tail (case 2, median = 488 nt) arrangements (PWilcoxon << 0.05; note that all three possible pairwise case comparisons result in highly significant differences as judged by the nonparametric Wilcoxon rank-sum test.) Thus, upstream regions may create an effective constraint such that overlapping cis-regulatory regions are avoided. The intergenic spaces between two neighboring genes in tail-to-tail orientation are shortest, in agreement with the notion that they may be sequences with fewer functional elements requiring any designated sequence space.
Inspecting the relative frequencies of the three different orientations as a function of distance between neighboring genes, we found that at intergenic region lengths of around 400 to 500 nt, the relative frequencies of the three different orientations change significantly (Fig. 1), which can be interpreted as an effective promoter region blocking. At shorter distances, the frequency of collinear orientation (case 1) drops significantly, and at shorter than 250 nt, head-to-head arrangements (case 3) decrease sharply in favor of tail-to-tail orientations (case 2). Thus, while shared promoters do occur, their frequency is reduced consistent with the notion of an effective blocking effect of promoters on the closest possible intergene distance. In summary, the statistics of intergenic distances suggest that single promoters may effectively require 250 to 500 nt in Arabidopsis.
Characterization of Intergenic Regions Using SNP Data
Under the assumption that promoter sequences are more conserved than other parts of the intergenic regions, the available SNP data from the Arabidopsis 1,001-genomes project used here may also provide a handle on the question of an effective average promoter length.
In total, 846,164 polymorphic sites exhibiting SNPs were found in intergenic regions of up to 3,000 nt upstream of TSSs across 349 Arabidopsis accessions relative to the Columbia-0 (Col-0) reference genome, of which 91.1% are biallelic, 8.5% are triallelic, and 0.4% are tetraallelic SNPs. Consequently, the overall SNP density of the intergenic regions was determined as 0.0189 SNPs per nt, equating to one polymorphic site occurring every 53 positions on average. The SNP density along the intergenic sequences relative to the TSS was found to be lower near the TSS compared with positions farther away (Fig. 2). In the range from −1,500 to −500 nt, the SNP density is relatively constant at 0.0191 SNPs per nt. From −500 nt onward, the SNP density decreases sharply toward the TSS and reaches its minimum at the TSS, signifying it as the location of greatest conservation. Inside genes (i.e. in intragenic regions), the SNP density was found to be constant across the first 500 nt at about 0.015 SNPs per nt, corresponding to one polymorphic site every 66 positions on average. The SNP density profiles were similar for both collinear and antioriented neighboring genes.
Figure 2.

Average SNP density profile relative to the TSS. SNP densities (window size of 25 nt) were computed for all intergenic regions (black solid line) for case 1 (Fig. 1), SNPs only (collinear orientation of neighboring genes, where the genes have the same orientation; dark gray solid line), and case 3, SNPs only, in which genes are oriented head to head (light gray solid line). A logistic curve with f(x) = A + (B − A)/(1 + C × exp(−Dx)), where A = 0.019, B = 0.015, C = 0.223, and D = −0.0098, was fitted to the all-SNPs data set (black dashed line). Note that case 2 gene-gene orientations (Fig. 1) do not harbor any promoter segments and therefore were not considered here.
The density profiles reveal a background SNP density that is likely linked to regions of no or reduced functional significance (beyond −500 nt), whereas regions immediately upstream of the TSS are characterized by a reduced SNP frequency, likely caused by evolutionary constraints on the gene regulatory apparatus. The profiles also identify the TSS as a unique position with highest conservation. Given that the exact location of the TSS is uncertain or multiple TSSs may exist for a single gene (see “Discussion”), this result can be taken as confirmation that its location annotation is often correct and justifies all subsequent position-centered analyses of cis-elements.
Based on the statistic of gene layout as well as the density of polymorphic sites in gene-upstream regions, we determined the effective average promoter length in Arabidopsis to be 500 nt. Therefore, we will focus our analyses on this sequence interval in all following analyses.
Gene Ontology Enrichment Analysis Identifies Functions, Processes, and Components with Increased/Reduced Variability of Their Regulatory Regions
Sequence variation may be taken as evidence for adaptive processes or neutrality associated with the respective sequence regions, while sequence conservation signifies a preservation of function across different environments. To associate promoter sequence variability with the function of the respective downstream genes, we performed a Gene Ontology (GO) term enrichment analysis to identify those functions, processes, and cellular components that are associated with genes with increased or decreased SNP frequency in their promoter regions. After sorting all 32,507 Arabidopsis genes according to their SNP density in their respective upstream regions of up to −500 nt, we compared the top-1,000 genes (i.e. those genes with greatest variability of their promoter regions as judged by their high SNP density) with the bottom-1,000 genes with lowest SNP frequency (Table I). Genes involved in transcriptional regulation, signaling, and response to stress processes are characterized by increased promoter SNP frequencies, while those involved in housekeeping functions (e.g. protein metabolism, general binding, and DNA-RNA interaction [replication]) are associated with less variable promoter regions. Thus, regulatory regions of genes involved in responding to the environment, which will likely vary for the different Arabidopsis accessions, are most variable. Genes with as yet unknown function and process involvement were also found with increased SNP densities in their promoter regions. While this may reflect a true and potentially interesting variability for those uncharacterized genes, it may also be caused by gene annotation uncertainties such that gene assignments may be wrong, with SNP rates corresponding to sequence portions under no particular conservation pressure. With regard to location (GO component term), no significant terms were detected.
Table I. Enriched GO-slim annotations.
Annotations in the set of genes associated with the 1,000 most and least conserved intergenic upstream regions up to 500 nt upstream of the TSS are listed according to their corresponding P values obtained from a one-sided Fisher’s exact with correction for multiple testing (Benjamini and Hochberg, 1995). Only terms with P < 0.05 are listed.
| GO-Term Category | Conserved (Few SNPs) |
Variable (Many SNPs) |
||
|---|---|---|---|---|
| P | Term | P | Term | |
| Function | 5.27e-08 | Nucleic acid binding | 5.07e-03 | Unknown molecular functions |
| 2.11e-07 | DNA or RNA binding | 6.83e-03 | Kinase activity | |
| 5.41e-03 | Other binding | 8.64e-03 | Other molecular functions | |
| 3.52e-02 | Protein binding | |||
| 3.52e-02 | Transferase activity | |||
| Process | 3.79e-05 | Protein metabolism | 3.79e-05 | Unknown biological processes |
| 2.79e-02 | Other cellular processes | 1.28e-03 | Signal transduction | |
| 3.82e-02 | Response to stress | |||
Mapping of Known cis-Elements to Arabidopsis Promoter Regions
Combining the information contained in three public databases of plant-specific cis-regulatory elements, AGRIS (Davuluri et al., 2003), Athena (O’Connor et al., 2005), and PLACE (Higo et al., 1998), we identified a set of 144 nonredundant known Arabidopsis cis-elements ranging in length from 5 to 49 nt (Supplemental Table S1). Of the 144 nonredundant motifs, 136 mapped to the 500 nt upstream of the TSS for all Arabidopsis genes with the sequence taken from the Col-0 ecotype. Sixteen motifs are palindromic. In total, 540,944 individual motif mappings were found in the 500-nt upstream genomic regions considered to be the promoter.
Cis-Element Motif Mapping Sites Exhibit a Reduced SNP Density
First, we tested whether mapping sites of known Arabidopsis cis-element motifs are more conserved across the different Arabidopsis accessions than intergenic sequence regions that are not part of such mapping locations. Indeed, sequence positions belonging to mappings of known cis-element motifs are less polymorphic than surrounding positions (Table II). While the probability of a non-cis-element mapping site position being polymorphic was estimated as 0.0187 SNPs per nt, it was significantly lower (0.0167 SNPs per nt) for positions that belong to mappings of known cis-elements to the 500 nt upstream of the TSS (P = 5.1E-103, Fisher’s exact test), albeit the reduction of SNP frequency was relatively small (11%). Nonetheless, the notion that mapping sites of cis-elements are more conserved in order to remain functional appears supported by the data. This result also provides the rationale for trying to infer novel motifs as sequence n-mers with reduced SNP frequencies. A significant SNP frequency signal associated with cis-element motif mappings was detectable, even though not all, in fact only a minority, of those mappings sites of known cis-element motifs can be expected to be functional. Still, valid motifs may be discernible based on SNP profiles. However, because of the relatively small difference (11%), we imposed additional criteria based on location preferences to identify novel motifs more confidently (see below).
Table II. Contingency table of intramotif/intermotif SNP frequency counts.
Listed are the counts of sites (positions in the upstream region) known to be either polymorphic or nonpolymorphic and/or belonging to a mapped motif in promoter regions from −500 nt to the TSS. Fisher’s exact test reveals a significantly reduced SNP frequency within cis-element mapping sites relative to positions no part of any known cis-element motif mappings (P = 5.1E-103).
| Site Is Polymorphic | Site Belongs to a TFBS Motif Mapping Site |
||
|---|---|---|---|
| Yes | No | Sum | |
| Yes | 42,295 | 218,632 | 260,927 |
| No | 2,492,262 | 11,494,127 | 13,986,389 |
| Sum | 2,534,557 | 11,712,759 | 14,247,316 |
Next, we gathered the statistics of SNP mappings to individual cis-elements. Table III lists the 10 motifs found with lowest and highest SNP density among those motifs found at least 1,000 times in the considered Arabidopsis gene promoter intervals. (The complete table for all 144 known motifs is available as Supplemental Table S1.) With regard to process involvement (e.g. stress, growth, light-mediated regulation) of the individual motifs, no obvious classification of high versus low SNP density can be gleaned from the data.
Table III. Intramotif SNP density for 20 selected cis-element motifs from AGRIS, Athena, and PLACE.
