

GCR1 shares the fold and key functional motifs of class A, class B, and class E G protein-coupled receptors.
Abstract
Whether G protein-coupled receptors (GPCRs) exist in plants is a fundamental biological question. Interest in deorphanizing new GPCRs arises because of their importance in signaling. Within plants, this is controversial, as genome analysis has identified 56 putative GPCRs, including G protein-coupled receptor1 (GCR1), which is reportedly a remote homolog to class A, B, and E GPCRs. Of these, GCR2 is not a GPCR; more recently, it has been proposed that none are, not even GCR1. We have addressed this disparity between genome analysis and biological evidence through a structural bioinformatics study, involving fold recognition methods, from which only GCR1 emerges as a strong candidate. To further probe GCR1, we have developed a novel helix-alignment method, which has been benchmarked against the class A-class B-class F GPCR alignments. In addition, we have presented a mutually consistent set of alignments of GCR1 homologs to class A, class B, and class F GPCRs and shown that GCR1 is closer to class A and/or class B GPCRs than class A, class B, or class F GPCRs are to each other. To further probe GCR1, we have aligned transmembrane helix 3 of GCR1 to each of the six GPCR classes. Variability comparisons provide additional evidence that GCR1 homologs have the GPCR fold. From the alignments and a GCR1 comparative model, we have identified motifs that are common to GCR1, class A, B, and E GPCRs. We discuss the possibilities that emerge from this controversial evidence that GCR1 has a GPCR fold.
There has been much interest in the identification of novel G protein-coupled receptors (GPCRs) from genome analysis, initially from the human genome, because GPCRs are highly druggable therapeutic targets, and more recently from other genome studies, because GPCRs are vital signaling molecules in diverse organisms. Therefore, whether GPCRs exist in plants is a fundamental biological question.
Here, our focus on putative plant GPCRs was initiated with the characterization of G protein-coupled receptor1 (GCR1) as an orphan GPCR that binds to the plant G protein Guanine nucleotide-binding protein alpha-1 subunit (GPA1) and that is involved in the drought response (Hooley, 1999; Pandey and Assmann, 2004). This observation was followed by intense efforts to identify other plant GPCRs (Moriyama et al., 2006; Liu et al., 2007; Gookin et al., 2008; Pandey et al., 2009). For well-established GPCRs, there are two main classification systems. The GRAFS system (Fredriksson et al., 2003) described five classes of human GPCRs: Glutamate, Rhodopsin, Adhesion, Frizzled/Taste2, and Secretin. Others (Attwood and Findlay, 1994; Kolakowski, 1994) described six classes, namely A to E and the Frizzled GPCRs (class F), that additionally include class D (Eilers et al., 2005) found in fungi and class E cAMP receptors associated with Dictyostelium species (Williams et al., 2005); the Adhesion and Secretin receptors, which differ primarily in their N termini (Lagerström and Schiöth, 2008), together form class B. GCR1 is particularly interesting from a bioinformatics perspective, as it has identifiable but distant homology to class E, class B, and class A GPCRs (Pandey and Assmann, 2004), and so has been used to inform the medically important class A-class B GPCR alignment (Vohra et al., 2007, 2013). GCR1 and the other putative plant GPCRs do not naturally fall into the well-characterized GPCR classes, as presented at the GPCRDB (Horn et al., 2003; Vroling et al., 2011) or elsewhere, and so confirmation that GCR1 is a GPCR is difficult. Indeed, the pitfalls of GPCR identification are illustrated by the high profile (Liu et al., 2007) but erroneous identification of GCR2 as a plant GPCR. It has now been confirmed through crystallization that GCR2 is a lantibiotic cyclase-like protein (Chen et al., 2013), as predicted by our fold recognition studies (Illingworth et al., 2008).
We are particularly interested in these putative GPCRs to assess whether, as remote homologs, they may similarly be used to address the difficult issue of alignment between GPCR families. In this respect, only GCR1 is useful, as the fold recognition studies indicate that GCR1 is the most likely candidate to have a GPCR fold while the evidence for other plant GPCRs is at best minimal. While many methods have been used to align GPCRs from different classes (Frimurer and Bywater 1999; Sheikh et al., 1999; Bissantz et al., 2004; Miedlich et al., 2004; Eilers et al., 2005; Kratochwil et al., 2005; Dong et al., 2007; Coopman et al., 2011; Gregory et al., 2013), it has not been possible to validate these methods on GPCRs until recently. However, with the recent publication of the structure of the class B glucagon receptor (Siu et al., 2013), the class B corticotropin-releasing factor1 receptor (Hollenstein et al., 2013), and the class F human smoothened receptor (Wang et al., 2013) and the associated structural alignments between class A and these remote homologs, we have been able, to our knowledge for the first time, to successfully test our new method. This method is a variation on that used to produce a well-validated class A-class B alignment (Vohra et al., 2013), in which GCR1 was used as a bridge; in a follow-on article, the alignment formed the basis of a class B calcitonin receptor-like receptor (CLR) active model (Woolley et al., 2013) that was later shown to be in good agreement with the class B glucagon receptor x-ray crystal structure. Consequently, we have aligned the GCR1 homologs to class A, class B, and class F and have generated comparative models of active and inactive GCR1. From the alignment, with the assistance of the models, we have identified a number of motifs that are common to GCR1, class A, class B, and class E GPCRs, thus greatly increasing the evidence that GCR1 has a GPCR fold. In addition, we have provided further evidence that GCR1 homologs have the same fold as class A and class B GPCRs from variability analysis. Here, we imply that the difference between a GPCR and a protein with a GPCR fold is the lack of definitive experimental evidence of conventional signaling partners.
Some bioinformatics studies have suggested that there might be about 50 plant GPCRs, including those with large non-membrane domains (Supplemental Table S1) or those with sequence similarities to other plant proteins (Supplemental Table S2), but now it has been questioned whether there are any plant GPCRs (Urano et al., 2012, 2013; Bradford et al., 2013; Urano and Jones 2013), primarily because the plant G protein is self activating and does not need a guanine nucleotide-exchange factor (GEF). One of the presentations of putative plant GPCRs is based on a hidden Markov model, trained on several hundred seven-transmembrane helical (7TM) proteins taken from the GPCRDB (both well-characterized GPCRs and other 7TM proteins such as the mildew resistance locus O [MLO] proteins); the genes were tentatively assigned as GPCRs on the basis of seven predicted transmembrane helices (Moriyama et al., 2006). This assignment has been made against the background of the well-documented and now closed debate regarding whether the 7TM protein bacteriorhodopsin was a suitable template for modeling GPCRs (Hibert et al., 1993; Hoflack et al., 1994), most typified by the article of Hibert et al. (1993): “This is not a G protein coupled receptor.” Given that a number of distinct GPCR x-ray crystal structures have become available (Congreve et al., 2011; Katritch et al., 2013; Venkatakrishnan et al., 2013), it is now possible to analyze these putative plant GPCR sequences to assess whether, in the light of new structural information, they are more or less likely to be GPCRs and, thus, to move beyond the assumption implicit in Moriyama et al. (2006) that a receptor with seven transmembrane helices is a GPCR (e.g. bacteriorhodopsin has seven transmembrane helices but is not a GPCR; Hibert et al., 1993).
Here, our approach to analysis of the 56 putative plant GPCRs is to combine transmembrane structure prediction and sequence analysis with fold recognition methods. There are essentially two approaches to fold recognition, namely sequence-based methods, such as genTHREADER (Jones, 1999b), and empirical potential-based methods, such as Threader (Jones et al., 1992; Jones, 1998). The sequence-based methods have the advantage of speed and may be suitable for whole-genome analysis but may not readily identify remote homologs when the sequence identity is low. The empirical potential-based methods may be more efficient at identifying remote homologs but are generally not parameterized for membrane proteins. For this reason, we have taken a heuristic approach and have tested a variety of fold recognition methods to see if they correctly identify characteristic GPCR sequences from classes A to F while at the same time not incorrectly assigning bacteriorhodopsin and GCR2 as GPCRs. In particular, our focus is on fold recognition methods such as I-TASSER (Zhang, 2008; Roy et al., 2010) that have performed well in the critical assessment of protein structure prediction (CASP) fold recognition competitions (Moult et al., 2009). For proteins where the evidence that they are GPCRs was not convincing, the fold recognition (threading) results were used to give a preliminary indication of which other types of membrane proteins they could be; the most likely alternatives were ion channels or transporters. The significance of this study, therefore, is 4-fold. First, it adds clarity to the field of plant GPCRs by indicating from a wide range of evidence that only GCR1 is strongly predicted to have a GPCR fold. Second, it provides evidence that some of the other candidates are more likely to be transporters. Third, it indicates computational approaches that could be taken to follow up initial genome analysis studies to help avoid the confusion that has shrouded the plant GPCR field. Fourth, the new alignment method has given promising results on well-validated alignments in or below the “twilight zone” (Doolittle, 1986) and so could, with development, be used in other more general applications. In addition, we discuss the implications of these results that are difficult to reconcile with current knowledge of the mechanism of the Arabidopsis (Arabidopsis thaliana) G protein, GPA1.
RESULTS AND DISCUSSION
Control Sequences
The fold recognition results for the six control sequences are given in Supplemental Table S3. For each server, the negative controls bacteriorhodopsin and GCR2 were correctly identified as bacteriorhodopsin and a protein of the LanC synthase family, respectively. The class A sequences were readily identified because the templates in the database were also from class A. Classes B and E were also generally identified strongly, whereas classes C and F appear to be the most difficult to identify. Nevertheless, the I-TASSER, mgenTHREADER, LOMETS, HHpred, and Phyre methods all identified class C and F GPCRs with reasonable confidence, albeit in the top four hits rather than in the top ranked hit for a few cases. The general ranking of these methods for this problem appears to be I-TASSER > LOMETS > HHpred > FUGUE > Phyre > mgenTHREADER > MUSTER > genTHREADER. The score at which the first incorrect result occurs is an important marker. Some methods do not report an incorrect result (e.g. bacteriorhodopsin submitted to Phyre or GCR2 submitted to HHpred), and for these methods, the lowest score for a correct result also provides a useful guide; these scores are given in Supplemental Table S4. Thus, the Phyre results are deceptively good, as all sequences were identified at rank 1 with 100% certainty. However, Phyre also identified ion channels and transporters as lower ranked hits, with 95% certainty for class A GPCRs; for this reason, only Phyre results reported with 100% certainty are included in Tables I and II. LOMETS also reported two results that are below the level of certainty provided by the controls, and so these are also omitted from the results given in Tables I and II. The full set of Phyre results is given in Supplemental Table S5. The performance of I-TASSER, LOMETS, HHpred, FUGUE, and Phyre on this particular problem was superior to that of the other methods, so further analysis was restricted to these.
Table I. Threading results of putative plant GPCRs split into families of proteins that share discernible homology.
Putative plant GPCRs with GPCRs hit are denoted with single asterisks; no GPCR hits are denoted with double asterisks. Each hit has an associated interpretation, such as high, medium, or low, to indicate the expected reliability. Proteins predicted not to be 7TM proteins are underlined, and the expected number of transmembrane helices is given. Nonplant 7TM lung receptors are shown in italics. The following MtN3 proteins had no hits: At1g21460, At3g16690, At3g48740, and At5g13170. The following expressed protein family 2 proteins had no hits: At1g10660 and At5g62960. The following expressed protein family 3 protein had no hits: At3g09570. All of the expressed protein family 5 proteins had no hits: At3g63310 and At4g02690. All of the GNS1/SUR4 membrane family proteins had no hits: At1g75000, At3g06470, and At4g36830. All of the MLO family proteins had no hits: At1g11000, At1g26700, At1g24560, At2g33670, At2g44110, At4g24250, and At5g53760. Both of the GTG family proteins had no hits: GTG1 (nine-transmembrane helical) and GTG2 (nine-transmembrane helical). The groups are those reported by Moriyama et al. (2006).
| TAIR Locus Identifier | I-TASSER | LOMETS | HHpred | FUGUE | Phyre |
|---|---|---|---|---|---|
| Nodulin MtN3 family proteins (8/17; two hits/eight proteins) | |||||
| At3g28007 | ** | ** | ** | * (guess) | ** |
| At4g25010 | ** | ** | * (guess) | ** | ** |
| Expressed protein family 2 (one hit/three proteins) | |||||
| At2g47115 | ** | ** | * (uncertain) | ** | ** |
| Expressed protein family 3 (one hit/two proteins) | |||||
| At5g42090 | ** | ** | * (guess) | ** | ** |
| TOM3 family proteins | |||||
| At1g14530 | ** | ** | * (marginal) | ** | ** |
| At2g02180 | ** | ** | * (guess) | ** | ** |
| At4g21790 | ** | ** | * (marginal guess) | * (guess) | ** |
| Lung_7-TM_R | |||||
| At2g01070 | ** | * (high) | * (guess) | ** | * (high) |
| Q22938_CAEEL | * (high) | ** | * (high) | ** | ** |
| A8K285_HUMAN | ** | ** | * (guess) | ** | ** |
| YHB7_YEAST | ** | * (marginal) | * (high) | ** | ** |
Table II. Threading results of putative plant GPCRs split into groups of proteins that do not share discernible homology.
Putative plant GPCRs with GPCR hits are indicated with single asterisks; no GPCR hits are indicated by double asterisks . Each hit has an associated interpretation, such as high, medium, or low, to indicate the expected reliability. Proteins predicted not to be 7TM proteins are underlined, and the expected number of transmembrane helices is given. There were no hits for all of the expressed protein family 1 (At1g77220 and At4g21570), miscellaneous expressed protein family 4 (At1g49470 and At5g19870), Perl1-like family protein (At1g16560 and At5g62130), and the miscellaneous single members from big gene families (At1g71960, At3g01550, At5g23990, and At5g37310 [predicted approximately nine-transmembrane helical]). There were no hits for the following members of miscellaneous single-copy genes: At1g57680, At2g41610, At2g31440, At3g04970 (predicted six-transmembrane helical), and At3g26090 (RGS1). There were no hits for the following members of miscellaneous single members from small gene families: At3g19260, At2g35710 (predicted six-transmembrane helical), At2g16970 (predicted 12-transmembrane helical), At1g15620 (predicted five/six-transmembrane helical), At1g63110, and At4g36850.
| TAIR Locus Identifier | I-TASSER | LOMETS | HHpred | FUGUE | Phyre |
|---|---|---|---|---|---|
| Miscellaneous single-copy genes | |||||
| At1g48270 (GCR1) | * (high) | * (high) | * (high) | * (high) | * (high) |
| At3g59090 | ** | ** | ** | * (marginal) | ** |
| At4g20310 (four-transmembrane helical) | ** | ** | ** | * (guess) | ** |
| Miscellaneous single members from small gene families (eight) | |||||
| At5g27210 | ** | ** | ** | * (guess) | ** |
Transmembrane Helix Prediction
The 16 putative GPCR sequences (Moriyama et al., 2006; Pandey et al., 2009) that were not predicted to be 7TM proteins by TMHMM are recorded in Supplemental Table S6. Nine were subsequently predicted by more than two methods to be 7TM proteins; seven were predicted not be 7TM proteins by more than four methods (underlined in Tables I and II, with the probable number of transmembranes given in parentheses). Proteins At5g37310 and At5g62130 have been predicted by SPOCTOPUS and MEMSAT to have an N-terminal signal peptide instead of the first predicted transmembrane helix. Therefore, At5g37310 is assumed to have nine transmembranes instead of 10 transmembranes and At5g62130 may have seven transmembranes instead of eight transmembranes. The OCTOPUS server has only predicted At5g62960, a member of expressed protein family 2 (Table I), to have a reentrant loop (originating from inside, between residues 248 and 255, which are predicted to lie between transmembrane helix5 [TM5] and TM6). Since reentrant loops are not a common feature of GPCRs (except perhaps for extracellular loop2 [ECL2] in rhodopsin or the smoothened receptor), this may indicate a different superfamily such as transporters. G protein coupling preferences are given in Supplemental Table S7, but these are not helpful as discussed in Supplemental Text S1.
Fold Recognition
Overall Results
The fold recognition (threading) results for the putative GPCRs are given in Tables I and II. Threading hits from the I-TASSER server (a reliable server according to the CASP fold recognition competition; Moult et al., 2009) indicate that GCR1 is the only candidate that is strongly predicted to have a GPCR fold. The LOMETS server has predicted two putative plant GPCRs, GCR1 and At2g01070, with high confidence and two (At5g19870 and At5g13170) with low confidence. At2g01070 aligns with the lung 7TM receptors (Protein Families Database code PF06814), which have homologs in plants, invertebrates, fungi, and mammals. As yet, there is no evidence that these proteins are GPCRs, but A2g01070 was predicted by three servers. All members of the lung 7TM family have GPCR hits, including Q22938_CAEEL by I-TASSER, and all members were predicted to be GPCRs by HHpred, some with high confidence. The HHpred server has indicated that seven other putative plant GPCR sequences are likely to be GPCRs, albeit with low confidence except for GCR1. FUGUE indicates that six of the 54 sequences are likely to be GPCRs. However, apart from GCR1, the GPCR hits were reported below the cutoff. The Phyre server gave two GPCR hits with 100% confidence, GCR1 and At2g01070, which was also weakly predicted by HHpred (the Phyre homology search is driven by HHpred).
As a result of this analysis, there is additional evidence that 12 of 56 proteins could be GPCRs, since they have been identified by one or more fold recognition servers. There is also additional evidence that 44 of 56 proteins are less likely to be genuine GPCRs, since they were not identified as having a GPCR fold by any of the fold recognition servers, and for some of these the treading gave alternative indications (Supplemental Tables S8–S12). In addition, since only three of the seven MLO proteins are predicted (very weakly) to be GPCRs and the remaining four are not, it seems reasonable to assume that none of the MLO proteins are GPCRs (since a homologous family should all have the same identity). Urano and Jones (2013) dismissed the MLO proteins as GPCRs primarily because their role in conferring fungal resistance is independent of G proteins. Similarly, it is most likely that none of the Medicago truncatula Nodulin3 proteins are GPCRs, especially as MtN3 nonplant proteins are three-transmembrane helical proteins (Protein Families Database code PF03083) that have high similarity (approximately 85% identity) to TM1 to TM3 and TM5 to TM7 of their plant relatives. Such symmetry between TM1 to TM3 and TM5 to TM7 would not occur in a classic GPCR, so the MtN3 putative plant GPCRs are similarly unlikely to be GPCRs. We note that symmetry does occur in transporter families. The GPCR-type G (GTG) proteins GTG1 and GTG2 and the three groups of “expressed protein” families (Table I) are similarly ruled out by the homologous family argument. GTG1 and GTG2 are particularly suspect, as they seem to have the wrong number of transmembrane helices to be GPCRs. Within Table I, the only families where the fold recognition results indicate that they could be GPCRs is the tobamovirus multiplication protein3 (TOM3) family and the lung 7TM proteins, since every member of the family has been implicated and there is evidence from more than one server. For the proteins in Table II, we note that only GCR1 is predicted to be a GPCR by more than one method. In conclusion, the most likely GPCRs, besides GCR1, are At2g01070 and the TOM3 family. However, apart from GCR1, the results are far from conclusive. Four other possible GPCR candidates are listed in Table III, with the final list of nine proteins reduced from 12 by homology arguments.
Table III. The five most likely plant GPCRs ranked according to the strength of evidence from fold recognition, sequence comparison, and transmembrane helix prediction.
The first rank, GCR1, is clearly predicted to be GPCR. At2g01070 and TOM3 are possibly GPCRs on the basis of more than one piece of evidence. The remaining two hits have been implicated by just one piece of evidence.
| Rank | TAIR Locus Identifier/Family |
|---|---|
| 1 | At1g48270 (GCR1) |
| 2 | TOM3 |
| At2g01070 | |
| 3 | At5g27210 |
| At3g59090 |
GCR1 Fold Recognition Results
Despite the additional sequences in Table III, we present evidence here that GCR1 is the only candidate to have a GPCR fold. All 5-fold recognition methods matched GCR1 to the GPCR fold. Second, GCR1 was used as a bridge in the well-validated class A-class B GPCR alignment (Vohra et al., 2013) that has generated a CLR model (Woolley et al., 2013), in good agreement with subsequent experimental structures (Siu et al., 2013); this approach would probably have been ineffective if GCR1 did not have a GPCR fold. In “Conclusion,” we summarize nine additional observations (see below) consistent with the hypothesis that GCR1 has a GPCR fold.
However, the role of GCR1 as a GPCR has been questioned (Urano and Jones, 2013), first on the basis of a lack of homology to other GPCRs, second because of questions regarding its class E homologs, third because of doubts about the GCR1-G protein interaction, and finally because of the observation that plant G proteins do not require GPCRs to act as GEFs (Johnston et al., 2008; Urano and Jones, 2013).
Relatively few commentators doubt that class E homologs signal through G proteins (Janetopoulos et al., 2001; Ray et al., 2011; Krishnan et al., 2012; Yan et al., 2012). With regard to homology between GCR1 and other GPCRs, rather than the limited homology to TM3 and TM4 of class E GPCRs as claimed previously (Urano and Jones, 2013), earlier reports identified similarities to class A and class B GPCRs covering a much wider range (Josefsson and Rask, 1997; Plakidou-Dymock et al., 1998). We extend this work to show (below) that there is considerable homology to all eight helices of class A and class B GPCRs. While there has been difficulty in reproducing (Urano and Jones, 2013) the reported GCR1-GPA1 interaction (Pandey and Assmann, 2004), we show that GCR1 possesses motifs that would facilitate this interaction. The idea that GCR1 and G proteins can act independently (Chen et al., 2004) is not necessarily relevant, as this is a property of well-characterized GPCRs (Bockaert and Pin, 1999; Rajagopal et al., 2010b; Koval and Katanaev, 2011; Whalen et al., 2011). The final point, however (i.e. whether GCR1 is a GEF), is a most noteworthy point and will be discussed below. This point lies at the heart of the question of whether plants have GPCRs and can only be finalized by experiment. However, given that the function of GCR1 is currently unknown, it is important to assess what can be learned about GCR1 from structural bioinformatics so that these experiments can be planned more carefully based on the known interplay between structure and function.
The Class A-Class B-Class F Alignment
Illustrating the Method
Selected alignment results to illustrate the method are given in Figure 1. Figure 1A shows the number of “votes” for each of the 17 alternative TM3 class A-class B pairwise alignments, evaluated using both the Blosum62 matrix and the PHAT matrix. Here, the Blosum62 results show that alignment 0 (the alignment inferred by superposition of the x-ray structures) is the overwhelming choice, as the alternatives received very few votes. Alignment 0 is also an overwhelming choice for the PHAT matrix, but the preference of the 0 alignment over the alternatives is not quite so marked. The corresponding pairwise sequence alignment-based results for TM1, shown in Figure 1B, are more representative, in that alignment 0 receives the highest number of votes but other alignments also receive votes; for TM1, the PHAT matrix highlights the experimentally inferred alignment (0) more strongly than the Blosum62 matrix. Overall, the performance of the two matrices is very similar: PHAT gives a clearer preference for TM1 and TM2, while Blosum62 gives a clearer preference for TM3 and TM4; for TM5 to TM7, there is no clear pattern. Figure 1C shows the results for the class A-class B TM7 alignment; here, the method does not indicate a clear alignment choice but a number of different alignments are indicated. This situation can arise if the two multiple sequence alignments are too distant from each other, if the alignment contains gaps, or if the alignment region is too short and key motifs have been omitted, as here (for Fig. 1C, the alignment was terminated prior to position 7.52 because this region of TM7 is α-helical in class A and a 310 helix in class F, i.e. there are three amino acids per turn); under such circumstances other information may be required to determine the true alignment.
Figure 1.

Various alignment results to illustrate the method; each legend is valid until replaced by an alternative. A to C, Number of votes for each of the 17 alternative class A-class B pairwise alignments evaluated using the PHAT matrix (P, red, left) and the Blosum62 matrix (B, orange, right): TM3 (A), TM1 (B), and TM7 (C). D, Number of votes for the TM1 class A-class B pairwise alignments evaluated using hydrophobicity (H, green, left) and volume matrix (Vo, yellow, right). E, Maximum lagged correlation values for each alignment evaluated using entropy (S, purple, left) and variability (Va, cyan, right) for the class A-class B TM1 alignment. F, Correlation values of E scaled between 0 and 1.0 for the class B-class F TM2 alignment. G, Product scores for each of the TM3 class A-class B alignments. The product scores are as follows (left to right): P × H × Vo × S (red), P × Vo × S (green), P × H × S (yellow), P × H × Vo (white), and P × H × Vo × Va (purple). H, The product scores for each of the TM5 class A-class B alignments. The product scores are as follows: B × H × Vo × S (orange), B × Vo × S (green), B × H × S (yellow), B × H × Vo (white), and B × H × Vo × Va (purple). [See online article for color version of this figure.]
Figure 1D shows the number of votes for each of the 17 alternative TM3 class A-class B pairwise alignments, evaluated using both hydrophobicity and volume. For each of these measures, 0 is not the preferred alignment, but the 0 alignment nevertheless receives a reasonable number of votes. It is important to note that in about one-third of the cases where the alignment is known, hydrophobicity and volume gave a small number of votes to the correct alignment such that the scaled score was less than 0.5. While it is clearly important that the overall hydrophobicity profiles have a reasonable match, local variations arise between remote homologs, so hydrophobicity as used here may not always be appropriate. Only in one case out of 21 did both hydrophobicity and volume give a low number of votes to the correct alignment.