The top- and bottom-10 nonredundant cis-motifs, which were found at least 1,000 times in the sequence interval between −500 nt and the TSS with respect to the TSS, were selected according to their intramotif SNP density. Functional associations were taken from the ProFITS database (Ling et al., 2010). Motifs were required to not contain any ambiguity codes in their motif definitions, to ensure that detected intramotif SNPs correspond to true deviations from the motif and are not merely predefined acceptable variations of the same motif. For example, the motif CARGCW8GAT, sequence CWWWWWWWWG, was found with high SNP density likely caused by the loosely defined motif sequence alone. The motifs are listed with their motif sequences, including ambiguity codes, motif and SNP counts, and intramotif SNP density (i.e. the number of SNPs in a specific motif divided by the number of mappings of this motif times its length).
| SNP Status | Motif Name | Motif Sequence | Length | Motif Hits | No. of SNPs | SNP Density | ProFITS Annotation |
|---|---|---|---|---|---|---|---|
| SNP poor | G-box promoter motif | CACGTG | 6 | 3,675 | 248 | 0.011 | Fos-related oncoproteins |
| TELO-box promoter motif | AAACCCTAA | 9 | 2,020 | 249 | 0.014 | Activation of expression in root primordia | |
| UP2ATMSD | AAACCCTA | 8 | 3,473 | 391 | 0.014 | Axillary bud outgrowth | |
| GADOWNAT | ACGTGTC | 7 | 2,073 | 232 | 0.016 | GA-down-regulated genes found in Arabidopsis seed germination | |
| ANAERO3CONSENSUS | TCATCAC | 7 | 2,735 | 316 | 0.017 | Anaerobically induced transcription | |
| AtMYC2 BS in RD22 | CACATG | 6 | 7,315 | 729 | 0.017 | Dehydration-responsive gene; ABA induction, water stress | |
| MYCATERD1 | CATGTG | 6 | 7,315 | 729 | 0.017 | Water stress | |
| SORLIP5 | GAGTGAG | 7 | 1,523 | 179 | 0.017 | Light-Induced transcription | |
| LTRE promoter motif | ACCGACA | 7 | 899 | 114 | 0.018 | Putative low-temperature-responsive element | |
| AtMYB2 BS in RD22 | CTAACCA | 7 | 2,081 | 266 | 0.018 | Dehydration-responsive gene; ABA induction, water stress | |
| SNP rich | TATA-box motif | TATAAA | 6 | 42,104 | 5,663 | 0.022 | Process of transcription by RNA polymerase |
| T-box promoter motif | ACTTTG | 6 | 12,852 | 1788 | 0.023 | Light-activated transcription | |
| ANAERO2CONSENSUS | AGCAGC | 6 | 2,554 | 369 | 0.024 | Anaerobically induced transcription | |
| ASF1MOTIFCAMV | TGACG | 5 | 13,173 | 1,606 | 0.024 | Involved in transcriptional activation of several genes by auxin and/or salicylic acid; may be relevant to light regulation | |
| Bellringer BS1 IN AG | AAATTAAA | 8 | 8,137 | 1,620 | 0.025 | Floral meristem, carpel, and stamen development | |
| RAV1-B binding site motif | CACCTG | 6 | 1,975 | 299 | 0.025 | Auxin- and salicylic acid-induced transcription; light-regulated transcription | |
| LEAFYATAG | CCAATGT | 7 | 1,623 | 290 | 0.026 | Floral meristem development | |
| LTRECOREATCOR15 | CCGAC | 5 | 8,606 | 1,207 | 0.028 | ABA, cold, and drought stress-induced transcription | |
| MYB2AT | TAACTG | 6 | 5,479 | 991 | 0.030 | Water stress | |
| Hexamer promoter motif | CCGTCG | 6 | 1,855 | 370 | 0.033 | Mersitem development, histone regulation |
Anticorrelation of SNP and Occurrence Frequency Profiles in Upstream Regions
We established that cis-element sites are generally characterized by lowered SNP frequencies. Next, we investigated for every individual known cis-element motif whether there is a systematic trend that sequence regions, in which a particular motif is preferentially found, also exhibit lower SNP frequencies for that motif.
For every known motif and for upstream sequence intervals of different lengths, we computed the correlation of relative occurrence counts and SNP frequencies across overlapping sequence windows of length 50 nt and step size 10 nt. For 30 of the 136 nonredundant known cis-element motifs, significant negative correlations of fractional occurrences and SNP densities were found. Of those, 15 motifs exhibited inverse anticorrelations such that lowered occurrence counts coincided with increased SNP frequencies (Supplemental Table S2; Supplemental Fig. S1). Even though this is consistent with our assumption that, where there is no motif, SNP frequencies can be elevated, no positive confirmation of a true positional preference was evident. Hence, those motifs have been dismissed from further analysis (for further comments on this point, see “Discussion”). The remaining 15 motifs with significant anticorrelation in at least one of three considered upstream regions of different length and evidence of a true positional preference are listed in Table IV. Statistical significance was achieved more often in the longer considered intervals, likely because, in the shorter interval (−100.0), counts are naturally lower and the statistical power weaker.
Table IV. Occurrence-SNP density correlations for known cis-element motifs.
Nonredundant, known cis-element motifs with significant negative Pearson correlation coefficients between their occurrence and SNP density profiles in at least one of the considered upstream sequence ranges and corresponding q values are shown. Correlation coefficients were calculated for the sequence intervals (−500, 0), (−300, 0), (−100, 0) with a sliding window of 50 nt at an increment of 10 nt. Significant values are highlighted in boldface.
| Motif Name | Sequence Interval (−100, 0) |
Sequence Interval (−300, 0) |
Sequence Interval (−500, 0) |
|||
|---|---|---|---|---|---|---|
| Correlation Coefficient | q | Correlation Coefficient | q | Correlation Coefficient | q | |
| ABRE-like binding site motif | +0.729 | 5.28e-01 | 0.073 | 1.00e+00 | −0.503 | 2.95e-03 |
| ACGTABREMOTIFA2OSEM | +0.542 | 8.48e-01 | −0.393 | 1.47e-01 | −0.589 | 3.05e-04 |
| Box II promoter motif | −0.918 | 3.04e-01 | −0.520 | 3.19e-02 | −0.371 | 4.77e-02 |
| CGCGBOXAT | −0.992 | 5.88e-02 | −0.833 | 1.73e-05 | −0.466 | 7.84e-03 |
| DRE-like promoter motif | −0.886 | 3.53e-01 | −0.487 | 5.02e-02 | −0.663 | 2.05e-05 |
| DREB1A/CBF3 | −0.690 | 5.73e-01 | −0.349 | 2.30e-01 | −0.549 | 8.50e-04 |
| GADOWNAT | −0.316 | 1.00e+00 | −0.655 | 3.23e-03 | −0.693 | 6.57e-06 |
| LTRE promoter motif | −0.771 | 4.84e-01 | −0.587 | 1.35e-02 | −0.392 | 3.42e-02 |
| SORLIP2 | −0.816 | 4.50e-01 | −0.868 | 2.87e-06 | −0.693 | 6.57e-06 |
| TATA-box motif | −0.942 | 2.81e-01 | −0.708 | 7.71e-04 | −0.606 | 1.62e-04 |
| TELO-box promoter motif | −0.915 | 3.04e-01 | −0.566 | 1.59e-02 | −0.342 | 7.05e-02 |
| TGA1 binding site motif | −0.940 | 2.81e-01 | −0.569 | 1.59e-02 | −0.502 | 2.95e-03 |
| UP1ATMSD | −0.870 | 3.57e-01 | −0.817 | 2.24e-05 | −0.628 | 8.04e-05 |
| UP2ATMSD | −0.851 | 4.08e-01 | −0.671 | 2.21e-03 | −0.442 | 1.36e-02 |
| Z-box promoter motif | −0.868 | 3.57e-01 | −0.688 | 1.38e-03 | −0.355 | 6.09e-02 |
Figure 3 illustrates the actual occurrence and SNP frequency profiles across the gene-upstream sequence regions for the 15 cis-element motifs with significant occurrence-SNP density anticorrelation. Despite relatively large fluctuations, particularly of SNP frequencies, the significant anticorrelation of occurrence and SNP frequency profiles is evident. Almost all of the 15 motifs were found to exhibit a positional preference peak within the first 250 nt upstream of the TSS. As only 56 of the 136 motifs were detected more than 1,000 times in Arabidopsis promoter sequences allowing robust statistics, a significant number of known motifs (28%) passed our motif verification approach using occurrence-SNP anticorrelations. Among those are the UP1ATMSD and UP2ATMSD motifs, which are related to axillary bud growth (Tatematsu et al., 2005), and elements that provide abscisic acid (ABA) responsiveness, such as ABRE-like, ACGTABREMOTIFA2OS, and GADOWNAT (Huang et al., 2008). Thus, for those 15 cis-element motifs, their positional interval, in which they can be considered functional, is supported both by elevated occurrences and reduced SNP frequencies.
Figure 3.

Occurrence-polymorphicity profiles for selected known cis-element motifs. Relative motif occurrence and SNP frequency profiles in the sequence interval from −750 nt to 250 nt relative to the TSS for the 16 TFBSs with significant anticorrelations (Table IV) are shown. A sliding window of 50 nt and an increment of 10 nt were used. In order to improve visual clarity, both statistics were scaled to the same mean value. Thus, their absolute value is meaningless.
Analysis of SNP Distributions within cis-Element Motifs
The available SNP density allows analyses at even finer positional resolution by examining the distribution of polymorphisms across the individual motif positions of the 136 nonredundant known cis-element motifs with detected mappings in the Col-0 genome. Specifically, our goal was to identify those motifs with pronounced SNP frequency differences across their motif sequences. Positions that are important for the recognition by the respective transcription factors may be more conserved and thus will exhibit a low SNP frequency, while variable sites (high SNP frequency) are either not important or may modulate motif function.
According to the specified criterion (see “Materials and Methods”), eight cis-element motifs were identified as displaying large and significant SNP frequency differences across their individual motif positions (Fig. 4). The positions with noticeably higher SNP frequency may be less essential for recognition by the respective transcription factor, whereas the ones that are more essential may be those with low SNP frequency. For example, the SNP frequency associated with the central position of the GAREAT motif is significantly increased relative to the surrounding positions. Indeed, mutation of the barley (Hordeum vulgare) GAREAT motif from TAACAAAC to TAACCAAC still provides GA responsiveness, supporting the notion that the central position is not essential (Gubler et al., 1999). Similarly, all other identified positions with increased SNP frequency may tolerate base changes without disrupting the motif-specific function.
Figure 4.

Position-specific SNP frequency profiles of the eight known cis-element motifs detected with nonuniform SNP frequency distribution (Eq. 5).
The available polymorphism information may also help to better define known motifs. Including not only the motif mappings in the Col-0 genome (i.e. “horizontal mappings”) but also “vertically” across all 350 Arabidopsis accessions may lead to more refined position-specific weight matrices (PWMs) as the identity of all alleles across all accessions is known. Indeed, inspecting the obtained sequence logos associated with the computed PWMs suggests alternative motif consensus sequences for two motifs, ANAERO5CONSENSUS and E2FAT, as the presence of alternative alleles at various motif positions across the considered Arabidopsis accessions is noticeable (Fig. 5). While the current definition sequence of ANAERO5CONSENSUS reads as TTCCCTGTT, the SNP-based consensus motif suggests TTSCCYSTT instead. Similarly, the E2FAT motif may be captured better by the consensus YYKCCKCC than by TYTCCCGCC.
Figure 5.

Sequence logos generated using the R package seqLogo based on the computed PWMs for the two motifs ANAERO5CONSENSUS and E2FAT. PWMs were computed from all detected mappings of the motifs in the Col-0 genome to the (−500, TSS) gene-upstream intervals considering also the aligned sequences associated with the other 349 Arabidopsis accessions. Information content is given in bits. The literature-derived consensus sequence associated with the motif is given in the header of each graph.