The results of the maximum lagged correlation of entropy and variability are given in Figure 1, E and F, for two alignments. Figure 1E shows that entropy and variability are generally not as discriminating as hydrophobicity and volume, as more alignments tend to receive a high score. In Figure 1E, entropy gives a higher score for the 0 alignment, while in Figure 1F, scaled variability gives a higher score. Overall, the performance of these two measures is similar, but variability requires a larger number of sequences and so may be more difficult to calculate. Occasionally, the maximum lagged correlation of entropy can suggest an erroneous alternative alignment if strongly conserved residues align (Vohra et al., 2013). Figure 1G shows that the product scores for the class A-class B TM3 alignment all give overwhelming support to the correct (0) alignment. Figure 1H shows that the product scores for the class A-class B TM5 alignment all give support to the correct (0) alignment but also indicate alternative alignments. Here, we note that the scaled volume score is low, hence the high product score when this is omitted.
The Class A-Class B-Class F Alignment
The publication of the x-ray crystal structure of two class B GPCRs (Hollenstein et al., 2013; Siu et al., 2013) and a class F GPCR (Wang et al., 2013) provides the first opportunity to validate methods for aligning helices of remote GPCR homologs. Ideally, the method should reproduce the class A-class B, class A-class F, and class B-class F alignments (Fig. 2) for each helix in agreement with experiment. Since these alignments are difficult, particularly those involving class F, as shown by a blind modeling competition (R. Abagyan, unpublished data), a secondary criterion is that the method should generate consistent alignments (i.e. the class B-class F alignment should be consistent with the class A-class B and class A-class F alignments) and that this consistency could arise through the choice of an alternative alignment that receives a reasonable score. (Consistency provides a useful control in situations where the experimental alignment is not known.)
Figure 2.

Sequence alignment between GCR1 and the class A, class B, and class F template sequences; the color reflects the biophysical properties. The most conserved positions within each helix in class A are marked by a vertical bar and correspond to position 50. The residues are color coded according to their properties as follows: blue, positive; red, negative or small polar; purple, polar; cyan, polar aromatic; green, large hydrophobic; yellow, small hydrophobic. This corresponds to the Taylor scheme, as implemented in Jalview (Clamp et al., 2004). For clarity, some ungapped sequence sections have been truncated.
The alignments for TM1 to TM7 are given in Supplemental Figures S1 to S7. For all three TM1 alignments, alignment 0 receives a good score, and excellent results are obtained if hydrophobicity is omitted from the product for class F alignments. Supplemental Figure S1G shows that variability for the class A-class F and class B-class F 0 alignments fits better than that for the −3 and −7 alignments, respectively (indicated in Supplemental Fig. S1, D and F), as the latter have three minima outside of the shaded area, as opposed to one: the low variability should either be in internal regions or in external regions that are tightly packed against neighboring helices; the apparent violation for the class F 0 alignment at positions 1.38 and 1.43 fit into this latter category, but the other violations do not. For TM2, TM3, and TM4, excellent agreement with experiment is achieved for all three alignments.
For TM5 to TM7, the situation is a little more difficult, partly because there are gaps reported in the class A-class F alignments. However, the reported alignment places equivalent residues in very different environments (Wang et al., 2013), and alternative structural alignments place the gaps in different positions (data not shown).
For TM5, the gap in class F alignments is outside of our alignment window and so is not a problem. The correct alignment is obtained for class A-class B (the +4 alternative aligns the conserved Pro residues; Supplemental Fig. S5G also shows that it should be given a low weighting because it gives a minimum in variability in an external position [5.59]). The correct alignment is also given for class B-class F (where the Pro residues align). For class AF, the +4 alternative aligns the Pro residues, but this is not consistent with experiment or the class B-class F alignment; the +4 alternative alignment also gives several variability minima in external regions and, more worryingly, a variability maximum in internal position 5.64. Both the class A-class F 0 and −4 alignments are consistent with the alignments to class B (i.e. the following two sets of alignments are mutually consistent [but only the first is consistent with experiment]: AB: 0, AF: 0, BF: 0 and AB: 0, AF: −4, BF: −4. The class F 0 and −4 alignments are also largely consistent with the variability data, as the maxima and minima are generally in external and internal positions, respectively. Thus, this is an example of where additional information may be required to determine the alignment: the strongest scoring −4 alignment places a class F polar Lys at position 5.65, which is normally hydrophobic and required for G protein coupling (Vohra et al., 2013). For the 0 alternative, which has a reasonable score if volume is omitted, a conserved Leu aligns with position 5.65. Given that the class A-class B alignment is difficult (Vohra et al., 2013) and that class F is even more distant, these represent good results.
For TM6, the class A-class B alignment is reproduced well, but the class A-class F and class B-class F results are clearly not in line with the structural alignment. The simplest explanation for this is that the structural alignment places a gap in the middle of TM6 for class F and that our current alignment methods cannot easily deal with this problem. More significantly, Wang et al. (2013) place this gap at position 6.47, whereas we place this at 6.41; the lack of a clear correspondence over such a range no doubt contributes to the difficulty of the alignment. The four high-scoring class B-class F alignments (Supplemental Fig. S6E) and the lack of consistency between the alignments should alert the reader that there may be a problem, even in the absence of an experimental alignment.
For TM7, the class A-class B alignment is reproduced well. There is some uncertainty as to the alignment of the intracellular end of the smoothened receptor. Our structural alignment places Trp-535 in the same position as Y7.53 of the NPXXY motif; therefore, we place a gap at position 7.52 (Wang et al. [2013] place five gaps). Our class A-class F structural alignment, therefore, reproduces the alignment better (Supplemental Fig. S7G) if the right-hand window limit is reduced to position 7.51 from position 7.53 (Supplemental Fig. S7D). There is no need to shorten the window for the class A-class B alignment, as there is no gap; indeed, such shortening reduces the quality of the alignment (as shown in Fig. 1C), as part of the key NPXXY motif is missing. The class B-class F alignment is reproduced provided that hydrophobicity is omitted from the product (Supplemental Fig. S7F). Moreover, the class F variability for the +1 alignment has three high scores/maxima at internal positions (7.39, 7.42, and 7.49), as shown in Supplemental Figure S7H. The class F variability fits the topology except at position 7.53, but this is due to the change in conformation to a 310 helix. The variability for the class F −4 alignment is compatible with the topology.
Together, the results in Supplemental Figures S1 to S7 show that the method is capable of aligning GPCR transmembrane helices of remote homologs, especially where allowance is made for insight from structural information and where there are no gaps in the alignment window. For some helices, the method is very clear, but in general, the procedure is not a black box method, as some attention may need to be given to the role of hydrophobicity and volume and to the nature of the alternative alignments, which in some cases may be eliminated using variability. As in all alignments of remote homologs, care can be given to the alignment of motifs (Lesk, 2002); this has been done elsewhere for the class A-class B alignment, which has been well tested by mutagenesis studies (Vohra et al., 2013). While there are clearly limitations to the method, it should be appreciated that these are difficult alignments and that the Web version of a recent state-of-the-art method (Stamm et al., 2013) only correctly aligned a few of these 21 transmembrane helix pairs and did not align any pairs in a mutually consistent way; the stand-alone version offers more control and probably does much better. We will now apply the method to GCR1 homologs, which are not as difficult as the class A-class F and class B-class F alignments, as they are less firmly in the twilight zone (Doolittle, 1986) of approximately 18% to 25% identity.
The Alignment of GCR1 Homologs
For each helix where the class A-class B-class F structural alignment is well defined, the new alignment method generates a clear alignment in the sense that the alignment is (1) unambiguous, as there is a single main peak, (2) an equivalent alignment is given to class A, class B, and class F, and (3) there is no need to omit hydrophobicity or volume from the product, as all four measures support the preferred alignment. The exception is the TM6 class F alignment, which will be discussed below. The full results for the alignment are shown in Supplemental Figures S8 to S14, and these are summarized in Figure 3. Therefore, the individual alignments to the GCR1 homologs are better defined than the corresponding class A-class B-class F GPCR alignments, which are known from the structural alignments. The reason for this is probably that GCR1 homologs are generally closer to class A, class B, and class F GPCRs than these are to each other. Thus, Figure 4A and Supplemental Figure S15 show that the alignments involving GCR1 homologs generally have higher percentage identities, higher average matrix scores, and higher product scores than the alignments between the well-known GPCRs (class A, class B, and class F). Analysis of Figure 4A indicates why the alignments involving TM6 of class F are difficult: these alignments have the lowest percentage identities (e.g. 7% for the alignment to class A), resulting in some of the lowest PHAT matrix scores and the lowest product scores (Supplemental Fig. S15). (Some TM3 alignments also have low PHAT matrix scores, but for TM3, these are nevertheless much higher than the next scoring alignments, while for TM6, this is not the case.) To some extent, the structural alignment depends on how the superposition is carried out, but for TM6, class F residues have a greater tendency to point in different directions than their counterparts, even when the Cα atoms are in close proximity. In general, TM1 to TM4 show a closer structural superposition than TM5 to TM7, and this is in line with the greater sequence similarity shown by TM1 to TM4. The alignment of GCR1 to class A, class B, and class F GPCRs is shown in Figure 2.
Figure 3.

The product of the four scaled scores (PHAT matrix score × hydrophobicity × volume × entropy) for the alignment between class A GCR1 homologs (left, purple) and class B GCR1 homologs (right, cyan). The alignment corresponding to the 0 alignment is given in Figure 2. The legend given for TM1 is valid for all plots. [See online article for color version of this figure.]
Figure 4.

A, Mean percentage identity (%ID) between different GPCR families. The class A-class B, class A-class F, and class B-class F percentage identities are shown to the left of the vertical line in red, orange, and yellow, respectively; the percentage identities between class A, class B, and class F with the GCR1 homologs are shown to the right in green, blue, and cyan, respectively. The percentage identities for TM3 between GCR1 homologs and class C and class D GPCRs are 14.3% and 11.9%, respectively. B, Structural alignment (determined by modeler using all residues) between the inactive GCR1 (green), the class A dopamine D3 (blue), the class B glucagon (orange), and the class F smoothened (red) receptors looking toward TM1 to TM4. The root mean square deviation (RMSD) between minimized inactive GCR1 and the dopamine, glucagon, and smoothened receptors is 1.29, 2.07, and 3.33 Å, respectively. For comparison, the expected RMSDs between the α, β, χ, and δ class A GPCRs are 2.2 to 3.0 Å and that between class A and class B GPCRs is typically 2.7 to 3.3 Å (Hollenstein et al., 2013; Siu et al., 2013), so these RMSDs are of the expected magnitude. The RMSDs were calculated over the helical domain over the ranges 1.36 to 1.59, 2.40 to 2.58, 3.25 to 3.51, 4.45 to 4.62, 5.43 to 5.65, 6.33 to 6.43, and 7.43 to 7.53; shorter sections were used for TM6 and TM7 because of the known outward tilt in class B in this region. C, Snake diagram showing GCR1 features that characterize the GPCR fold. The Cys residues of the TM3-ECL2 disulfide bond are shown in yellow with black lettering. Motifs shared with class A and/or class B GPCRs are shown in red with white lettering. Group-conserved residues that have the same character in class A, class B, and GCR1 homologs are shown in cyan with dark blue lettering; other common group-conserved positions are shown in dark blue with cyan lettering. The TLH positional equivalent of the DRY motif is shown in orange (residues are only shown in one category). ICL1 and ICL2 are denoted in purple, as they are the same length as their class A and/or class B counterparts. ECL2 and ICL3 are shown in light blue, as they are the longest ECL and ICL, respectively. The ampipathic helix 8 is denoted by a light green background. Potential phosphorylation sites C terminal of the ampipathic helix are denoted by red lettering. The potential glycosylation site in ECL2 is also denoted by red lettering.
Alignment of GCR1 TM3 to All Known GPCR Classes
Supplemental Figure S10 also shows the alignment scores between class C and class D GPCRs with GCR1 homologs for TM3, which is the structural and functional hub for GPCRs (Venkatakrishnan et al., 2013). Therefore, it is noteworthy that the new method also gives a very strong signal for these two additional classes, provided that volume is excluded, as in the original method (Vohra et al., 2013) that has yielded a model of the CGRP class B GPCR in good agreement with the class B x-ray structure of the glucagon receptor (Woolley et al., 2013).
Consequently, Supplemental Figure S16 shows a TM3 alignment of GCR1 against all known GPCR classes. While this alignment shows a degree of diversity among the different GPCR classes, it is clear that TM3 of GCR1 also shares many similarities, particularly with class B, class E, and Frizzled/smoothened (e.g. the CY3.26 motif, the conserved W3.42, and the conserved aromatic residues at positions 3.33 and 3.51; Krishnan et al., 2012). The TM3 percentage identities between class C and class D with the GCR1 homologs are 14.3% and 11.9%, respectively, giving rise to mean PHAT matrix scores of −3.6 and −6.2, suggesting that GCR1 homologs lie closer to class A, class B, and class F GPCRs than to class C or the class D (fungal) GPCRs.
Variability
The variability for class A, class B, and GCR1 homologs is shown in Figure 5; class F was omitted from this analysis because of the greater divergence in sequence and structure, despite the high percentage identity to some helices. For each helix, the pattern of variability for the GCR1 homologs is very similar to that for the class A and class B sequences. For each helix, there is essentially a repeating pattern, with low variability at the internal or buried positions (e.g. positions 1.46 and 1.50 on helix 1) and high variability at the external exposed positions (e.g. positions 6.41 and 6.46 in helix 6). For such exposed positions, the maximum for GCR1 homologs generally coincides with the exposed region and usually aligns with that for class A or class B or both.
Figure 5.

Variability for each of the seven transmembrane helices. The variability for class A GPCRs (A, solid lines) and class B GPCRs (B, dotted lines) is shown in black; the variability for GCR1 homologs is shown in orange (G). Shading indicates the internal or buried positions (which should have low variability). For TM1, TM5, and TM6, the variability for the alternative +3, +4, and −6 GCR1 alignments (G′) is shown with orange dashes. Position 7.34 is a restricted external position in many receptors, hence its low variability. The different helix lengths shown reflect both the natural helix length and the length over which a common conformation can be expected (Vohra et al., 2013). Although positions 3.35 to 3.40 in TM3 are nominally internal, they still show a maximum variability in line with the helix periodicity. [See online article for color version of this figure.]
The magnitude of the variability is partly a reflection of the number of subsets used, but within each helix, the qualitative patterns are generally the same for all three classes, and these patterns are distinct from those for other helices. There are a small number of exceptions to the general internal/external pattern, but the deviations are small and comparable to those observed for class B, and these mainly arise from low variability at external positions that are nevertheless restrained by steric interactions with neighboring helices (e.g. position 5.57). In summary, the overall picture to arise from the variability is that the GCR1 homologs share the GPCR fold, since the distinct patterns result from the GPCR fold. The TM6 −4 alternative alignment (which derives from the class F alignment) is clearly incompatible with the fold because of the high variability at internal position 6.48; the variability patterns for alternative +3 and +4 alignments for TM1 and TM5, respectively, do not match the class A and class B patterns as well as the 0 alignment does but cannot be eliminated as the mismatch is not too severe.
GCR1 Motifs
Analysis of the sequence alignments (Fig. 2; Supplemental Fig. S16) and the GCR1 comparative models (Fig. 4B; available from ftp.essex.ac.uk/pub/oyster/GCR1_2013/GCR1_models.tar.gz) has identified a number of motifs that are common between class A and class B GPCRs and the GCR1 homologs, as shown in Table IV. The most notable motif is the disulfide bond between the top of TM3 and ECL2, which is present in almost all GPCRs and is characteristic of the fold, regardless of the class, as illustrated by the conservation of C3.25 shown in Figure 2 and Supplemental Figure S16. ECL2 is the longest extracellular loop in GCR1, and this too is a typical feature of the GPCR fold (Venkatakrishnan et al., 2013). The conserved WCW motif in ECL2 occurs in a similar position to the class B ECL2 CW motif, as shown in Supplemental Figure S17. In addition, GCR1 has a potential sodium-binding site that lies between TM2, TM3, and TM7, identified by simulations (Selent et al., 2010) and crystallography (Liu et al., 2012), that is only found in class A GPCRs. The other motif that is only found in class A GPCRs is Y5.58, which is involved in stabilizing the active GPCR conformation (White et al., 2012). GCR1 homologs share an FxxP motif on TM5 with both class A and class B GPCRs, but in class A GPCRs, this is displaced by one turn of the helix.
Table IV. Key motifs conserved in class A, class B, and GCR1/class E GPCRs.
Lowercase letters indicate that the GCR1 residues are not conserved.
| Transmembrane | Class A Motifs | Class B Motifs | GCR1 Motif | GCR1/Class E Family Motifs | Probable Function (Where Known) |
|---|---|---|---|---|---|
| IL1 | K1.61KLHxxxN | R1.61KLHxxxN | KELRkfsF | K1.61ELRxxx[F/N] | Stability |
| TM2 | NLxxxD2.50 | NLxxxF2.50 | YLalsD2.50 | YLxxxD2.50 | Structure |
| TM2-7 | D2.50, S3.39, W6.48, N7.45, S7.46 | –b | D2.50, S3.39, W6.48 | D2.50, [S/D]3.39, W6.48 | Sodium-binding site |
| TM3 | CK3.26 | CK3.26 | CY | CY3.26 | Structure: disulfide bond to ECL2 |
| DRY motif | D/ER3.50Y/W | R2.39…H2.43…E3.46 | –a | –a | Activation |
| TM3 | DR3.50Y | YL3.50H | TLH | TL3.50[Y/H] | Activation |
| TM4 | W4.50 | W4.50 | W | W4.50 | Structure |
| ECL2 | CW | WCW | WCW | ||
| TM5 | FxxP5.50 | FxxP5.46 | FxxP5.46 | FxxP5.46 | Structure |
| TM5 | Y5.58 | –b | Y5.58 | Y5.58 | Activation |
| TM5 | IxxL5.65 | IXXL5.65 | VXXI5.65 | VXXL5.65 | Activation |
| TM6 | KxxK6.35 | KxxK6.35 | KvlN | Kxx[K/N]6.35 | Activation |
| TM6 | CWxP6.50 | P6.42…TY6.48 | P6.41...SWaF | P6.41...W6.48x[F/P] | Activation |
| R3.50, E6.30 | R2.39, T6.37 | –a | –a | Ionic lock | |
| TM7 | NP7.50xxY | VA7.50VLY | NS7.50xxY | NS7.50xxY | Activation |
| H8 | EF8.50xxxL | EV8.50xxxL | SV8.50xxxI | SV8.50xxxI | Stability |
See text. bMotif not observed in class B.
Given the conserved L at position 1.63, it appears that GCR1/class E shares the novel KKLH motif on intracellular loop1 (ICL1), albeit in a modified form, with the consensus being KELR, which interacts with a polar/hydrophobic motif on helix 8 (SVxxxI in GCR1, EFxxxF in class A, and EVxxxL in class B); this motif is difficult to align (Roy et al., 2013) but came to light in ungapped interclass helix alignments (Vohra et al., 2013). The length of the intracellular loops may also be highly relevant. In the β2-adrenergic receptor-stimulatory G protein (β2-AR-Gs) complex, both ICL1 and ICL2 interact with the G protein. Analysis of the alignments in the PRINTS database shows that ICL1 is the same length in the majority of class B and GCR1 sequences, while ICL1 is the same length in 68% of PRINTS class A sequences and all but 12% have the same length to within one residue; ICL2 is the same length in class B and GCR1 (bar 9% of PRINTS class B sequences). In common with many GPCRs, ICL3 of GCR1 is the longest intracellular loop.
The class A EFxxxF motif is part of the ampiphilic helix 8 that runs parallel to the membrane plane, as shown by most GPCR crystal structures, the exceptions being CXCR4 (Wu et al., 2010), which has positive residues (hence, a repulsive interaction at positions 1.61, 1.62, and 8.49), the neurotensin NTSR1, where the thermostabilized construct is inactive even though it is an “active” agonist-bound structure (White et al., 2012), and the class B corticotropin-releasing factor receptor1, where H8 was truncated (Hollenstein et al., 2013). The structural motif is present in class B and F GPCRs, as illustrated by the glucagon (Siu et al., 2013) and the smoothened receptor (Wang et al., 2013) structures; therefore, it is a structural feature characteristic of GPCRs. The signature for an ampipathic helix 8 is strong in class B, class D, class F, and plant GPCRs, as can be seen from the sequence alignments at the PRINTS database (Supplemental Fig. S18). The C-terminal region beyond the ampipathic helix of GCR1 is rich in Ser and Thr residues, as could be expected by analogy to other GPCR classes, and a number of Ser/Thr kinases exist in Arabidopsis that have homology to mammalian GPCR kinases, However, there is less evidence for plant analogs of arrestin, which binds phosphorylated GPCRs in mammalian systems, so other proteins could be involved in GCR1 internalization (Urano et al., 2013). There is a consensus glycosylation site, Nx[S/T], in ECL2. While N-glycosylation in the N terminus is common, 32% of GPCRs have at least one glycosylation site in ECL2, and 85% of these are between the top of TM4 and the conserved Cys (Wheatley et al., 2012).
For the alignment of remote homologs, reliance solely on alignment scores and/or statistics is unwise; rather, it is important to identify common motifs (Lesk, 2002). In summary, a number of common motifs have been identified. Many of these reside in regions associated with receptor activation and G protein binding. These motifs are prime candidates for experiments to investigate the possibility that the similarity that GCR1 shares with its class A and class B cousins underlies an ability to interact with heterotrimeric G proteins irrespective of any GEF or other regulatory action.
The DRY Motif
The two most important class A activation microswitches are DRY3.51 on TM3 and NPXXY7.53 on TM7. The second microswitch has readily identifiable counterparts in both class B (VAVLY7.53) and GCR1 (NSIAY7.53), but the DRY3.51 motif raises difficulties, since the class B positional equivalent (YLH3.51) is not as important in activation as its class A counterpart and the class B DRY3.51 functional equivalent, which also involves charged residues, is disjointed, as it is distributed between TM2 and TM3 (Frimurer and Bywater, 1999; Vohra et al., 2013). Supplemental Figure S16 shows that contiguous charged/aromatic residues are also missing from TM3 positions 3.49 to 3.51 in class C, class D, class E, GCR1, and class F GPCRs. Consequently, in these GPCRs, it is highly likely that the DRY3.51 functionally equivalent motif may use different positions and take an alternative form that could involve polar rather than charged residues. The class A GPCR-Gs interaction is mediated by positive residues on the GPCR, most notably R3.50, but the C-terminal peptide of Gs is not rich in negative residues. On the assumption that the DRY3.51 functionally equivalent motif donates a hydrogen bond to the G protein, possible GCR1 candidate residues could include Arg-1073.52, Arg-481.64, and Lys-492.37, which could adopt the right conformation given minor conformational changes to ICL1. Of these, Lys-492.37 is the most likely, as it could also form an ionic lock with Glu-2116.30 in the inactive structures. However, the C-terminal part of the plant G protein (GPA1_ARATH) has three consecutive Arg residues (373–375) that may mediate the GPCR-G protein interaction, so the use of positive residues by class A and class B GPCRs may not be followed by other classes. In our model, Ser-512.39 is the only residue in TM2 and TM3 making interactions to charged residues in the GAP1 C terminus. Such a small polar residue would seem an unlikely alternative, but Rosenkilde et al. (2005) describe a constitutively active virus-encoded GPCR containing a DTW3.51 motif. With regard to a possible ionic lock involving Glu-2116.30, Glu is not highly conserved at position 6.30 in GCR1/class E, but neither is it highly conserved in class A. Given the potential role of Thr in the class B polar lock (Vohra et al., 2013), Glu-2116.30 could also form a potential polar lock with Thr-1083.53.
The uncertainty in analyzing these potential interactions arises because of difficulties in modeling loops (Goldfeld et al., 2011), but this is not a major issue with regard to whether GCR1 has a GPCR-specific 7TM fold, since many of the motifs listed in Table IV and shown in Figure 4C reside within the helices, not the loops. While comparative models may be useful for giving an overall picture of GPCR interactions (Taddese et al., 2012, 2013), they are certainly not completely reliable and so are better used for indicating possible candidate residues for mutagenesis experiments than for providing a definitive identification of all key residues. For these reasons, and because of the lack of mutagenesis data, Table IV does not specify a GCR1/class E functional equivalent of the DRY motif or an ionic/polar lock.
Group-Conserved Residues
The positions of the 24 helical group-conserved residues (Eilers et al., 2005) that are common to class A, class B, and GCR1/class E are given in Supplemental Table S13. Each individual helix arrangement appears to be nonrandom, with 14 of the group-conserved residues being of the same type in all three classes.
Alignment Quality
The quality scores for the alignment between GCR1 homologs and class A or class B, and the comparative score for random sequences, are given in Figure 6. For each helix, with the possible exception of TM6 for class A, the score for the alignment between GCR1 homologs and class A, and particularly class B, is such that very few of the alternative alignments give a lower score. It appears that the distribution of entropy in each pair of aligned helices is not random, and it is reassuring that similar results are obtained for each helix. However, it is not possible to extend this analysis to the whole alignment, since the evolution of one helix may not be entirely independent of that of another helix.