Besides the two discussed examples, the SNP-based PWMs and associated sequence logos did not deviate significantly from the known consensus motifs for any of the other remaining motifs (data not shown), even for the eight motifs with high SNP frequency contrast (Fig. 4). This result can be taken as confirmation that the consensus motifs are accurate for most motifs. However, both the SNP density and the frequency of alternative alleles across the 350 accessions may still be too low to substantially affect allele counts. Indeed, on average, only about 2% of all individual motif positions (across all mapping sites) were detected as polymorphic in the data set investigated here (Supplemental Table S1).
Identification of New Candidate cis-Element Hexamer Motifs
A central goal of this study was to identify novel candidate cis-regulatory motifs by finding sequence hexamers with reduced SNP frequencies and signs of positional preferences. In short (for details, see “Materials and Methods”), we computed for all 2,080 possible sequence hexamers, comprising 2,016 hexamer sequences representing both a forward and the associated reverse complement sequence treated as functionally identical and an additional set of 64 palindromic hexamers, how polymorphic they are in the 500 nt immediately upstream of the TSS relative to their background SNP density (Eq. 6) in the region farther upstream (−1,500 to −500 nt). The 10% of motifs with the largest drop in SNP density in the TSS-proximal regions relative to background were then filtered further to identify those that also exhibit a significant negative correlation between their SNP rates and their positional occurrences. Thus, we specifically looked for those hexamer motifs that show signs of positional preferences that are also supported by decreased SNP frequency at those preferred positions as an indication of functional relevance, as was done for known cis-element motifs above.
Of the 208 top-10% motifs with lowest SNP density relative to background, 71 motifs were found with significant negative correlations between their position frequency and SNP density in at least one of three considered upstream interval regions. Twenty-six of these candidate hexamers were identified as already included in the sequence definitions of known Arabidopsis cis-motifs (Supplemental Table S3). Twenty-eight candidate hexamers, even though displaying anticorrelation, were discarded because the correlation was due to the inverse of the desired amplitude changes, such that their SNP densities were found increased in those places where their relative occurrence was decreased. As in the case of known cis-element motifs (see above), those hexamer motifs were discarded for lack of clear-cut evidence of a confirmed positional preference (Supplemental Table S4; Supplemental Fig. S2). The 17 remaining candidate motifs are listed in Table V. The SNP frequency and positional preference profiles for the 17 novel candidate hexamer motifs are shown in Figure 6. A significant anticorrelation of occurrence and SNP frequency profiles for those candidate hexamers is evident. The preferred positional interval of those motifs appears to lie within 250 nt to the TSS. Especially hexamers 1, 2, 3, 5, 6, and 16 show a pronounced positional preference.
Table V. Candidate hexamer motifs with significant negative correlation of occurrences and SNP densities in at least one of the three considered sequence intervals with corresponding q < 0.05 (P value corrected for multiple testing).
Pearson correlation coefficients were calculated for the sequence intervals (−500, 0), (−300, 0), and (−100, 0) with a sliding window of 50 nt at an increment of 10 nt. Hexamers are ordered by their q values in the range (−300, 0), numbered consecutively, and significant values are highlighted in boldface.
| No. | Hexamer Sequence | Sequence Interval (−100, 0) |
Sequence Interval (−300, 0) |
Sequence Interval (−500, 0) |
|||
|---|---|---|---|---|---|---|---|
| Correlation Coefficient | q | Correlation Coefficient | q | Correlation Coefficient | q | ||
| 1 | ACCGGT | −0.932 | 1.97e-01 | −0.921 | 7.24e-09 | −0.772 | 3.79e-08 |
| 2 | AACCGG | −0.902 | 2.03e-01 | −0.842 | 4.59e-06 | −0.765 | 3.91e-08 |
| 3 | AAACCG | −0.712 | 3.97e-01 | −0.832 | 7.65e-06 | −0.880 | 3.83e-13 |
| 4 | CGGGTC | −0.977 | 9.36e-02 | −0.798 | 3.10e-05 | −0.716 | 8.50e-07 |
| 5 | AAGGCC | −0.993 | 5.20e-02 | −0.780 | 5.28e-05 | −0.472 | 4.74e-03 |
| 6 | GGCCTA | −0.865 | 2.24e-01 | −0.773 | 6.96e-05 | −0.505 | 2.11e-03 |
| 7 | AGGGTA | −0.102 | 9.33e-01 | −0.768 | 8.07e-05 | −0.661 | 1.06e-05 |
| 8 | CGACCC | −0.963 | 1.59e-01 | −0.749 | 1.36e-04 | −0.583 | 1.76e-04 |
| 9 | ACGGCC | 0.366 | 7.39e-01 | −0.743 | 1.69e-04 | −0.632 | 3.15e-05 |
| 10 | GGGACC | −0.746 | 3.78e-01 | −0.706 | 4.76e-04 | −0.549 | 5.75e-04 |
| 11 | GACCCA | 0.429 | 6.71e-01 | −0.697 | 6.23e-04 | −0.623 | 4.50e-05 |
| 12 | CGGTTC | −0.861 | 2.31e-01 | −0.694 | 6.63e-04 | −0.579 | 2.02e-04 |
| 13 | ACCCGA | −0.912 | 2.03e-01 | −0.651 | 1.99e-03 | −0.660 | 1.06e-05 |
| 14 | ACCGAA | −0.982 | 7.68e-02 | −0.634 | 2.92e-03 | −0.765 | 3.91e-08 |
| 15 | AACCCG | −0.898 | 2.03e-01 | −0.624 | 3.40e-03 | −0.373 | 3.18e-02 |
| 16 | ATAGAG | −0.897 | 2.03e-01 | −0.616 | 4.13e-03 | −0.606 | 7.75e-05 |
| 17 | AACGGC | 0.827 | 2.70e-01 | −0.592 | 6.49e-03 | 0.121 | 5.56e-01 |
Figure 6.

Relative motif occurrence and SNP frequency profiles in a sequence interval from −750 nt to 250 nt relative to the TSS for the 17 significantly anticorrelated candidate hexamers, which are not already contained in known TFBSs, and keeping the order from Table V. A sliding window of 50 nt at an increment of 10 nt was used. In order to improve visual clarity, both statistics were scaled to the same mean value, rendering their absolute values meaningless.
Because the individual hexamers may represent nested submotifs of an actually larger parent motif, the 17 hexamers were clustered hierarchically according to their sequence similarity using the Levenshtein edit distance to create consensus motifs (Fig. 7; see “Materials and Methods”). This approach resulted in five candidate motifs with the consensus sequences ATAGAG, ACCSRW, WMGGCC, GGGWCS, and CHGGKTH (Supplemental Table S5).
Figure 7.

Dendrogram of candidate hexamers and associated GO annotations. Levenshtein edit distances were calculated using a nucleotide substitution matrix (match = 0, mismatch = 1). The set of 17 motifs was subdivided into five distinct clusters with correspondingly created multiple sequence alignments (MSA) and visualized as sequence logos (Gupta et al., 2007) based on the observed distances and concordance of GO annotations (C, component; P, process; no function term turned out significant) of the respective downstream genes associated with every individual motif. Hexamer 17 was excluded from the alignment process due to the deviating GO term associations.
A GO term enrichment analysis was performed for the genes whose upstream sequences contain each individual hexamer compared with all other Arabidopsis genes. Hexamers 5, 6, and 9, all found in the same clustering subtree, indeed are likely to belong to the same candidate motif, as they are enriched in the upstream regions of genes that are coding for ribosome-associated components, functions, and processes. Because hexamer 17 was found to be associated with differing GO terms (plasmodesma, microtubule, and nucleosome), we decided to exclude it in the WMGGCC cluster. Positive functional associations were found for additional hexamer motifs (e.g. hexamers 2, 3, and 12 belonging to the CHGGKTH consensus cluster may potentially be chloroplast associated; Supplemental Table S6). Thus, at least for some of the novel motifs, a likely functional association can be postulated, providing evidence for their validity as well as making them interesting motifs for further follow-up studies.
We checked whether the novel motifs found in Arabidopsis may have matches or similarities to any cis-motifs described in other plant species or predicted in Arabidopsis based on alternative computational methods using the AtCOECiS database (Vandepoele et al., 2009; Supplemental Table S7). AtCOECiS includes a set of 866 candidate 8-mer Arabidopsis motifs derived from a computational prediction approach based on sequence conservation in orthologous Arabidopsis and poplar gene promoters and coexpression analysis in Arabidopsis (Vandepoele et al., 2009). Exact matches to longer motifs in other plant species, which themselves are not found in Arabidopsis gene promoters, were identified for motif 5 (−284MOTIFZMSBE1 motif in maize), motif 10 (NDEGMSAUR motif in soybean [Glycine max]), and motif 17 (IDE2HVIDS2 motif in barley). Motif 17 mapped to the GLUTAACAOS motif from rice (Oryza sativa) that was found in 10 Arabidopsis gene promoters as reported by AtCOECiS, but without coexpression evidence in Arabidopsis. Motif 10 was found to also map to the GGTCCCATGMSAUR motif reported in rice, with 212 incidences found in Arabidopsis promoter regions, three of which were also found to be coexpressed. Motif 17 also mapped to a computationally predicted candidate 8-mer Arabidopsis motif (AACGGCTA) with a supposed role in cell cycle regulation and a kinase regulatory activity. Thus, this motif was predicted before based on conservation in poplar and coexpression analysis and was discovered here again by following the presented SNP-based approach.
Using the TOMTOM tool (Gupta et al., 2007), we compared the candidate motifs with a large set of motifs from several nonplant species such as S. cerevisiae, Mus musculus, Drosophila melanogaster, and Homo sapiens. Although this comparison did not reveal any significant hits at q values less than 0.05, borderline significant and visually convincing motif hits were indeed found (Supplemental Table S8). Similarly, for most other TOMTOM hits and using the respectively annotated transcription factors binding to those motifs in the respective species as query sequences, orthologous transcription factor proteins in Arabidopsis were found (Supplemental Table S8). Thus, a number of lead sequences and their cognate transcription factors warranting follow-up studies may have been identified by our study.
Validation of Novel Motifs as Judged by Coexpression Regulation
As it is the defining feature of cis-regulatory elements to influence the expression of their respective downstream genes, the validity of a novel motif can be effectively judged by comparing the expression of genes harboring or not harboring the respective motif. For a functionally relevant motif, it can be expected that genes carrying it display positively correlated gene expression behavior. Indeed, for all but one of the 17 individual novel motifs, pairwise correlated coexpression was increased among genes harboring the respective motif (Table VI) compared with those transcripts whose genes do not harbor the motif in their promoter region. Only hexamer 16 (equivalent to consensus motif ATAGAG) did not show the expected increased coexpression. However, the probability of only one motif out of 17 showing the opposite trend (correlation is smaller in the motif set) is 1.4E-04, based on a binomial distribution with assumed 0.5 prior outcome probability (decrease versus increase). Thus, in addition to the significant differences per motif, the whole motif set proved statistically significant.