Figure 6.

A helix-by-helix quality assessment of the alignment of GCR1 homologs. The score from Equation 1 for the alignment between GCR1 homologs and class A and class B GPCRs is denoted by bottom (red) and top (blue) arrows denoted A:G and B:G, respectively. A histogram of the scores from Equation 1 between GCR1 homologs and the 198 comparator sequences is also given; the number of comparator scores that are higher than the class A or class B scores are shown in parentheses beside each arrow. [See online article for color version of this figure.]
What Is the True Identity of the Non-GPCRs?
The fold recognition results indicate that a number of proteins, namely the MtN3 family, At2g16970, and At1g71960, are likely to be transporters; this is discussed further in Supplemental Text S1 and Supplemental Tables S9-S12).
The GPA1-GCR1-GEF Dilemma
The observation that GPA1 is self-activating, in that it readily binds GTP rather than requiring a GPCR to catalyze the exchange of GDP for GTP (Johnston et al., 2007), has led to the suggestion that the activity of GPA1 is regulated by RGS (for regulator of G protein signaling; a GTPase-accelerating protein) rather than by a GPCR, although some plants lack RGS (Urano et al., 2012). Hence, it has been implied that GPA1 does not require a GEF and, therefore, that GCR1 is not a GPCR, since it is not required to regulate GPA1.
In contrast, GCR1 has been shown to interact with GPA1 by both a split-ubiquitin method and coimmunoprecipitation (Pandey and Assmann, 2004). GCR1 was also predicted to have a GPCR fold by the fold recognition methods, and this implies much more than a collection of seven randomly packed transmembrane helices; rather, the GPCR fold implies very specific helix lengths, tilts, rotations, and helix-helix interactions. The GPCR 7TM fold is usually accompanied by an eighth helix. Other additional evidence that GCR1 shares the GPCR fold comes from the alignments and variability, the experimentally validated class A-class B alignment, the identification of GPCR motifs, analysis of the loop lengths, and the alignment of group-conserved residues. Consequently, GCR1’s status as a GPCR cannot be dismissed merely because it does not behave as a GEF in current experiments. It appears, then, that the two contrasting observations must be held in tension until a resolution of the apparent contradiction is uncovered. This resolution may reside in the complexity of the plant signaling apparatus.
The issue of whether GPCRs are GEFs is not new to the GPCR field. Indeed, for many years, this question was used as an objection to recognizing class F Frizzled receptors as GPCRs. This objection was overturned by biochemical evidence (Koval and Katanaev, 2011; Malbon, 2011) and, more recently, by an x-ray crystal structure (Wang et al., 2013). In addition, the idea that GPA1 does not need a GEF does not necessarily mean that GCR1 is not a GEF, even if GPA1 is the only Gα-subunit in Arabidopsis.
Given that GCR1 has a GPCR fold, it would be interesting to see whether GCR1 behaves as a GEF in a chimeric GPA1 in which the part of the helical domain responsible for GPA1 self-activation was replaced by a corresponding part from a non-self-activating G protein (Jones et al., 2011); since GCR1 may interact more readily with a GDP-bound form of GPA1, this may also help to reconcile conflicting reports regarding whether GCR1 does indeed interact with GPA1 (Pandey and Assmann, 2004; Urano et al., 2013).
If GCR1 was ultimately found not to couple to G proteins in any circumstances, this would be particularly interesting, given that it has the features expected of a bona fide GPCR in terms of fold and motifs.
Decoy GPCRs
GPCRs are not always defined by their GEF activity, as GPCRs also promote G protein-independent signaling with conventional signaling partners (Bockaert and Pin, 1999; Rajagopal et al., 2010b; Koval and Katanaev, 2011; Whalen et al., 2011). This is illustrated, for example, by the “decoy” GPCRs. Decoy GPCRs have the expected GPCR motifs and are considered part of the GPCR family but do not signal through G proteins. These include C5L2, a C5a anaphylatoxin chemotactic receptor, and CXCR7 (for C-X-C chemokine receptor type7) (Okinaga et al., 2003; Chen et al., 2007; Rajagopal et al., 2010a). CXCR7 has all the motifs given in Table IV, except that the KKLH motif appears as KTTG, the KxxK6.32 motif appears as SSRK, and there is no obvious ionic lock. Wild-type H2LC has DLC3.51 instead of the DRY motif, but the G protein coupling is restored if this is mutated to DRC; in other respects, it is a chemokine GPCR. If GCR1 does not couple to G proteins, then GCR1 could also be designated as a decoy GPCR. However, unlike CXCR7 (Rajagopal et al., 2010a), it probably does not have the option of signaling through arrestin, so it would be interesting to identify any G protein-independent signaling pathways of GCR1.
CONCLUSION
Here, we have presented a novel perspective on the likelihood that the putative plant GPCRs derived from genome analysis are genuine GPCRs using heuristic fold recognition methods. Only GCR1 emerges as a strong GPCR candidate, and for approximately six other proteins (the TOM3 family, At2g01070, At5g27210, and At3g59090) there are additional indications, beyond seven transmembrane helices, that they could have a GPCR fold, but these indications are weak. For some candidate GPCRs (approximately 37), there is little consensus regarding their true identity, while for others (approximately 10), it is more likely that they are transporters. Thus, to predict GPCRs, the identification of seven hydrophobic regions is only the first step (Urano and Jones, 2013). We have shown that it is important to also consider fold and motifs (Krishnan et al., 2012) to distinguish between GPCRs and other proteins that may share a 7TM scaffold.
Eleven pieces of evidence are relevant to the debate as to whether GCR1 has a GPCR fold. (1) All 5-fold recognition methods matched GCR1 to the GPCR fold. (2) GCR1 homologs were used as a bridge in the experimentally validated class A-class B GPCR alignment (Vohra et al., 2013). (3) The alignment method has been validated on the class A-class B-class F alignments. (4) The alignments of the GCR1 homologs to class A, class B, and class F GPCRs are clear and mutually consistent (excluding TM6 class F). (5) The helix-helix alignments involving GCR1 homologs have a higher similarity (Fig. 4A) than the well-established class A-class B-class F GPCR alignments. (6) Patterns of variability on all seven helices are consistent with the GPCR fold. (7) The alignment has identified 15 motifs that GCR1 shares with class A and class B GPCRs, including the diagnostic disulfide bond between TM3 and ECL2. (8) GCR1 has an ampipathic eighth helix, which is characteristic of GPCRs and which has Ser and Thr residues in the expected positions. (9) The lengths of ICL1 and ICL2 in GCR1 are largely identical to those of their class A and/or class B counterparts. (10) The lengths of ECL2 and ICL3 relative to the other loops are as expected for a GPCR. (11) The alignment of any given individual helix appears to be nonrandom. Together, this evidence validates the use of GCR1 as an intermediate sequence in the class A-class B alignment.
This creates an interesting dilemma when seen against the issues of heterotrimeric G protein regulation raised by Urano and Jones (2013), which suggest that GCR1 is not a GPCR, primarily because GPA1 and other similar G proteins in lower organisms do not need a GEF (Urano et al., 2012, 2013; Bradford et al., 2013; Urano and Jones, 2013). Whether GCR1 is ultimately confirmed as a G protein-interacting protein (as the Frizzled-smoothened GPCRs were after a long debate; Koval and Katanaev, 2011; Malbon, 2011) remains to be seen. If it is not confirmed as a GPCR, then it raises a very interesting question regarding the function of a protein that has the fold and expected motifs of a bona fide GPCR. Thus, if GCR1 has a function that is not well known for GPCRs, then other well-accepted GPCRs may possibly have similar hitherto unknown functions.
MATERIALS AND METHODS
Transmembrane Helix Prediction
The putative plant GPCRs were taken from Moriyama et al. (2006) via the Kyoto Encyclopedia of Genes and Genomes Web site (Kanehisa and Goto, 2000; Kanehisa et al., 2010), along with the sequences of GTG1 and GTG2 (Pandey et al., 2009), which are also putative plant GPCRs. Transmembrane helix prediction was carried out using TMHMM 2.0 (Krogh et al., 2001; Möller et al., 2001), since we have found this to be reliable for GPCRs in general and for our controls in particular (see below). This was carried out for two purposes. First, to confirm that the sequences were indeed predicted to be 7TM proteins, and second, to identify large extracellular or cytosolic domains that could be separated to increase the efficiency of subsequent fold recognition steps carried out on the individual domains (Supplemental Table S1). Since TMHMM is not 100% reliable (Melén et al., 2003; Inoue et al., 2005; Kahsay et al., 2005), we have used other well-regarded methods, such as HMMTOP (Tusnády and Simon 1998, 2001), MEMSAT3 (Jones et al., 1994; Jones, 2007), and TOPpred2 (Claros and von Heijne, 1994), on proteins predicted to have other than 7TM helices. The TMLOOP (Viklund et al., 2006) and OCTOPUS and SPOCTOPUS (Viklund and Elofsson, 2008; Viklund et al., 2008) servers were used to predict reentrant loops and signal peptides, since reentrant loops are not a common feature of GPCRs, except perhaps for ECL2 in rhodopsin, which is weakly reentrant (Palczewski et al., 2000), and signal peptides can present as transmembrane helices. However, it should be noted that reentrant loop prediction is currently not very reliable.
Sequence Similarities
Because some of the proteins (e.g. MtN3) form distinct homologous groups (Supplemental Table S2), the results were analyzed in the light of results for other members of the same family, as they are either all GPCRs or all not GPCRs. Moreover, if they are not GPCRs, then they should all belong to the same alternative family.
Fold Recognition
The well-characterized GPCRs and related sequences used as positive controls were A4D2G4_HUMAN (olfactory 2, class A), Q8IV17_HUMAN (secretin receptor, class B), B0UXY7_HUMAN (GABAB subtype1, class C), Q6TMC6_COPCI (pheromone receptor, class D), CAR3_DICDI (cAMP receptor 3, class E), and FRIZ2_DROME (frizzled 2, class F). The negative controls were AAU04564.1 Halobiforma lacisalsi (bacteriorhodopsin) and AT2G20770 (Arabidopsis [Arabidopsis thaliana] GCL2 GCR2). Bacteriorhodopsin is a 7TM protein but not a GPCR, while GCR2 has seven hydrophobic helical motifs that are almost long enough to span the membrane, so that its homologs were initially erroneously identified as GPCRs (Illingworth et al., 2008). These sequences were submitted to the following fold recognition servers: I-TASSER (Roy et al., 2010), FUGUE (Shi et al., 2001), Phyre (Bennett-Lovsey et al., 2008), genTHREADER (Jones, 1999a), mgenTHREADER (McGuffin and Jones, 2003), HHpred (Söding et al., 2005), LOMETS (Wu and Zhang, 2007), and MUSTER (Wu and Zhang, 2008). Generally, each server gives a key metric, such as a Z-score, and an associated interpretation, such as “high,” “medium,” or “low,” that indicates the expected reliability of the result. It is understood that fold recognition methods do not necessarily give the correct fold as the highest ranked hit, so we have looked for the correct fold from the controls to be given in the top 10 hits.
Transmembrane Helix Alignment
The alignment between GCR1/class E and class A and class B GPCRs was previously determined on a helix-by-helix basis by combining (via a product of scaled scores) the results of a profile alignment with maximum lagged correlation. The alignment was evaluated over a well-defined transmembrane region in which the internal/external character of the residues was invariant over a number of class A GPCR structures; the profile contained a flank of eight residues on either side of this region (Taddese et al., 2012), but flanks of 15 residues were investigated to ensure that eight was sufficient; the averaging and scaling of the individual alignment scores or correlation coefficients between 0 and 1 ensured that the noise was minimized and allowed the correct alignment to appear above the noise (Vohra et al., 2013). Here, we have replaced the profile alignment with an ungapped pairwise alignment of all possible pairs of sequences of each class; the best alignment for a given helix was taken as the most common alignment (Fig. 1). Here, the alignments were scored using the PHAT substitution matrix (Ng et al., 2000) that was specifically derived for transmembrane helices; they were also scored using the widely used Blosum62 substitution matrix (Henikoff and Henikoff, 1993) to check that the results are not unduly sensitive to the choice of matrix; Blosum62 was used in the previous study. (There are problems with the derivation of the Blosum62 matrix, but these actually serve to enhance its performance in searches [Styczynski et al., 2008].) For the hydrophobicity, we have carried out maximum lagged correlation, not of the average hydrophobicity, as previously, but for every pair of sequences (one from each class), with the best alignment for each pair of sequences given by the highest correlation coefficient. The best alignment was again taken as the most common alignment (i.e. the one that received the most votes; Fig. 1). In addition, we have included amino acid volume (Sandberg et al., 1998) as an additional property that was treated in the same way as the hydrophobicity. Thus, for the substitution matrix, for hydrophobicity and for volume, each ungapped alignment (−1, 0, +1, etc.) received a number of votes according to the number of times that it received the highest score. However, in the subsequent step, it was the number of votes that were scaled between 0 and 1 rather than the scores. Entropy is a property of every sequence in the alignment, so we have retained the maximum lagged correlation of the entropy. As before, the scores were averaged over the forward and backward alignments, scaled between 0 and 1, and the four scores were multiplied together to give an overall score that gives an indication of the preferred alignment. Each of the four methods may indicate a different alignment, but the benefit of scaling the measures between 0 and 1 and multiplying them together is that alignments receiving little support are suppressed while alignments receiving multiple support are enhanced. For remote homologs, some measures may occasionally receive a score near 0, and to check for this, the product was also generated three more times, with hydrophobicity, volume, or entropy omitted.