Table VI. Relevance of novel motifs for coexpression regulation.
Pairwise correlations as quantified by the Pearson correlation coefficients, r, of logarithmic, quantile-normalized ATH1 expression values associated with 5,295 NASCArray samples between transcripts of genes harboring a given motif compared with transcript pairs whose genes are not containing this motif in their promoter sequence up to where computed are shown. Here, promoter regions up to 250 nt upstream of the TSS were considered because of the characteristic peak locations (Figs. 3 and 6). Motifs are sorted by Cohen’s d (Eq. 7) score, measuring the difference of the mean correlation values relative to the average of the sd associated with the two gene pair sets (effect size; see “Materials and Methods”). Cohen’s d values greater than 5% are highlighted in boldface. For the consensus motif WMGGCC, the analysis was also performed considering only the 427 genes with the GO term component annotation “ribosome,” as the associated component sequences were found enriched in genes annotated to this compartment. Similarly, for the motifs CHGGKTH and WYSGGT, the analysis was performed considering only the 2,862 genes with GO term component “chloroplast” annotations, as well as the 1,086 mitochondrial genes for motif GGGWCS and the 3,391 GO term endomembrane system genes for motif ATAGAG (Fig. 7), with the respective GO terms indicating the corresponding compartment/organelle localization of the nucleus-encoded genes. For every motif, from all transcripts whose associated genes are not containing the respective motif, 15% were chosen at random to render the set sizes similar compared with the motif-containing set as well as to reduce computation time. In the case of the compartment-specific analyses, all alternative transcripts with the same component annotation were used, as the sets were smaller. Wilcoxon P values were obtained for the two respective r value distributions from the motif-containing or motif-not containing gene transcript pairs. *Computations performed for shuffled versions of the motif; **motifs with significant z scores (Eq. 8) relative to shuffled motif versions (P < 0.05; see “Materials and Methods”).
| Motif | Motif Contained in n Gene Promoter Regions (ATH1 Transcripts) | r, Motif Gene Set | r, Others | Wilcoxon P | Effect Size, Cohen’s d |
|---|---|---|---|---|---|
| 5 | 2,367 (1,678) | 0.0354 | 0.0023 | 0 | 0.186 |
| 6 | 1,847 (1,328) | 0.0343 | 0.0021 | 0 | 0.181 |
| 4 | 695 (494) | 0.0263 | 0.0027 | 0 | 0.135 |
| 2 | 2,628 (1,862) | 0.0242 | 0.0016 | 0 | 0.129 |
| 13 | 870 (588) | 0.0242 | 0.0026 | 0 | 0.125 |
| 8 | 661 (432) | 0.0205 | 0.0025 | 7.7E-181 | 0.103 |
| 1 | 1,212 (881) | 0.0207 | 0.0033 | 0 | 0.101 |
| 15 | 888 (620) | 0.0204 | 0.0028 | 0 | 0.101 |
| 9 | 521 (393) | 0.0204 | 0.0033 | 7.8E-73 | 0.098 |
| 12 | 2,148 (1,496) | 0.0189 | 0.0022 | 0 | 0.095 |
| 3 | 4,230 (2,906) | 0.0145 | 0.0014 | 0 | 0.073 |
| 14 | 3,005 (2,078) | 0.0123 | 0.0024 | 0 | 0.067 |
| 17 | 2,063 (1,357) | 0.0116 | 0.0022 | 0 | 0.054 |
| 10 | 907 (628) | 0.0090 | 0.0027 | 2.8E-04 | 0.036 |
| 7 | 1,313 (901) | 0.0076 | 0.0029 | 4.8E-50 | 0.027 |
| 11 | 2,126 (1,488) | 0.0054 | 0.0026 | 4.0E-02 | 0.015 |
| 16 | 3,561 (2,284) | 0.0006 | 0.0041 | 1.6E-176 | −0.020 |
| Consensus motifs | |||||
| WMGGCC | 5,278 (3,851) | 0.0309 | 0.0019 | 0 | 0.152 (0.027 ± 0.023)*,** |
| Ribosomal genes | 238 (192) | 0.4269 | 0.1739 | 0 | 0.781 (0.03 ± 0.31)*,** |
| CHGGKTH | 7,003 (4,573) | 0.011 | 0.002 | 0 | 0.053 (0.009 ± 0.015)*,** |
| Chloroplast genes | 697 (603) | 0.139 | 0.114 | 1.8E-273 | 0.081 (-0.013 ± 0.067)*,** |
| ACCSRW | 12,968 (8,517) | 0.006 | 0.0016 | 9.4E-195 | 0.025 (0.001 ± 0.016)* |
| Chloroplast genes | 1,246 (1,072) | 0.1387 | 0.1061 | 0 | 0.1044 (0.001 ± 0.08)* |
| GGGWCS | 3,099 (2,050) | 0.0050 | 0.0027 | 1.5E-02 | 0.013 (0.04 ± 0.03)* |
| Mitochondrial genes | 117 (94) | 0.0842 | 0.0542 | 3.7E-20 | 0.146 (0.09 ± 0.13)* |
| ATAGAG | 3,561 (2,347) | 0.0006 | 0.0034 | 6.8E-169 | −0.017 (-0.004 ± 0.007)* |
| Endomembrane system | 463 (284) | 0.0171 | 0.0084 | 1.3E-08 | 0.05 (0.006 ± 0.016)*,** |
| Fifteen known cis-element motifs (Fig. 3; Table IV) | |||||
| TELO-box promoter motif | 1,208 (756) | 0.0584 | 0.0027 | 0 | 0.322 |
| UP1ATMSD | 2,641 (2,073) | 0.0559 | 0.0021 | 0 | 0.290 |
| UP2ATMSD | 2,068 (1,279) | 0.0527 | 0.0023 | 0 | 0.284 |
| SORLIP2 | 4,995 (3,714) | 0.0345 | 0.0014 | 0 | 0.172 |
| DREB1A_CBF3 | 752 (482) | 0.0198 | 0.0030 | 5.5E-254 | 0.097 |
| GADOWNAT | 1,266 (995) | 0.0156 | 0.0026 | 0 | 0.075 |
| Z-box promoter motif | 242 (180) | 0.0148 | 0.0027 | 1.1E-16 | 0.069 |
| TGA1 binding site motif | 469 (356) | 0.0132 | 0.0027 | 1.4E-11 | 0.060 |
| DRE-like promoter motif | 1,274 (866) | 0.0126 | 0.0032 | 1.6E-221 | 0.054 |
| LTRECOREATCOR15 | 4,232 (2,808) | 0.0104 | 0.0019 | 0 | 0.048 |
| ACGTABREMOTIFA2OSEM | 2,252 (1,756) | 0.0119 | 0.0034 | 1.8E-166 | 0.047 |
| LTRE promoter motif | 494 (341) | 0.0105 | 0.0026 | 3.6E-27 | 0.046 |
| ABRE-like binding site motif | 2,972 (2,272) | 0.0098 | 0.0033 | 8.7E-144 | 0.036 |
| Box II promoter motif | 4,578 (3,015) | 0.0066 | 0.0024 | 2.5E-273 | 0.024 |
| TATA-box motif | 14,844 (9,606) | 0.0024 | 0.0057 | 2.2E-202 | −0.019 |
Because of the large numbers (N × [N − 1]/2 correlation values per set containing N genes), even small differences are judged significant, but they may not translate into differences of relevant magnitude. Thirteen motifs resulted in greater than 5% differences of the mean correlation coefficients relative to the average associated sd (effect size judged by Cohen’s d; see “Materials and Methods,” Eq. 7), with the largest relative difference obtained for hexamer motif 5 (17.6%). Similarly, for the five generated consensus motifs, all but one showed the desired increased pairwise correlation, with the motif WMGGCC producing the largest relative difference of 15.2% as judged by Cohen’s d.
For comparison, we also performed the coexpression analysis for the 15 known Arabidopsis cis-motifs exhibiting the same SNP density-positional preference anticorrelation profile on which the detection of novel hexamer motifs was based (Fig. 3; Table IV). Nine of the 15 motifs were found with greater than 5% Cohen’s d effect size difference of correlated gene expression behavior within gene sets containing them versus sets not containing them (Table VI). Fourteen known motifs resulted in positive Cohen’s d values (i.e. confirmed the expected positive effect on an increased correlated gene expression). For the TATA box only, a small negative effect was measured, possibly reflecting its function as a general rather than specific promoter element (Roeder, 1996). Four known motifs (TELO-box, UP1ATMSD, UP2ATMSD, and SORLIP2) were detected with greater than 10% Cohen’s d effect size difference. Thus, even though the expression-based evidence for the known motifs was stronger than for our consensus motifs (one consensus motif [WMGGCC] found with greater than 10% effect size), the effect sizes associated with the individual hexamer motifs, in particular, were observed to fall into a similar range (Cohen’s d of approximately 10%) as for known motifs (Table VI).
The component sequences making up all five generated consensus sequences were partially found to be associated with particular cellular compartments, as judged by GO component term annotations (Fig. 7). Thus, the found motifs may act as specific expression modulators of genes associated with those compartments. For every consensus motif, we repeated the correlation analysis, but considering only genes carrying the specific component annotation. For all motifs, a significant increase of coexpression signal was detected (Table VI). Evidence to act as a component-specific signal was particularly strong (Cohen’s d = 78.1%) for consensus motif WMGGCC, whose member sequences suggest a ribosome-associated role (Fig. 7). Further inspecting the specific GO terms associated with those genes associated with the general term “ribosome,” we found that especially genes annotated as “cytosolic large subunit” when using a more detailed GO annotation level were found enriched among the gene set carrying this consensus motif (P = 3.5E-04, Fisher’s exact test).
Because of its short length and ambiguous sequence (three ambiguity codes), consensus motif ACCSRW was found in very many (12,968) promoter regions. Unless this motifs acts as a constitutive and general core promoter element, this large number of influenced genes may be unreasonable. Nonetheless, pairwise correlation among the 12,968 genes (8,517 transcripts covered on the ATH1 chip) is higher than among the remaining genes (Cohen’s d = 2.5%). From the GO enrichment analysis (Fig. 7), it was established that the constituent hexamer motifs associated with this consensus motif were preferentially associated with chloroplast-targeted Arabidopsis genes. Therefore, this motif may exert its function in particular in genes targeted toward the chloroplast. Indeed, by only considering the 2,616 annotated chloroplast genes and comparing the average pairwise correlation levels of the 975 genes with this particular consensus motif with the remaining chloroplast genes, the magnitude of correlation difference increased to 10%.