For comparative purposes, we have also generated the alignments using AlignMe in default mode via the Web (Stamm et al., 2013). For these alignments to be a fair comparison, the first profile (e.g. class A) included the 16 flanking residues while the second profile (e.g. class B) omitted these; the reverse alignment (i.e. omitting the flanking residues for class A) was also performed; standard profile alignments were also performed.
We have also used the new method to assess the TM3 alignment between GCR1/class E and all other GPCR classes, as defined at the GPCRDB (www.gpcr.org/7TM_old/); we focused on TM3 since its alignment is more straightforward than that of other helices for class A-class B-GCR1/class E, so it seemed reasonable to expect this to carry over to other GPCR classes (Vohra et al., 2013). The class C, class D, and class F multiple sequence alignments for TM3 included 464, 39, and 107 sequences and were prepared as described previously (Vohra et al., 2013); class F GPCRs, like class A and class B, are believed to have evolved from class E GPCRs (Chabbert et al., 2012; Krishnan et al., 2012) and have some homology to GCR1 (Pandey and Assmann, 2004). In addition to the combined alignment for GCR1/class E, we also repeated the work using sequences in the GCR1 plant group (PR02000), as defined by the PRINTS database (Attwood et al., 2012); despite the small number of unique sequences (nine), this gave essentially the same results, except for class C. For convenience, the PRINTS class A (PR00327) and class B (PR00249) alignments were also analyzed.
Our use of the numbering system of Ballesteros and Weinstein (1995) is defined elsewhere (Vohra et al., 2013).
Variability
The α-carbon template of Baldwin et al. (1997), derived primarily from the rhodopsin electron cryomicroscopy map, was widely used until the first GPCR x-ray crystal structure was published (Palczewski et al., 2000); its reliability was, in part, due to the incorporation of variability. Variability is very sensitive to the microenvironment of a residue within the helical bundle; hence, it is able to report on the fold: families with a similar fold should have similar patterns in variability. Most notably, external residues should have high variability, while internal residues should have low variability. Our method for determining variability was given previously (Vohra et al., 2013). For the GCR1 homologs, analysis of 191 sequences on a helix-by-helix basis resulted in 42, 47, 19, 40, 49, 30, and 34 subsets for TM1 to TM7, respectively. We have compared the variability with that of class A and class B GPCRs, which was reported previously (Vohra et al., 2013). In addition, variability was used as an alternative to entropy in the transmembrane helix alignments.
Alignment Quality
For the alignment of remote GPCR homologs, it has been proposed that while equivalent residues may differ in identity and properties, the positions of functionally important residues are likely to be conserved (Frimurer and Bywater, 1999); this “cold-spot” method has formed the basis of many class B and class C GPCR models. Here, an indication that a given helix alignment could have arisen by chance was assessed as follows:
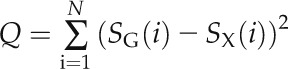 |
(1) |
where SG(i) is the entropy of position i in a given GCR1/class E helix and SX(i) is the entropy of the corresponding class A or class B residues; the sum is evaluated over the N helical residues of the alignment window. This was compared with the distribution of values generated when the target sequences were compared with other potentially relevant sequences. These latter were observed sequences taken from other helices and from other classes (i.e. for TM1 of GCR1 homologs, the “random” or rather comparator sequences were taken from TM2 to TM7 of classes A, B, and F). This choice ensured that the comparison was with sequences possessing relevant properties such as hydrophobicity, secondary structure, periodicity, and conservation. For this purpose, the midpoints of the comparator helices were aligned to the midpoints of the reference helix and a total of 11 sequences generated by shifting the comparator helix by ± five or fewer residues (to give a reasonable number of residues with an even radial distribution). There are caveats in this approach. First, we have assumed that the functional residues can be equated with low entropy (although the mathematic approach uses all entropy values). Second, the validity of the cold-spot method has not been fully validated. Third, the comparator helices may be distantly related by evolution. (An alternative approach to this problem involving group-conserved residues [Eilers et al., 2005] is described in Supplemental Table S13, but the entropy-based approach is superior because it uses the full range of conservation data for all residues in the helix.)
GCR1 Comparative Model
The Arabidopsis GCR1 (The Arabidopsis Information Resource [TAIR] locus identifier At1g48270; 288 amino acids) sequence was obtained from the Kyoto Encyclopedia of Genes and Genomes Web site (Kanehisa and Goto, 2000; Kanehisa et al., 2006, 2010). Nine class A x-ray crystal structures of the β1-AR (Protein Data Bank code 2VT4; Warne et al., 2008), rhodopsin (1U19; Palczewski et al., 2000), the adenosineA2AR (3EML; Jaakola et al., 2008), dopamine D3R (3PBL; Chien et al., 2010), muscarinic M2R (3OUN; Haga et al., 2012), histamine H1R (3RZE; Shimamura et al., 2011), sphingosine S1P1R (3V2W; Hanson et al., 2012), the chemokine CXCR4 (30DU; Wu et al., 2010), protease-activated receptor1 (3VW7; Zhang et al., 2012), two class B crystal structures of the corticotropin-releasing factor1 receptor (4K5Y; Hollenstein et al., 2013) and the glucagon receptors (4L6R; Siu et al., 2013), and the class F structure of the smoothened receptor (4JKV; Wang et al., 2013) were used as templates (for the alignment, see Fig. 2; this was originally derived from a structural superposition of the structures using modeler and is consistent with the transmembrane helix alignment; see below). Here, we used multiple templates to generate a single inactive GCR1 model because it is generally appreciated that the use of multiple templates results in better comparative models (Taddese et al., 2013). For class F, alternative TM6 and TM7 alignments can be derived depending on how the structural alignment is performed, but these alternatives did not affect the modeler results (data not shown), presumably because of the low percentage identity to class F GPCRs in TM6 and TM7.
The models were ranked according to their Discrete Optimized Protein Energy assessment scores (Eswar et al., 2006). From these highest scoring models, the one with the least amount of helical distortion in the transmembrane region was selected using the secondary structural assessment as implemented in VMD (Humphrey et al., 1996). This essentially amounted to ensuring that the distortion due to the class A TM2 Pro was not transmitted to the GCR1 models. The inactive models were also selected on the basis of an ECL2 conformation that was similar to that in one of the class B GPCR structures, since GCR1 homologs share the ECL2 CW motif with class B GPCRs. The intracellular and extracellular loops were refined in modeler using the modeler loopmodel function, and the structure with the lowest Discrete Optimized Protein Energy score was selected. A similar loop-refinement strategy, combined with experimental mutagenesis, gave rise to the prediction of a CLR ECL2 conformation that was later shown to be similar to that in the glucagon receptor (Woolley et al., 2013). However, it must be stressed that loop modeling is difficult and that major indeterminations will reside in the loop regions. The models were used in conjunction with the alignments and class A and class B structures to identify common motifs.
Sequence data from this article can be found in the GenBank/EMBL data libraries under accession numbers.
Supplemental Data
The following materials are available in the online version of this article.
Supplemental Figure S1. The class A-class B-class F TM1 alignment.
Supplemental Figure S2. The class A-class B-class F TM2 alignment.
Supplemental Figure S3. The class A-class B-class F TM3 alignment.
Supplemental Figure S4. The class A-class B-class F TM4 alignment.
Supplemental Figure S5. The class A-class B-class F TM5 alignment.
Supplemental Figure S6. The class A-class B-class F TM6 alignment.
Supplemental Figure S7. The class A-class B-class F TM7 alignment.
Supplemental Figure S8. The class A-class B-class F GCR1 homologues TM1 alignment.
Supplemental Figure S9. The class A-class B-class F GCR1 homologues TM2 alignment.
Supplemental Figure S10. The class A-class B-class F GCR1 homologues TM3 alignment.
Supplemental Figure S11. The class A-class B-class F GCR1 homologues TM4 alignment.
Supplemental Figure S12. The class A-class B-class F GCR1 homologues TM5 alignment.
Supplemental Figure S13. The class A-class B-class F GCR1 homologues TM6 alignment.
Supplemental Figure S14. The class A-class B-class F GCR1 homologues TM7 alignment.
Supplemental Figure S15. A comparison of the distance between families (PHAT, product scores).
Supplemental Figure S16. The TM3 alignment for all known GPCR classes.
Supplemental Figure S17. ECL2 alignment for class B human and plant sequences.
Supplemental Figure S18. The ampiphatic nature of helix 8.
Supplemental Table S1. Proteins predicted to have large extracellular or cytosolic domains.
Supplemental Table S2. Putative Arabidopsis plant family sequence similarities.
Supplemental Table S3. Performance of selected fold recognition servers on control sequence.
Supplemental Table S4. Lowest/highest score for which good/poor results have been obtained.
Supplemental Table S5. The full set of results from the Phyre server.
Supplemental Table S6. Proteins predicted by TMHMM not to be 7TM proteins.
Supplemental Table S7. G-protein coupling preferences, calculated using PRED-COUPLE.
Supplemental Table S8. The 15 proteins that are least likely to be GCPRs.
Supplemental Table S9. Conclusive threading to small domains for single sequence.
Supplemental Table S10. Summary of MtN3 threading results.
Supplemental Table S11. High confidence threading hits for the sequence At2g16970.
Supplemental Table S12. High confidence threading hits for the sequence At1g71960.
Supplemental Table S13. Group-conserved residues that are conserved in all 3 classes.
Supplemental Text S1. Basic sequence analysis, GPCR-specific websites, and additional information.