Similarly, for consensus motif CHGGKTH, whose member hexamer sequences also suggest a chloroplast association, the magnitude of coexpression increased to 8.1% in the chloroplast set compared with 5.5% when considering all genes. Likewise, for motif GGGWCS, with a possible mitochondrial role, Cohen’s d increased from 1.3% to 14.6% when only considering mitochondrial genes. Even for the motif ATAGAG, which resulted in a negative Cohen’s d when considering all genes, it was found associated with a positive score of 5% when considering only genes associated with the endomembrane system.
To account for possible nucleotide composition biases, the coexpression analyses were repeated for all possible permutations of the five reported consensus motifs (Table VI, with Cohen’s d in parentheses). For three consensus motifs and compartment restrictions resulting in effect sizes greater than 5%, the actual effect size was significantly larger than for the set of exhaustively shuffled motif versions. Only for consensus motif GGGWCS and its hypothesized association with mitochondria and for motif ACCSRW, with a predicted chloroplast-related location and function, significance could not be established, despite the affirmative difference of the mean d values. Thus, compositional bias can be excluded as the source of the found positive association of motif presence and increased correlated gene expression.
Note that the compartment-specific annotation was applied to both the motif containing as well as not containing genes. Thus, the increase in correlation is not merely an effect of compartment association but an indication of the specific role of the tested novel motifs.
Thus, functional relevance was established for essentially all identified novel motifs based on an analysis of coexpression. In particular, the consensus motif WMGGCC, with ribosome-associated constituent hexamer motifs, was found associated with a pronounced correlated expression of genes harboring this motif, in particular those of the large cytosolic subunit. Furthermore, this analysis also suggests a chloroplast-specific function of the consensus motifs ACCSRW and CHGGKTH and a mitochondria-associated function for the consensus motif GGGWCS.
DISCUSSION
Enabled by novel sequencing technologies, the goal of comprehensively identifying all genetic variations in a population at affordable costs and at nucleotide-level resolution is within reach. The promise of this deep genetic variant information has been seen primarily in the context of identifying genetic causes for phenotypic variations via association studies (Hirschhorn and Daly, 2005). Based on the already available large SNP data sets for selected species, tools and services are being developed that facilitate such association studies also for the plant model species Arabidopsis (Childs et al., 2012; Grimm et al., 2012; Seren et al., 2012). Here, we aimed at exploiting the available SNP data obtained from the Arabidopsis 1,001-genomes project with the different objective to profile known cis-element motifs in gene promoter sequences and to identify novel motifs that may act as cis-regulatory elements. The assumption that functional motifs are more conserved than nonfunctional sequences formed the rationale of this study. We found that, indeed, known cis-element motifs harbor fewer SNPs than their supposedly nonfunctional sequence counterparts. We showed that SNP density profiles allowed providing an effective average length estimate of 500 nt for functional gene promoters in Arabidopsis and that they can be used to identify preferred motif locations. Most importantly, our analysis yielded 17 novel Arabidopsis hexamer motifs, collapsing into five unique candidate promoter elements that await experimental verification. Of the 17 individual motifs, three are supported by similar motifs in other plant species, and one confirms an earlier computational prediction employing an alternative approach. Given that the current set of known Arabidopsis motifs comprises 144 nonredundant elements, potentially adding five (or up to 17 individual hexamer motifs) can be considered significant. The validity of the found motifs has been verified by testing for positive coexpression of the downstream genes (Table VI). Based on the expression results as well as on functional annotation for the respective downstream genes, for some of the novel motifs a specific functional association can be postulated, providing evidence for their validity as well as making them interesting candidates for experimental follow-up studies. Most noticeably, consensus motif WMGGCC was found to be specifically and very strongly associated with the large cytosolic subunit proteins of ribosomes and not with other ribosomal genes (e.g. chloroplast targeted). To our knowledge, there are currently only two motifs reported to be specifically associated with ribosomal gene expression regulation (TELO-box AAACCCTAA and site II motif TGGGCC/T; Trémousaygue et al., 2003; McIntosh and Bonham-Smith, 2005; McIntosh et al., 2011). Thus, the found consensus motif WMGGCC, which bears some resemblance to the site II motif, may constitute an important addition to the set of ribosomal gene regulators and may open novel avenues for ribosomal gene expression regulation research.
Critical for the success of this study, we had to assume that the density of SNPs is high enough already to warrant the characterization of and search for motifs. Even though the density of frequent SNPs was estimated at only 0.0187 SNPs per nt in gene promoter sequences, we effectively increased the SNP density by searching for polymorphisms not only at a single locus but by inspecting all mapping locations of a given motif across all gene promoters, irrespective of the identity and function of the associated gene, in whose upstream sequence regions the motif was found. While it can be assumed that the function of genes harboring the same upstream motif is similar, this need not necessarily be the case, given also the chance of false-positive motif mappings. However, as there is no immediate criterion by which to distinguish true from false mappings, we tolerated the chance of false-positive mappings in favor of increased statistical support. In addition, by limiting the analysis to the 500 nt upstream, which we showed here to be most conserved, the number of false-positive mappings can be expected to be effectively reduced.
As a second step, we then searched vertically across all Arabidopsis accessions at each location. Thus, we assumed that motif positions are conserved. It is possible, however, that insertions/deletions may have led to a change of motif position relative to the TSS in other Arabidopsis accessions. Furthermore, cis-elements may get lost altogether in evolution and be replaced by novel motifs, a process referred to as “turnover,” which has been described to occur at appreciable frequencies across different eukaryotic genomes (Dermitzakis and Clark, 2002). Thus, a strict vertical sequence comparison across different Arabidopsis accessions may have resulted in false SNP frequencies in those cases. However, as the recognition of cis-elements by transcription factors was shown to tolerate substantial changes despite their short length (Spivakov et al., 2012), deciding whether a motif is effectively lost or has moved to a new location by bioinformatics means alone is challenging. The large tolerance with respect to nucleotide changes may also explain the small absolute difference of SNP density within cis-element sites compared with non-cis-element sites found here. Despite being very significant, the absolute difference was found to be small (0.0167 SNPs per nt for cis-element sites versus 0.0187 SNPs per nt for non-cis-element sites).
On the latter point of the seemingly small difference in SNP density within and outside cis-element motifs (11% difference), one has to bear in mind that the genomes associated with the accessions used here are closely related by ancestry and diverged only relatively recently in evolution. Therefore, the overwhelming number of sites can be expected to be unchanged. Once available, a large set of more divergent species/ecotypes should be used to confirm the findings reported here. Furthermore, promoter regions are constrained by factors other than transcription factor binding alone. For example, it was shown that promoter regions possess special structural properties (melting temperature, curvature, bendability, and stability) that are conserved in evolution (Kanhere and Bansal, 2005) and that have been shown to influence gene expression, for example, via determining nucleosome occupancies of promoter regions (Miele et al., 2008). Thus, promoter regions are, in general, more conserved (Fig. 2), likely explaining also the only small difference in SNP density within and outside motifs. Furthermore, SNPs, by definition, result in changes that are tolerated; otherwise, the polymorphism and associated carrier plants would have been lost. Nonetheless, preservation of function is most easily achieved, arguing teleologically, by sequence conservation once a working motif has been “invented” by nature. Indeed, even though SNP effects can be expected to be largely neutral, a significantly lowered SNP density was found in known motifs (Table II).
While there certainly is the risk of including false-positive motif mappings, within a single accession and across multiple accessions, adding noise to the SNP statistic, no systematic influence should be expected. Furthermore, pronounced and statistically significant motif position-specific SNP frequency differences were found (Fig. 3), suggesting that true signals have been detected rather than random associations alone.
Even though higher SNP numbers would have been desirable for the purpose of this study, we still decided to inspect frequent SNPs only (i.e. those with minor allele frequency greater than 5% across the 350 accessions). This effectively reduced the rate of false-positive SNPs and furthermore included only those SNPs that got fixed in several Arabidopsis accessions, as they are either tolerated or may even lead to beneficial adaptive changes in the respective ecotypes, thus associating SNPs with functional adaptation processes to changing environments and rendering the results more biologically meaningful. As a test for robustness of our findings, we computed the SNP density difference within versus outside cis-motifs under the threshold criterion that a position is called polymorphic if at least a single accession shows an alternative allele relative to all others at this position. As expected, the SNP count increases substantially (to 1.020 million, with 322,000 polymorphic sites supported only by a single accession deviating from all others, in comparison with the 260,000 polymorphic sites for the 5% threshold used in the study). With regard to cis-motif SNP density, the difference amounted to a 7.4% reduced SNP density (P = 1.6E-173) within motifs relative to outside regions when the single accession difference was applied. Thus, qualitatively, the same result of reduced diversity within motifs was obtained.
A number of analyses presented here, such as the position/conservation correlation results, required the location of the TSS to be known and assumed that only a single TSS exists per gene. However, determining the exact location of the TSS is notoriously difficult and often simply corresponds to the 5′-most sequence ever detected for a particular gene transcript. Also, alternative TSSs have been described for many Arabidopsis genes (Alexandrov et al., 2006). Nonetheless, we found a pronounced SNP frequency drop right at the site of the surmised TSS (Fig. 2), indicating that, while some TSS assignments may be wrong and multiple TSSs would have to be considered, in the statistical sense, the correct TSS position was used most of the time.
Notwithstanding the uncertainty with regard to the TSS location, the SNP density profile as a function of distance from the assumed TSS resulted in an estimate of 500 nt for an effective average promoter length (Fig. 2). Individual promoter lengths will vary, and cis-elements will also be found functional farther away from the TSS; nonetheless, this effective promoter length estimate may be useful in large-scale screens for novel cis-elements in Arabidopsis.
Our result of lower SNP density immediately upstream of the TSS (Fig. 2) stands in contradiction to results obtained in human (Guo and Jamison, 2005), where an increased SNP density near the TSS compared with regions farther upstream was found. Furthermore, those authors reported increased variability in TFBSs compared with nonbinding sites. However, our results are in agreement with the basic assumption made in genomic research in that functional elements are conserved. Furthermore, as intragenic regions are conserved also in human, the results reported by Guo and Jamison (2005) would generate a sharp discontinuity near the TSS. By contrast, our SNP profiles smoothly connect intergenic and intragenic regions with regard to SNP density, rendering our results biologically plausible. Thus, resolving this discrepancy may require another independent investigation.