Glossary
- GPCR
G protein-coupled receptor
- CLR
calcitonin receptor-like receptor
- GEF
guanine nucleotide-exchange factor
- 7TM
seven-transmembrane helical
- TAIR
The Arabidopsis Information Resource
- RMSD
root mean square deviation
References
- Attwood TK, Coletta A, Muirhead G, Pavlopoulou A, Philippou PB, Popov I, Romá-Mateo C, Theodosiou A, Mitchell AL. (2012) The PRINTS database: a fine-grained protein sequence annotation and analysis resource—its status in 2012. Database (Oxford) 2012: bas019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Attwood TK, Findlay JB. (1994) Fingerprinting G-protein-coupled receptors. Protein Eng 7: 195–203 [DOI] [PubMed] [Google Scholar]
- Baldwin JM, Schertler GF, Unger VM. (1997) An alpha-carbon template for the transmembrane helices in the rhodopsin family of G-protein-coupled receptors. J Mol Biol 272: 144–164 [DOI] [PubMed] [Google Scholar]
- Ballesteros JA, Weinstein H. (1995) Integrated methods for the construction of three-dimensional models and computational probing of structure-function relationships in G-protein coupled receptors. Methods Neurosci 25: 366–428 [Google Scholar]
- Bennett-Lovsey RM, Herbert AD, Sternberg MJ, Kelley LA. (2008) Exploring the extremes of sequence/structure space with ensemble fold recognition in the program Phyre. Proteins 70: 611–625 [DOI] [PubMed] [Google Scholar]
- Bissantz C, Logean A, Rognan D. (2004) High-throughput modeling of human G-protein coupled receptors: amino acid sequence alignment, three-dimensional model building, and receptor library screening. J Chem Inf Comput Sci 44: 1162–1176 [DOI] [PubMed] [Google Scholar]
- Bockaert J, Pin JP. (1999) Molecular tinkering of G protein-coupled receptors: an evolutionary success. EMBO J 18: 1723–1729 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bradford W, Buckholz A, Morton J, Price C, Jones AM, Urano D. (2013) Eukaryotic G protein signaling evolved to require G protein-coupled receptors for activation. Sci Signal 6: ra37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chabbert M, Castel H, Pele J, Deville J, Legendre R, Rodien P. (2012) Evolution of class A G-protein-coupled receptors: implications for molecular modeling. Curr Med Chem 19: 1110–1118 [DOI] [PubMed] [Google Scholar]
- Chen JG, Pandey S, Huang JR, Alonso JM, Ecker JR, Assmann SM, Jones AM. (2004) GCR1 can act independently of heterotrimeric G-protein in response to brassinosteroids and gibberellins in Arabidopsis seed germination. Plant Physiol 135: 907–915 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen JH, Guo J, Chen JG, Nair SK (2013) Crystal structure of Arabidopsis GCR2. RCSB Protein Data Bank. http://www.rcsb.org/pdb/explore/explore.do?structureId=3t33 (October 30, 2013)
- Chen NJ, Mirtsos C, Suh D, Lu YC, Lin WJ, McKerlie C, Lee T, Baribault H, Tian H, Yeh WC. (2007) C5L2 is critical for the biological activities of the anaphylatoxins C5a and C3a. Nature 446: 203–207 [DOI] [PubMed] [Google Scholar]
- Chien EY, Liu W, Zhao Q, Katritch V, Han GW, Hanson MA, Shi L, Newman AH, Javitch JA, Cherezov V, et al. (2010) Structure of the human dopamine D3 receptor in complex with a D2/D3 selective antagonist. Science 330: 1091–1095 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clamp M, Cuff J, Searle SM, Barton GJ. (2004) The Jalview Java alignment editor. Bioinformatics 20: 426–427 [DOI] [PubMed] [Google Scholar]
- Claros MG, von Heijne G. (1994) TopPred II: an improved software for membrane protein structure predictions. Comput Appl Biosci 10: 685–686 [DOI] [PubMed] [Google Scholar]
- Congreve M, Langmead CJ, Mason JS, Marshall FH. (2011) Progress in structure based drug design for G protein-coupled receptors. J Med Chem 54: 4283–4311 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coopman K, Wallis R, Robb G, Brown AJ, Wilkinson GF, Timms D, Willars GB. (2011) Residues within the transmembrane domain of the glucagon-like peptide-1 receptor involved in ligand binding and receptor activation: modelling the ligand-bound receptor. Mol Endocrinol 25: 1804–1818 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong M, Lam PC, Gao F, Hosohata K, Pinon DI, Sexton PM, Abagyan R, Miller LJ. (2007) Molecular approximations between residues 21 and 23 of secretin and its receptor: development of a model for peptide docking with the amino terminus of the secretin receptor. Mol Pharmacol 72: 280–290 [DOI] [PubMed] [Google Scholar]
- Doolittle RF (1986) Of URFs and ORFs: A Primer on How to Analyze Derived Amino Acid Sequences. University Science Books, Mill Valley, CA [Google Scholar]
- Eilers M, Hornak V, Smith SO, Konopka JB. (2005) Comparison of class A and D G protein-coupled receptors: common features in structure and activation. Biochemistry 44: 8959–8975 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eswar N, Webb B, Marti-Renom MA, Madhusudhan MS, Eramian D, Shen MY, Pieper U, Sali A (2006) Comparative protein structure modeling using Modeller. Curr Protoc Bioinformatics Chapter 5: Unit 5.6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fredriksson R, Lagerström MC, Lundin LG, Schiöth HB. (2003) The G-protein-coupled receptors in the human genome form five main families: phylogenetic analysis, paralogon groups, and fingerprints. Mol Pharmacol 63: 1256–1272 [DOI] [PubMed] [Google Scholar]
- Frimurer TM, Bywater RP. (1999) Structure of the integral membrane domain of the GLP1 receptor. Proteins 35: 375–386 [PubMed] [Google Scholar]
- Goldfeld DA, Zhu K, Beuming T, Friesner RA. (2011) Successful prediction of the intra- and extracellular loops of four G-protein-coupled receptors. Proc Natl Acad Sci USA 108: 8275–8280 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gookin TE, Kim J, Assmann SM. (2008) Whole proteome identification of plant candidate G-protein coupled receptors in Arabidopsis, rice, and poplar: computational prediction and in-vivo protein coupling. Genome Biol 9: R120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gregory KJ, Nguyen ED, Reiff SD, Squire EF, Stauffer SR, Lindsley CW, Meiler J, Conn PJ. (2013) Probing the metabotropic glutamate receptor 5 (mGlu5) positive allosteric modulator (PAM) binding pocket: discovery of point mutations that engender a “molecular switch” in PAM pharmacology. Mol Pharmacol 83: 991–1006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haga K, Kruse AC, Asada H, Yurugi-Kobayashi T, Shiroishi M, Zhang C, Weis WI, Okada T, Kobilka BK, Haga T, et al. (2012) Structure of the human M2 muscarinic acetylcholine receptor bound to an antagonist. Nature 482: 547–551 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanson MA, Roth CB, Jo E, Griffith MT, Scott FL, Reinhart G, Desale H, Clemons B, Cahalan SM, Schuerer SC, et al. (2012) Crystal structure of a lipid G protein-coupled receptor. Science 335: 851–855 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henikoff S, Henikoff JG. (1993) Performance evaluation of amino acid substitution matrices. Proteins 17: 49–61 [DOI] [PubMed] [Google Scholar]
- Hibert MF, Trumpp-Kallmeyer S, Hoflack J, Bruinvels A. (1993) This is not a G protein-coupled receptor. Trends Pharmacol Sci 14: 7–12 [DOI] [PubMed] [Google Scholar]
- Hoflack J, Trumpp-Kallmeyer S, Hibert M. (1994) Re-evaluation of bacteriorhodopsin as a model for G protein-coupled receptors. Trends Pharmacol Sci 15: 7–9 [DOI] [PubMed] [Google Scholar]
- Hollenstein K, Kean J, Bortolato A, Cheng RK, Doré AS, Jazayeri A, Cooke RM, Weir M, Marshall FH. (2013) Structure of class B GPCR corticotropin-releasing factor receptor 1. Nature 499: 438–443 [DOI] [PubMed] [Google Scholar]
- Hooley R. (1999) A role for G proteins in plant hormone signalling? Plant Physiol Biochem 37: 393–402 [Google Scholar]
- Horn F, Bettler E, Oliveira L, Campagne F, Cohen FE, Vriend G. (2003) GPCRDB information system for G protein-coupled receptors. Nucleic Acids Res 31: 294–297 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Humphrey W, Dalke A, Schulten K. (1996) VMD: visual molecular dynamics. J Mol Graph 14: 33–38 [DOI] [PubMed] [Google Scholar]
- Illingworth CJ, Parkes KE, Snell CR, Mullineaux PM, Reynolds CA. (2008) Criteria for confirming sequence periodicity identified by Fourier transform analysis: application to GCR2, a candidate plant GPCR? Biophys Chem 133: 28–35 [DOI] [PubMed] [Google Scholar]
- Inoue Y, Yamazaki Y, Shimizu T. (2005) How accurately can we discriminate G-protein-coupled receptors as 7-tms TM protein sequences from other sequences? Biochem Biophys Res Commun 338: 1542–1546 [DOI] [PubMed] [Google Scholar]
- Jaakola VP, Griffith MT, Hanson MA, Cherezov V, Chien EY, Lane JR, Ijzerman AP, Stevens RC. (2008) The 2.6 angstrom crystal structure of a human A2A adenosine receptor bound to an antagonist. Science 322: 1211–1217 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janetopoulos C, Jin T, Devreotes P. (2001) Receptor-mediated activation of heterotrimeric G-proteins in living cells. Science 291: 2408–2411 [DOI] [PubMed] [Google Scholar]
- Johnston CA, Taylor JP, Gao Y, Kimple AJ, Grigston JC, Chen JG, Siderovski DP, Jones AM, Willard FS. (2007) GTPase acceleration as the rate-limiting step in Arabidopsis G protein-coupled sugar signaling. Proc Natl Acad Sci USA 104: 17317–17322 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnston CA, Willard MD, Kimple AJ, Siderovski DP, Willard FS. (2008) A sweet cycle for Arabidopsis G-proteins: recent discoveries and controversies in plant G-protein signal transduction. Plant Signal Behav 3: 1067–1076 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones DT (1998) THREADER: protein sequence threading by double dynamic programming. In SL Salzberg, DB Searls, S Kasif, eds, Computational Methods in Molecular Biology. Elsevier, Amsterdam, pp 285–311 [Google Scholar]
- Jones DT. (1999a) GenTHREADER: an efficient and reliable protein fold recognition method for genomic sequences. J Mol Biol 287: 797–815 [DOI] [PubMed] [Google Scholar]
- Jones DT. (1999b) Protein secondary structure prediction based on position-specific scoring matrices. J Mol Biol 292: 195–202 [DOI] [PubMed] [Google Scholar]
- Jones DT. (2007) Improving the accuracy of transmembrane protein topology prediction using evolutionary information. Bioinformatics 23: 538–544 [DOI] [PubMed] [Google Scholar]
- Jones DT, Taylor WR, Thornton JM. (1992) A new approach to protein fold recognition. Nature 358: 86–89 [DOI] [PubMed] [Google Scholar]
- Jones DT, Taylor WR, Thornton JM. (1994) A model recognition approach to the prediction of all-helical membrane protein structure and topology. Biochemistry 33: 3038–3049 [DOI] [PubMed] [Google Scholar]
- Jones JC, Duffy JW, Machius M, Temple BR, Dohlman HG, Jones AM. (2011) The crystal structure of a self-activating G protein alpha subunit reveals its distinct mechanism of signal initiation. Sci Signal 4: ra8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Josefsson LG, Rask L. (1997) Cloning of a putative G-protein-coupled receptor from Arabidopsis thaliana. Eur J Biochem 249: 415–420 [DOI] [PubMed] [Google Scholar]
- Kahsay RY, Gao G, Liao L. (2005) An improved hidden Markov model for transmembrane protein detection and topology prediction and its applications to complete genomes. Bioinformatics 21: 1853–1858 [DOI] [PubMed] [Google Scholar]
- Kanehisa M, Goto S. (2000) KEGG: Kyoto Encyclopedia of Genes and Genomes. Nucleic Acids Res 28: 27–30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanehisa M, Goto S, Furumichi M, Tanabe M, Hirakawa M. (2010) KEGG for representation and analysis of molecular networks involving diseases and drugs. Nucleic Acids Res 38: D355–D360 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanehisa M, Goto S, Hattori M, Aoki-Kinoshita KF, Itoh M, Kawashima S, Katayama T, Araki M, Hirakawa M. (2006) From genomics to chemical genomics: new developments in KEGG. Nucleic Acids Res 34: D354–D357 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katritch V, Cherezov V, Stevens RC. (2013) Structure-function of the G protein-coupled receptor superfamily. Annu Rev Pharmacol Toxicol 53: 531–556 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kolakowski LF., Jr (1994) GCRDb: a G-protein-coupled receptor database. Receptors Channels 2: 1–7 [PubMed] [Google Scholar]
- Koval A, Katanaev VL. (2011) Wnt3a stimulation elicits G-protein-coupled receptor properties of mammalian Frizzled proteins. Biochem J 433: 435–440 [DOI] [PubMed] [Google Scholar]
- Kratochwil NA, Malherbe P, Lindemann L, Ebeling M, Hoener MC, Mühlemann A, Porter RH, Stahl M, Gerber PR. (2005) An automated system for the analysis of G protein-coupled receptor transmembrane binding pockets: alignment, receptor-based pharmacophores, and their application. J Chem Inf Model 45: 1324–1336 [DOI] [PubMed] [Google Scholar]
- Krishnan A, Almén MS, Fredriksson R, Schiöth HB. (2012) The origin of GPCRs: identification of mammalian like Rhodopsin, Adhesion, Glutamate and Frizzled GPCRs in fungi. PLoS ONE 7: e29817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krogh A, Larsson B, von Heijne G, Sonnhammer EL. (2001) Predicting transmembrane protein topology with a hidden Markov model: application to complete genomes. J Mol Biol 305: 567–580 [DOI] [PubMed] [Google Scholar]