We demonstrated that the available SNP information may help to identify the locations in which motifs are truly functional as those where elevated mapping frequencies coincide with reduced SNP frequencies. Indeed, for 15 of the 136 known nonredundant cis-element motifs (57 with counts greater than 1,000), such significant anticorrelations were found. While this can be seen as a powerful additional confirmation of the validity of those motifs, the fact that many motifs did not noticeably follow the expected behavior may raise concerns. Either the exact location does not matter for those motifs, or the SNP density proved insufficient to reveal a significant anticorrelation. Alternatively, for some motifs, the high motif variability may actually reflect evolved adaptive changes, and thus high SNP densities may be “desired” rather than avoided. Thus, the question of SNP-inferred location preferences needs to be revisited once an even larger SNP set is available. Furthermore, our approach will also produce false negatives. In fact, upon visual inspection of the rejected 15 motifs with significant anticorrelation, a number of them may have been considered confirmed had the decision been based on a different verification scheme. For example, focusing more on local optima rather than on global correlations may have resulted in more positive confirmations (Supplemental Fig. S1). Similarly, the true set of novel hexamer motifs may be larger than reported here. However, because of the very short length of the candidate motifs (6 nt), a double-evidence-based identification scheme seemed indicated to reduce the number of false positives.
CONCLUSION
Our study demonstrates the potential of polymorphism information in the context of characterizing and identifying functional genomic elements. The quantity of sequence and associated SNP information proves large enough already to permit meaningful studies on motif characterization and allows detecting novel motifs, at least for those species for which broad sequencing projects have been initiated, such as Arabidopsis and human. Similarly, it can also be applied to all other types of regulatory sequence motifs, such as enhancer elements, whose successful identification based also on polymorphism information has been demonstrated before (Dyer and Rosenberg, 2000). As the number of detected polymorphisms will increase at a rapid pace and many more species will likely be screened at similar precision and depth, the approach pursued here can be expected to provide a fruitful avenue for identifying novel regulatory motifs and to help contribute to a comprehensive cataloging and understanding of the regulatory code in the genomes of living organisms.
MATERIALS AND METHODS
Genomic Sequence Information
Nuclear genome sequence, gene mapping position, and annotation information of Arabidopsis (Arabidopsis thaliana), Col-0 ecotype, was downloaded from The Arabidopsis Information Resource (TAIR), version 10 (http://www.arabidopsis.org; Rhee et al., 2003). Mitochondria- and chloroplast-encoded genes and associated intergenic regions, cis-elements, etc., were not considered. Gene-upstream regions were defined as the 3,000 nt 5′ upstream of every gene considering also the respective strand assignment (i.e. sequences were reverse complemented and relative sequence positions were adjusted if a gene was found to map on the opposite strand relative to the sequence strand deposited in TAIR). Upstream regions were truncated at the 3′ terminus of the next upstream gene (or 5′ start site, if the upstream gene was found on the opposite strand). Furthermore, upstream regions were required to be at least 10 nt long. Thus, regions of overlapping genes and upstream sequences were excluded. For comparison, intragenic regions of up to 500 nt in the 3′ direction of the TSS were also considered. In total, 32,507 upstream regions were considered for SNP and motif analyses. Note that upstream regions may overlap in cases of head-to-head orientations of neighboring genes. Then, the corresponding regions were analyzed twice, but considering the orientation relative to the associated gene. TSSs were taken as annotated in TAIR (i.e. the 5′-most position of each gene model was taken as the TSS).
SNP Data Set
SNP information was obtained from the released Arabidopsis 1,001-genomes project data (http://www.1001genomes.org/; Cao et al., 2011; Schneeberger et al., 2011; Long et al., 2012; Schmitz et al., 2012). As of April 2012, over 8.5 million polymorphic sites corresponding to SNPs in other accessions relative to the reference Col-0 accession were identified in Arabidopsis based on genomic sequence information for 349 accessions (350 accessions including Col-0). To exclude possible sequencing errors and to only include SNPs with appreciable frequency across different Arabidopsis accessions, only those polymorphic sites with minor allele frequency of greater than 5% were included in the analysis. A total of 2.4 million SNPs contained in the initial set were excluded, as they did not pass this frequency threshold criterion. Ambiguous base calls (e.g. “N” [leading to the exclusion of 250,000 SNPs]) as well as insertions and deletions were not considered. In total, 1,080,084 SNPs were located in the scanned sequence position range from −3,000 to +500 nt relative to the TSS. The majority, 846,164, of those SNPs were found in the intergenic regions. Note that we refer to polymorphic sites and SNPs interchangeably (i.e. the term SNP is also used to refer to a polymorphic site throughout this article). Note that throughout this paper, the terms “SNP frequency” and “SNP density” are used interchangeably and refer to the frequency of polymorphic sites in a given interval of genomic sequence. The minor allele frequency threshold is kept constant at 5% with one exception (1%), to test for the effect of minor allele frequency on the reported statistic.
GO Term Enrichment Analysis
GO annotations (GO-slim and the detailed GO terms) for all Arabidopsis genes were retrieved from TAIR, comprising 15 (377) GO-slim (GO-detailed) terms for the categories cellular component, 16 (1,027) terms for molecular function, and 13 (2,128) terms for biological process. GO term enrichment or depletion in a gene set relative to another was tested by applying Fisher’s exact test with subsequent multiple testing correction applied to the obtained P values (Benjamini and Hochberg, 1995).
Mapping of Known Arabidopsis cis-Element Motifs
Known transcription binding site (cis-element) motifs were downloaded from three public databases, AGRIS (Davuluri et al., 2003), Athena (O’Connor et al., 2005), and PLACE (Higo et al., 1998). Based on all motifs derived from these three resources, a nonredundant motif set was generated by eliminating redundant motifs as defined by identical sequences. Motifs with the same name but different sequences were kept as alternative versions of the same motif but given a version number, such as v2, v3, etc. In total, 144 nonredundant motifs of length ranging from five to 49 nucleotides were considered in the analysis (Supplemental Table S1).
Sequence regions from −3,000 to +500 nt relative to the TSS for all considered upstream and intragenic regions and using the sequence of the Col-0 ecotype of Arabidopsis were scanned for those motifs via exact match identification in forward- and reverse-complement orientation. In cases where motif definitions contained ambiguity codes, thus allowing several bases at a particular position, the respective alternative bases were tolerated. Thus, the most stringent mapping criterion possible (exact match) was used.
Correlation Analysis of Motif Position and SNP Frequency
In order to detect positional preferences of motifs in upstream regions that are furthermore characterized by reduced SNP frequencies, relative motif occurrences and SNP densities were correlated as a function of distance from the TSS. The relative occurrence, Pm,i, of a motif m was calculated as the frequency count of that motif at a specific positional interval i divided by the sum of the motif counts over all valid positional intervals i = 1,...,n. To account for different interval counts due to truncated promoters, this relative frequency was normalized by the interval frequency count I at a specific positional interval i divided by the sum of the interval frequency counts over all intervals i = 1,...,n:
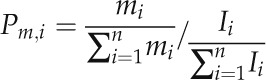 |
(1) |
The normalized SNP density per positional interval, SDm,i, was computed as the number of SNPs, S, in a specific motif m at the given positional interval I divided by its occurrence M in that interval multiplied by its length L such that:
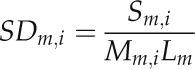 |
(2) |
The number of SNPs was determined based on all found mappings of a particular motif at the considered positional interval across all gene promoter sequences. Positional intervals were treated as sliding windows of length 50 nt and a step size (positional increment) of 10 nt (i.e. the windows were overlapping). The Pearson correlation coefficient, r, between the Pm and SDm measures was calculated for three different sequence intervals, (−500, 0), (−300, 0), and (−100, 0), upstream relative to the TSS. Motifs with associated r ≤ −0.5 and associated q < 0.05, with q values corresponding to the false discovery rate-corrected P values (Benjamini and Hochberg, 1995), were considered to exhibit a significant position/SNP density anticorrelation. We operated under the assumption that valid novel motifs are signified by pronounced positional preferences and simultaneous decreases of SNP frequency. Therefore, motifs with lowered positional and simultaneously increased SNP frequency counts were eliminated despite the detected anticorrelation. The total number of tests for known motifs was 136 and 208 for novel candidate hexamers, respectively.
SNP Frequency Distributions within cis-Elements
The position-specific SNP frequency, SFp,m, across all sequence positions in a single cis-element motif was calculated by determining how often a particular position p in a motif m of length n was found to be polymorphic across all its mappings to Arabidopsis promoter regions (interval from −500 nt to the TSS), resulting in the count Sp,m for every motif position p and dividing it by the total count for all sequence positions P = 1,...,n such that:
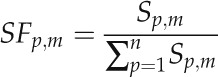 |
(3) |
To obtain reliable results, only motifs with an average Sp,m of 10 or greater per motif position compiled from all identified motif mappings were considered. sd values for the relative position-specific counts SFp,m, were estimated according to a Poisson distribution assumed for the individual position counts as:
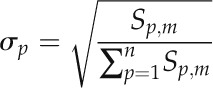 |
(4) |
Large-SNP-contrast motifs were identified as those with large differences between their individual position-specific relative frequencies defined as:
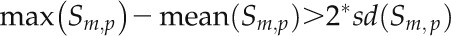 |
(5) |
where sd refers to the sd of the SNP frequency across all motif positions (i.e. motifs containing positions [“hot spots”] with considerably increased SNP frequencies relative to other positions in the motif).
Novel Hexamer Motifs
Potentially novel cis-regulatory motifs were identified by detecting hexameric sequence motifs exhibiting decreased SNP frequencies correlated with increased position-specific frequency (i.e. motifs with pronounced and SNP-supported positional preferences followed by a motif clustering based on sequence and annotation information). All analyses were confined to the 500 nt upstream of the TSS (i.e. the putative gene promoter regions).
Assuming that forward- and reverse-complement motifs are functionally equivalent, we collapsed the set of all 4,096 different hexamers to a set of 2,080 hexamers, which is composed of 2,016 hexamer pairs including their forward and reverse complements and 64 palindromic hexamers. For all 2,080 hexamers, SNP densities were calculated as defined by Equation 2 for two sequence region intervals, one representing the background distribution and ranging from −1,500 to −500 nt (referred to as poly1500) and one consisting of −500 to 0 nt (poly500) relative to the TSS, representing the promoter regions in which to find novel candidate motifs. Ratios of poly500 to poly1500 SNP densities were computed to detect motifs with increased conservation levels near the TSS. Positional-preference correlation analysis was performed to further filter the set of candidate motifs for those that exhibit a position-conservation correlation pattern as explained above.
Candidate hexamer motifs mapping exactly (considering also the respective ambiguity assignments) to the set of 144 known Arabidopsis motifs were removed. Likewise, motifs with known motifs of length five or shorter mapping to them were discarded as well. Trivial motif hits to six or longer consecutive segments of ambiguous bases as contained in eight known motifs were not considered as valid matches and the associated candidate hexamer motifs retained (e.g. hits to VOZATVPP, CCNNNNNNNNNNNNCCACG, or the AGATCONSENSUS motif, TTWCCWWWWNNGGWW).