- Lagerström MC, Schiöth HB. (2008) Structural diversity of G protein-coupled receptors and significance for drug discovery. Nat Rev Drug Discov 7: 339–357 [DOI] [PubMed] [Google Scholar]
- Lesk AM (2002) Introduction to Bioinformatics. Oxford University Press, Oxford [Google Scholar]
- Liu W, Chun E, Thompson AA, Chubukov P, Xu F, Katritch V, Han GW, Roth CB, Heitman LH, IJzerman AP, et al. (2012) Structural basis for allosteric regulation of GPCRs by sodium ions. Science 337: 232–236 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu XG, Yue YL, Li B, Nie YL, Li W, Wu WH, Ma LG. (2007) A G protein-coupled receptor is a plasma membrane receptor for the plant hormone abscisic acid. Science 315: 1712–1716 [DOI] [PubMed] [Google Scholar]
- Malbon CC. (2011) Wnt signalling: the case of the ‘missing’ G-protein. Biochem J 433: e3–e5 [DOI] [PubMed] [Google Scholar]
- McGuffin LJ, Jones DT. (2003) Improvement of the GenTHREADER method for genomic fold recognition. Bioinformatics 19: 874–881 [DOI] [PubMed] [Google Scholar]
- Melén K, Krogh A, von Heijne G. (2003) Reliability measures for membrane protein topology prediction algorithms. J Mol Biol 327: 735–744 [DOI] [PubMed] [Google Scholar]
- Miedlich SU, Gama L, Seuwen K, Wolf RM, Breitwieser GE. (2004) Homology modeling of the transmembrane domain of the human calcium sensing receptor and localization of an allosteric binding site. J Biol Chem 279: 7254–7263 [DOI] [PubMed] [Google Scholar]
- Möller S, Croning MD, Apweiler R. (2001) Evaluation of methods for the prediction of membrane spanning regions. Bioinformatics 17: 646–653 [DOI] [PubMed] [Google Scholar]
- Moriyama EN, Strope PK, Opiyo SO, Chen Z, Jones AM. (2006) Mining the Arabidopsis thaliana genome for highly-divergent seven transmembrane receptors. Genome Biol 7: R96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moult J, Fidelis K, Kryshtafovych A, Rost B, Tramontano A. (2009) Critical assessment of methods of protein structure prediction: round VIII. Proteins (Suppl 9) 77: 1–4 [DOI] [PubMed] [Google Scholar]
- Ng PC, Henikoff JG, Henikoff S. (2000) PHAT: a transmembrane-specific substitution matrix. Predicted hydrophobic and transmembrane. Bioinformatics 16: 760–766 [DOI] [PubMed] [Google Scholar]
- Okinaga S, Slattery D, Humbles A, Zsengeller Z, Morteau O, Kinrade MB, Brodbeck RM, Krause JE, Choe HR, Gerard NP, et al. (2003) C5L2, a nonsignaling C5A binding protein. Biochemistry 42: 9406–9415 [DOI] [PubMed] [Google Scholar]
- Palczewski K, Kumasaka T, Hori T, Behnke CA, Motoshima H, Fox BA, Le Trong I, Teller DC, Okada T, Stenkamp RE, et al. (2000) Crystal structure of rhodopsin: a G protein-coupled receptor. Science 289: 739–745 [DOI] [PubMed] [Google Scholar]
- Pandey S, Assmann SM. (2004) The Arabidopsis putative G protein-coupled receptor GCR1 interacts with the G protein alpha subunit GPA1 and regulates abscisic acid signaling. Plant Cell 16: 1616–1632 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pandey S, Nelson DC, Assmann SM. (2009) Two novel GPCR-type G proteins are abscisic acid receptors in Arabidopsis. Cell 136: 136–148 [DOI] [PubMed] [Google Scholar]
- Plakidou-Dymock S, Dymock D, Hooley R. (1998) A higher plant seven-transmembrane receptor that influences sensitivity to cytokinins. Curr Biol 8: 315–324 [DOI] [PubMed] [Google Scholar]
- Rajagopal S, Kim J, Ahn S, Craig S, Lam CM, Gerard NP, Gerard C, Lefkowitz RJ. (2010a) Beta-arrestin- but not G protein-mediated signaling by the “decoy” receptor CXCR7. Proc Natl Acad Sci USA 107: 628–632 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rajagopal S, Rajagopal K, Lefkowitz RJ. (2010b) Teaching old receptors new tricks: biasing seven-transmembrane receptors. Nat Rev Drug Discov 9: 373–386 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ray S, Chen Y, Ayoung J, Hanna R, Brazill D. (2011) Phospholipase D controls Dictyostelium development by regulating G protein signaling. Cell Signal 23: 335–343 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenkilde MM, Kledal TN, Schwartz TW. (2005) High constitutive activity of a virus-encoded seven transmembrane receptor in the absence of the conserved DRY motif (Asp-Arg-Tyr) in transmembrane helix 3. Mol Pharmacol 68: 11–19 [DOI] [PubMed] [Google Scholar]
- Roy A, Kucukural A, Zhang Y. (2010) I-TASSER: a unified platform for automated protein structure and function prediction. Nat Protoc 5: 725–738 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roy A, Taddese B, Vohra S, Thimmaraju PK, Illingworth CJ, Simpson LM, Mukherjee K, Reynolds CA, Chintapalli SV. (March 25, 2013) Identifying subset errors in multiple sequence alignments. J Biomol Struct Dyn http://dx.doi.org/10.1080/07391102.2013.770371 [DOI] [PubMed] [Google Scholar]
- Sandberg M, Eriksson L, Jonsson J, Sjöström M, Wold S. (1998) New chemical descriptors relevant for the design of biologically active peptides: a multivariate characterization of 87 amino acids. J Med Chem 41: 2481–2491 [DOI] [PubMed] [Google Scholar]
- Selent J, Sanz F, Pastor M, De Fabritiis G. (2010) Induced effects of sodium ions on dopaminergic G-protein coupled receptors. PLoS Comput Biol 6: e1000884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheikh SP, Vilardarga JP, Baranski TJ, Lichtarge O, Iiri T, Meng EC, Nissenson RA, Bourne HR. (1999) Similar structures and shared switch mechanisms of the beta2-adrenoceptor and the parathyroid hormone receptor: Zn(II) bridges between helices III and VI block activation. J Biol Chem 274: 17033–17041 [DOI] [PubMed] [Google Scholar]
- Shi J, Blundell TL, Mizuguchi K. (2001) FUGUE: sequence-structure homology recognition using environment-specific substitution tables and structure-dependent gap penalties. J Mol Biol 310: 243–257 [DOI] [PubMed] [Google Scholar]
- Shimamura T, Shiroishi M, Weyand S, Tsujimoto H, Winter G, Katritch V, Abagyan R, Cherezov V, Liu W, Han GW, et al. (2011) Structure of the human histamine H1 receptor complex with doxepin. Nature 475: 65–70 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siu FY, He M, de Graaf C, Han GW, Yang D, Zhang Z, Zhou C, Xu Q, Wacker D, Joseph JS, et al. (2013) Structure of the human glucagon class B G-protein-coupled receptor. Nature 499: 444–449 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Söding J, Biegert A, Lupas AN. (2005) The HHpred interactive server for protein homology detection and structure prediction. Nucleic Acids Res 33: W244–W248 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stamm M, Staritzbichler R, Khafizov K, Forrest LR. (2013) Alignment of helical membrane protein sequences using AlignMe. PLoS ONE 8: e57731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Styczynski MP, Jensen KL, Rigoutsos I, Stephanopoulos G. (2008) BLOSUM62 miscalculations improve search performance. Nat Biotechnol 26: 274–275 [DOI] [PubMed] [Google Scholar]
- Taddese B, Simpson LM, Wall ID, Blaney FE, Kidley NJ, Clark HS, Smith RE, Upton GJ, Gouldson PR, Psaroudakis G, et al. (2012) G-protein-coupled receptor dynamics: dimerization and activation models compared with experiment. Biochem Soc Trans 40: 394–399 [DOI] [PubMed] [Google Scholar]
- Taddese B, Simpson LM, Wall ID, Blaney FE, Reynolds CA. (2013) Modeling active GPCR conformations. Methods Enzymol 522: 21–35 [DOI] [PubMed] [Google Scholar]
- Tusnády GE, Simon I. (1998) Principles governing amino acid composition of integral membrane proteins: application to topology prediction. J Mol Biol 283: 489–506 [DOI] [PubMed] [Google Scholar]
- Tusnády GE, Simon I. (2001) The HMMTOP transmembrane topology prediction server. Bioinformatics 17: 849–850 [DOI] [PubMed] [Google Scholar]
- Urano D, Chen JG, Botella JR, Jones AM. (2013) Heterotrimeric G protein signalling in the plant kingdom. Open Biol 3: 120186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Urano D, Jones AM. (2013) “Round up the usual suspects”: a comment on nonexistent plant G protein-coupled receptors. Plant Physiol 161: 1097–1102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Urano D, Jones JC, Wang H, Matthews M, Bradford W, Bennetzen JL, Jones AM. (2012) G protein activation without a GEF in the plant kingdom. PLoS Genet 8: e1002756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Venkatakrishnan AJ, Deupi X, Lebon G, Tate CG, Schertler GF, Babu MM. (2013) Molecular signatures of G-protein-coupled receptors. Nature 494: 185–194 [DOI] [PubMed] [Google Scholar]
- Viklund H, Bernsel A, Skwark M, Elofsson A. (2008) SPOCTOPUS: a combined predictor of signal peptides and membrane protein topology. Bioinformatics 24: 2928–2929 [DOI] [PubMed] [Google Scholar]
- Viklund H, Elofsson A. (2008) OCTOPUS: improving topology prediction by two-track ANN-based preference scores and an extended topological grammar. Bioinformatics 24: 1662–1668 [DOI] [PubMed] [Google Scholar]
- Viklund H, Granseth E, Elofsson A. (2006) Structural classification and prediction of reentrant regions in alpha-helical transmembrane proteins: application to complete genomes. J Mol Biol 361: 591–603 [DOI] [PubMed] [Google Scholar]
- Vohra S, Chintapalli SV, Illingworth CJ, Reeves PJ, Mullineaux PM, Clark HS, Dean MK, Upton GJ, Reynolds CA. (2007) Computational studies of family A and family B GPCRs. Biochem Soc Trans 35: 749–754 [DOI] [PubMed] [Google Scholar]
- Vohra S, Taddese B, Conner AC, Poyner DR, Hay DL, Barwell J, Reeves PJ, Upton GJ, Reynolds CA. (2013) Similarity between class A and class B G-protein-coupled receptors exemplified through calcitonin gene-related peptide receptor modelling and mutagenesis studies. J R Soc Interface 10: 20120846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vroling B, Sanders M, Baakman C, Borrmann A, Verhoeven S, Klomp J, Oliveira L, de Vlieg J, Vriend G. (2011) GPCRDB: information system for G protein-coupled receptors. Nucleic Acids Res 39: D309–D319 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang C, Wu H, Katritch V, Han GW, Huang XP, Liu W, Siu FY, Roth BL, Cherezov V, Stevens RC. (2013) Structure of the human smoothened receptor bound to an antitumour agent. Nature 497: 338–343 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warne T, Serrano-Vega MJ, Baker JG, Moukhametzianov R, Edwards PC, Henderson R, Leslie AG, Tate CG, Schertler GF. (2008) Structure of a beta1-adrenergic G-protein-coupled receptor. Nature 454: 486–491 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whalen EJ, Rajagopal S, Lefkowitz RJ. (2011) Therapeutic potential of β-arrestin- and G protein-biased agonists. Trends Mol Med 17: 126–139 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wheatley M, Wootten D, Conner M, Simms J, Kendrick R, Logan R, Poyner D, Barwell J. (2012) Lifting the lid on GPCRs: the role of extracellular loops. Br J Pharmacol 165: 1688–1703 [DOI] [PMC free article] [PubMed] [Google Scholar]
- White JF, Noinaj N, Shibata Y, Love J, Kloss B, Xu F, Gvozdenovic-Jeremic J, Shah P, Shiloach J, Tate CG, et al. (2012) Structure of the agonist-bound neurotensin receptor. Nature 490: 508–513 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams JG, Noegel AA, Eichinger L. (2005) Manifestations of multicellularity: Dictyostelium reports in. Trends Genet 21: 392–398 [DOI] [PubMed] [Google Scholar]
- Woolley MJ, Watkins HA, Taddese B, Karakullukcu ZG, Barwell J, Smith KJ, Hay DL, Poyner DR, Reynolds CA, Conner AC. (2013) The role of ECL2 in CGRP receptor activation: a combined modelling and experimental approach. J R Soc Interface 10: 20130589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu B, Chien EY, Mol CD, Fenalti G, Liu W, Katritch V, Abagyan R, Brooun A, Wells P, Bi FC, et al. (2010) Structures of the CXCR4 chemokine GPCR with small-molecule and cyclic peptide antagonists. Science 330: 1066–1071 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu S, Zhang Y. (2007) LOMETS: a local meta-threading-server for protein structure prediction. Nucleic Acids Res 35: 3375–3382 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu S, Zhang Y. (2008) MUSTER: improving protein sequence profile-profile alignments by using multiple sources of structure information. Proteins 72: 547–556 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan J, Mihaylov V, Xu X, Brzostowski JA, Li H, Liu L, Veenstra TD, Parent CA, Jin T. (2012) A Gβγ effector, ElmoE, transduces GPCR signaling to the actin network during chemotaxis. Dev Cell 22: 92–103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang C, Srinivasan Y, Arlow DH, Fung JJ, Palmer D, Zheng Y, Green HF, Pandey A, Dror RO, Shaw DE, et al. (2012) High-resolution crystal structure of human protease-activated receptor 1. Nature 492: 387–392 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y. (2008) I-TASSER server for protein 3D structure prediction. BMC Bioinformatics 9: 40. [DOI] [PMC free article] [PubMed] [Google Scholar]