The remaining candidate hexamer motifs were clustered based on the pairwise Levenshtein edit distance applying an exchange score (0 = match, 1 = mismatch). Based on the distances between groups as well as the concordance of the GO annotations of the respective downstream genes associated with every individual motif, the individual motifs were clustered together. Candidate motifs belonging to the same cluster were aligned to construct a consensus motif using ClustalW2 (Thompson et al., 2002). The TOMTOM tool of the MEME Suite (http://meme.nbcr.net/meme/cgi-bin/tomtom.cgi; Gupta et al., 2007) was used to test for the novelty of candidate hexamer motifs relative to a large set of motifs from many species comprising 476 cis-element motifs contained in the JASPAR (Bryne et al., 2008) and 386 cis-elements contained in the UNIprobe (Newburger and Bulyk, 2009) databases. Transcription factors annotated to bind to the TOMTOM motifs in the respective species identified to be similar to the candidate Arabidopsis motifs were searched for orthologs in Arabidopsis using the BLAST server at TAIR.
Gene Expression Analysis
Gene expression information in Arabidopsis across a large number of sample conditions was obtained from NASCArray (Craigon et al., 2004). In total, 5,295 ATH1-Affymetrix gene chip data sets were downloaded as a precompiled data table, log-transformed, and jointly normalized applying a quantile normalization using the “normalize.quantile” routine from the “preprocessCore” R package. Evidence of correlated expression behavior was assessed by computing the pairwise Pearson correlation coefficient, r, of all 5,295 quantile-normalized expression values associated with two genes drawn from a set. Only the upper triangle of the correlation matrix was used, and self-correlations (diagonal elements) were excluded. For every candidate motif, all genes were partitioned into a set of genes harboring the motif and compared with the set of genes not harboring the respective motif. The significance of the difference of the coexpression patterns within the two gene sets was judged by the respective average intraset correlation coefficients and by applying the nonparametric Wilcoxon rank-sum test of the two distributions of r values. The magnitude of the obtained differences was assessed by comparing the obtained difference in mean r values with the mean sd associated with the two distributions by computing the Cohen’s d (Cooper and Hedges, 1994):
 |
(7) |
where r is the Pearson correlation coefficient between two gene transcripts belonging to the same set, σ is the respective sd associated with the two sets, n1 and n2 are the respective set sizes, and < …> denotes the average. In effect, the Cohen’s d expresses the observed differences of mean values as a fraction relative to the associated sd. Thus, d = 1 would indicate that the difference of the means is as large as the average sd for both sets.
For the five detected consensus motifs, the correlation analysis was repeated for shuffled motif versions to account for possible nucleotide composition biases. For each motif, all possible permutations (forward and reverse complements) were created followed by determining, for every shuffled motif version individually, all genes harboring it. Subsequently, the two resulting gene sets (shuffled motif containing and not containing) were tested for within-set coexpression as described above and repeated for every motif permutation. Based on the obtained mean and sd for Cohen’s d associated with the shuffled motifs, a z score was computed to quantify whether the Cohen’s d for the actual motif is significantly different compared with shuffled motif versions. z scores were computed as:
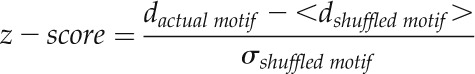 |
(8) |
Absolute z scores greater than 1.96, equivalent to P < 0.05 assuming normal distribution, were marked as significant.
Supplemental Data
The following materials are available in the online version of this article.
Supplemental Figure S1. SNP-density and location profiles for excluded known motifs.
Supplemental Figure S2. SNP-density and location profiles for excluded hexamer motifs.
Supplemental Table S1. Complete list of known cis-element motifs.
Supplemental Table S2. Excluded Known cis-element motifs with significant location and SNP profiles.
Supplemental Table S3. Comparison of known and hexamer motifs.
Supplemental Table S4. Excluded hexamer motifs.
Supplemental Table S5. Multiple sequence alignment of hexamer motifs.
Supplemental Table S6. GO-term annotations of genes downstream of candidate motifs.
Supplemental Table S7. Matches of candidate motifs with literature-described motifs.
Supplemental Table S8. TOMTOM-hits for consensus motifs.
Acknowledgments
We thank Liam Childs for providing the preprocessed Arabidopsis SNP information files and Christoph Thieme for providing a helpful test statistic.
Glossary
- TFBS
transcription factor-binding site
- nt
nucleotides
- SNP
single-nucleotide polymorphism
- TSS
transcription start site
- GO
Gene Ontology
- Col-0
Columbia-0
- ABA
abscisic acid
- PWMs
position-specific weight matrices
- TAIR
The Arabidopsis Information Resource
References
- Alexandrov NN, Troukhan ME, Brover VV, Tatarinova T, Flavell RB, Feldmann KA. (2006) Features of Arabidopsis genes and genome discovered using full-length cDNAs. Plant Mol Biol 60: 69–85 [DOI] [PubMed] [Google Scholar]
- Bailey TL, Boden M, Buske FA, Frith M, Grant CE, Clementi L, Ren J, Li WW, Noble WS. (2009) MEME SUITE: tools for motif discovery and searching. Nucleic Acids Res 37: W202–W208 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benjamini Y, Hochberg Y. (1995) Controlling the false discovery rate: a practical and powerful approach to multiple testing. J R Stat Soc B 57: 289–300 [Google Scholar]
- Blanchette M, Tompa M. (2002) Discovery of regulatory elements by a computational method for phylogenetic footprinting. Genome Res 12: 739–748 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blanchette M, Tompa M. (2003) FootPrinter: a program designed for phylogenetic footprinting. Nucleic Acids Res 31: 3840–3842 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bronner C, Achour M, Chataigneau T, Schini-Kerth VB. (2011) Epigenetic control of gene transcription. In A Giordano, M Mascaluso, eds, Cancer Epigenetics. John Wiley & Sons, New York, pp 57–99 [Google Scholar]
- Bryne JC, Valen E, Tang MH, Marstrand T, Winther O, da Piedade I, Krogh A, Lenhard B, Sandelin A. (2008) JASPAR, the open access database of transcription factor-binding profiles: new content and tools in the 2008 update. Nucleic Acids Res 36: D102–D106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bussemaker HJ, Li H, Siggia ED. (2001) Regulatory element detection using correlation with expression. Nat Genet 27: 167–171 [DOI] [PubMed] [Google Scholar]
- Cao J, Schneeberger K, Ossowski S, Günther T, Bender S, Fitz J, Koenig D, Lanz C, Stegle O, Lippert C, et al. (2011) Whole-genome sequencing of multiple Arabidopsis thaliana populations. Nat Genet 43: 956–963 [DOI] [PubMed] [Google Scholar]
- Chekulaeva M, Filipowicz W. (2009) Mechanisms of miRNA-mediated post-transcriptional regulation in animal cells. Curr Opin Cell Biol 21: 452–460 [DOI] [PubMed] [Google Scholar]
- Childs LH, Lisec J, Walther D. (2012) Matapax: an online high-throughput genome-wide association study pipeline. Plant Physiol 158: 1534–1541 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Childs LH, Witucka-Wall H, Günther T, Sulpice R, Korff MV, Stitt M, Walther D, Schmid KJ, Altmann T. (2010) Single feature polymorphism (SFP)-based selective sweep identification and association mapping of growth-related metabolic traits in Arabidopsis thaliana. BMC Genomics 11: 188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooper H, Hedges LV. (1994) The Handbook of Research Synthesis and Meta-Analysis. Russell Sage Foundation, New York [Google Scholar]
- Craigon DJ, James N, Okyere J, Higgins J, Jotham J, May S. (2004) NASCArrays: a repository for microarray data generated by NASC’s transcriptomics service. Nucleic Acids Res 32: D575–D577 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Das MK, Dai HK. (2007) A survey of DNA motif finding algorithms. BMC Bioinformatics (Suppl 7) 8: S21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davuluri RV, Sun H, Palaniswamy SK, Matthews N, Molina C, Kurtz M, Grotewold E. (2003) AGRIS: Arabidopsis Gene Regulatory Information Server, an information resource of Arabidopsis cis-regulatory elements and transcription factors. BMC Bioinformatics 4: 25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dermitzakis ET, Clark AG. (2002) Evolution of transcription factor binding sites in mammalian gene regulatory regions: conservation and turnover. Mol Biol Evol 19: 1114–1121 [DOI] [PubMed] [Google Scholar]
- D’Haeseleer P. (2006) How does DNA sequence motif discovery work? Nat Biotechnol 24: 959–961 [DOI] [PubMed] [Google Scholar]
- Dyer KD, Rosenberg HF. (2000) Shared features of transcription: mutational analysis of the eosinophil/basophil Charcot-Leyden crystal protein gene promoter. J Leukoc Biol 67: 691–698 [DOI] [PubMed] [Google Scholar]
- Filipowicz W, Bhattacharyya SN, Sonenberg N. (2008) Mechanisms of post-transcriptional regulation by microRNAs: are the answers in sight? Nat Rev Genet 9: 102–114 [DOI] [PubMed] [Google Scholar]
- Grimm D, Greshake B, Kleeberger S, Lipper C, Stegle O, Schölkopf B, Weigel D, Borgwardt K. (2012) easyGWAS: an integrated interspecies platform for performing genome-wide association studies. arXiv: 12124788 [Google Scholar]
- Gubler F, Raventos D, Keys M, Watts R, Mundy J, Jacobsen JV. (1999) Target genes and regulatory domains of the GAMYB transcriptional activator in cereal aleurone. Plant J 17: 1–9 [DOI] [PubMed] [Google Scholar]
- Guo Y, Jamison DC. (2005) The distribution of SNPs in human gene regulatory regions. BMC Genomics 6: 140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gupta S, Stamatoyannopoulos JA, Bailey TL, Noble WS. (2007) Quantifying similarity between motifs. Genome Biol 8: R24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haberer G, Mader MT, Kosarev P, Spannagl M, Yang L, Mayer KF. (2006) Large-scale cis-element detection by analysis of correlated expression and sequence conservation between Arabidopsis and Brassica oleracea. Plant Physiol 142: 1589–1602 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hatfield GW, Benham CJ. (2002) DNA topology-mediated control of global gene expression in Escherichia coli. Annu Rev Genet 36: 175–203 [DOI] [PubMed] [Google Scholar]
- Higo K, Ugawa Y, Iwamoto M, Higo H. (1998) PLACE: a database of plant cis-acting regulatory DNA elements. Nucleic Acids Res 26: 358–359 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Higo K, Ugawa Y, Iwamoto M, Korenaga T. (1999) Plant cis-acting regulatory DNA elements (PLACE) database: 1999. Nucleic Acids Res 27: 297–300 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirschhorn JN, Daly MJ. (2005) Genome-wide association studies for common diseases and complex traits. Nat Rev Genet 6: 95–108 [DOI] [PubMed] [Google Scholar]
- Horton MW, Hancock AM, Huang YS, Toomajian C, Atwell S, Auton A, Muliyati NW, Platt A, Sperone FG, Vilhjálmsson BJ, et al. (2012) Genome-wide patterns of genetic variation in worldwide Arabidopsis thaliana accessions from the RegMap panel. Nat Genet 44: 212–216 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang D, Wu W, Abrams SR, Cutler AJ. (2008) The relationship of drought-related gene expression in Arabidopsis thaliana to hormonal and environmental factors. J Exp Bot 59: 2991–3007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jen CH, Michalopoulos I, Westhead DR, Meyer P. (2005) Natural antisense transcripts with coding capacity in Arabidopsis may have a regulatory role that is not linked to double-stranded RNA degradation. Genome Biol 6: R51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanhere A, Bansal M. (2005) Structural properties of promoters: similarities and differences between prokaryotes and eukaryotes. Nucleic Acids Res 33: 3165–3175 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karlić R, Chung HR, Lasserre J, Vlahovicek K, Vingron M. (2010) Histone modification levels are predictive for gene expression. Proc Natl Acad Sci USA 107: 2926–2931 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kato M, Hata N, Banerjee N, Futcher B, Zhang MQ. (2004) Identifying combinatorial regulation of transcription factors and binding motifs. Genome Biol 5: R56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kellis M, Patterson N, Endrizzi M, Birren B, Lander ES. (2003) Sequencing and comparison of yeast species to identify genes and regulatory elements. Nature 423: 241–254 [DOI] [PubMed] [Google Scholar]
- Kharchenko PV, Tolstorukov MY, Park PJ. (2008) Design and analysis of ChIP-seq experiments for DNA-binding proteins. Nat Biotechnol 26: 1351–1359 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiełbasa SM, Korbel JO, Beule D, Schuchhardt J, Herzel H. (2001) Combining frequency and positional information to predict transcription factor binding sites. Bioinformatics 17: 1019–1026 [DOI] [PubMed] [Google Scholar]
- Kollias G, Hurst J, deBoer E, Grosveld F. (1987) The human beta-globin gene contains a downstream developmental specific enhancer. Nucleic Acids Res 15: 5739–5747 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li YY, Yu H, Guo ZM, Guo TQ, Tu K, Li YX. (2006) Systematic analysis of head-to-head gene organization: evolutionary conservation and potential biological relevance. PLoS Comput Biol 2: e74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ling Y, Du Z, Zhang Z, Su Z. (2010) ProFITS of maize: a database of protein families involved in the transduction of signalling in the maize genome. BMC Genomics 11: 580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Linhart C, Halperin Y, Shamir R. (2008) Transcription factor and microRNA motif discovery: the Amadeus platform and a compendium of metazoan target sets. Genome Res 18: 1180–1189 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Long Q, Rabanal FA, Meng D, Huber CD, Farlow A, Platzer A, Zhang Q, Vilhjalmsson BJ, Korte A, Nizhynska V, et al. (2013) Massive genomic variation and strong selection in Arabidopsis thaliana lines from Sweden. Nat Genet 45: 884–890 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McIntosh KB, Bonham-Smith PC. (2005) The two ribosomal protein L23A genes are differentially transcribed in Arabidopsis thaliana. Genome 48: 443–454 [DOI] [PubMed] [Google Scholar]
- McIntosh KB, Degenhardt RF, Bonham-Smith PC. (2011) Sequence context for transcription and translation of the Arabidopsis RPL23aA and RPL23aB paralogs. Genome 54: 738–751 [DOI] [PubMed] [Google Scholar]
- Mellor J. (2006) Dynamic nucleosomes and gene transcription. Trends Genet 22: 320–329 [DOI] [PubMed] [Google Scholar]
- Miele V, Vaillant C, d’Aubenton-Carafa Y, Thermes C, Grange T. (2008) DNA physical properties determine nucleosome occupancy from yeast to fly. Nucleic Acids Res 36: 3746–3756 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moses AM, Chiang DY, Kellis M, Lander ES, Eisen MB. (2003) Position specific variation in the rate of evolution in transcription factor binding sites. BMC Evol Biol 3: 19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newburger DE, Bulyk ML. (2009) UniPROBE: an online database of protein binding microarray data on protein-DNA interactions. Nucleic Acids Res 37: D77–D82 [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Connor TR, Dyreson C, Wyrick JJ. (2005) Athena: a resource for rapid visualization and systematic analysis of Arabidopsis promoter sequences. Bioinformatics 21: 4411–4413 [DOI] [PubMed] [Google Scholar]
- Pavesi G, Mereghetti P, Mauri G, Pesole G. (2004) Weeder Web: discovery of transcription factor binding sites in a set of sequences from co-regulated genes. Nucleic Acids Res 32: W199–W203 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Razin A, Kantor B. (2005) DNA methylation in epigenetic control of gene expression. Prog Mol Subcell Biol 38: 151–167 [DOI] [PubMed] [Google Scholar]
- Rhee SY, Beavis W, Berardini TZ, Chen G, Dixon D, Doyle A, Garcia-Hernandez M, Huala E, Lander G, Montoya M, et al. (2003) The Arabidopsis Information Resource (TAIR): a model organism database providing a centralized, curated gateway to Arabidopsis biology, research materials and community. Nucleic Acids Res 31: 224–228 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roeder RG. (1996) The role of general initiation factors in transcription by RNA polymerase II. Trends Biochem Sci 21: 327–335 [PubMed] [Google Scholar]
- Sandelin A, Alkema W, Engström P, Wasserman WW, Lenhard B. (2004) JASPAR: an open-access database for eukaryotic transcription factor binding profiles. Nucleic Acids Res 32: D91–D94 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmitz RJ, Schultz MD, Urich MA, Nery JR, Pelizzola M, Libiger O, Alix A, McCosh RB, Chen H, Schork NJ, et al. (2013) Patterns of population epigenomic diversity. Nature 495: 193–198 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneeberger K, Ossowski S, Ott F, Klein JD, Wang X, Lanz C, Smith LM, Cao J, Fitz J, Warthmann N, et al. (2011) Reference-guided assembly of four diverse Arabidopsis thaliana genomes. Proc Natl Acad Sci USA 108: 10249–10254 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seren U, Vilhjálmsson BJ, Horton MW, Meng D, Forai P, Huang YS, Long Q, Segura V, Nordborg M. (2012) GWAPP: a Web application for genome-wide association mapping in Arabidopsis. Plant Cell 24: 4793–4805 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sinha S, Tompa M. (2002) Discovery of novel transcription factor binding sites by statistical overrepresentation. Nucleic Acids Res 30: 5549–5560 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siva N. (2008) 1000 genomes project. Nat Biotechnol 26: 256. [DOI] [PubMed] [Google Scholar]
- Smith AD, Sumazin P, Das D, Zhang MQ. (2005) Mining ChIP-chip data for transcription factor and cofactor binding sites. Bioinformatics (Suppl 1) 21: i403–i412 [DOI] [PubMed] [Google Scholar]
- Spivakov M, Akhtar J, Kheradpour P, Beal K, Girardot C, Koscielny G, Herrero J, Kellis M, Furlong EE, Birney E. (2012) Analysis of variation at transcription factor binding sites in Drosophila and humans. Genome Biol 13: R49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stormo GD. (2000) DNA binding sites: representation and discovery. Bioinformatics 16: 16–23 [DOI] [PubMed] [Google Scholar]
- Tatematsu K, Ward S, Leyser O, Kamiya Y, Nambara E. (2005) Identification of cis-elements that regulate gene expression during initiation of axillary bud outgrowth in Arabidopsis. Plant Physiol 138: 757–766 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson JD, Gibson TJ, Higgins DG (2002) Multiple sequence alignment using ClustalW and ClustalX. Curr Protoc Bioinformatics Chapter 2: Unit 2.3 [DOI] [PubMed] [Google Scholar]
- Trémousaygue D, Garnier L, Bardet C, Dabos P, Hervé C, Lescure B. (2003) Internal telomeric repeats and ‘TCP domain’ protein-binding sites co-operate to regulate gene expression in Arabidopsis thaliana cycling cells. Plant J 33: 957–966 [DOI] [PubMed] [Google Scholar]
- Valouev A, Johnson DS, Sundquist A, Medina C, Anton E, Batzoglou S, Myers RM, Sidow A. (2008) Genome-wide analysis of transcription factor binding sites based on ChIP-Seq data. Nat Methods 5: 829–834 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vandepoele K, Quimbaya M, Casneuf T, De Veylder L, Van de Peer Y. (2009) Unraveling transcriptional control in Arabidopsis using cis-regulatory elements and coexpression networks. Plant Physiol 150: 535–546 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waleev T, Shtokalo D, Konovalova T, Voss N, Cheremushkin E, Stegmaier P, Kel-Margoulis O, Wingender E, Kel A. (2006) Composite Module Analyst: identification of transcription factor binding site combinations using genetic algorithm. Nucleic Acids Res 34: W541–W545 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Q, Wan L, Li D, Zhu L, Qian M, Deng M. (2009) Searching for bidirectional promoters in Arabidopsis thaliana. BMC Bioinformatics (Suppl 1) 10: S29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wasserman WW, Palumbo M, Thompson W, Fickett JW, Lawrence CE. (2000) Human-mouse genome comparisons to locate regulatory sites. Nat Genet 26: 225–228 [DOI] [PubMed] [Google Scholar]
- Wasserman WW, Sandelin A. (2004) Applied bioinformatics for the identification of regulatory elements. Nat Rev Genet 5: 276–287 [DOI] [PubMed] [Google Scholar]
- Wingender E, Dietze P, Karas H, Knüppel R. (1996) TRANSFAC: a database on transcription factors and their DNA binding sites. Nucleic Acids Res 24: 238–241 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wray GA, Hahn MW, Abouheif E, Balhoff JP, Pizer M, Rockman MV, Romano LA. (2003) The evolution of transcriptional regulation in eukaryotes. Mol Biol Evol 20: 1377–1419 [DOI] [PubMed] [Google Scholar]
- Xie X, Lu J, Kulbokas EJ, Golub TR, Mootha V, Lindblad-Toh K, Lander ES, Kellis M. (2005) Systematic discovery of regulatory motifs in human promoters and 3′ UTRs by comparison of several mammals. Nature 434: 338–345 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamamoto YY, Obokata J. (2008) ppdb: a plant promoter database. Nucleic Acids Res 36: D977–D981 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu Z, Shendure J, Church GM. (2005) Discovering functional transcription-factor combinations in the human cell cycle. Genome Res 15: 848–855 [DOI] [PMC free article] [PubMed] [Google Scholar]