

Abstract
RNA-guided endonucleases (RGENs), derived from the prokaryotic adaptive immune system known as CRISPR/Cas, enable targeted genome engineering in cells and organisms. RGENs are ribonucleoproteins that consist of guide RNA and Cas9, a protein component originated from Streptococcus pyogenes. These enzymes cleave chromosomal DNA, whose sequence is complementary, to guide RNA in a targeted manner, producing site-specific DNA double-strand breaks (DSBs), the repair of which gives rise to targeted genome modifications. Despite broad interest in RGEN-mediated genome editing, these nucleases are limited by off-target mutations and unwanted chromosomal translocations associated with off-target DNA cleavages. Here, we show that off-target effects of RGENs can be reduced below the detection limits of deep sequencing by choosing unique target sequences in the genome and modifying both guide RNA and Cas9. We found that both the composition and structure of guide RNA can affect RGEN activities in cells to reduce off-target effects. RGENs efficiently discriminated on-target sites from off-target sites that differ by two bases. Furthermore, exome sequencing analysis showed that no off-target mutations were induced by two RGENs in four clonal populations of mutant cells. In addition, paired Cas9 nickases, composed of D10A Cas9 and guide RNA, which generate two single-strand breaks (SSBs) or nicks on different DNA strands, were highly specific in human cells, avoiding off-target mutations without sacrificing genome-editing efficiency. Interestingly, paired nickases induced chromosomal deletions in a targeted manner without causing unwanted translocations. Our results highlight the importance of choosing unique target sequences and optimizing guide RNA and Cas9 to avoid or reduce RGEN-induced off-target mutations.
Programmable nucleases, which include zinc finger nucleases (ZFNs) (Bibikova et al. 2003; Urnov et al. 2005; Kim et al. 2011), transcription activator-like effector (TALE) nucleases (TALENs) (Miller et al. 2011; Kim et al. 2013a), and RNA-guided endonucleases (RGENs) (Cho et al. 2013a; Cong et al. 2013; Hwang et al. 2013; Jinek et al. 2013; Mali et al. 2013b), enable efficient genome editing in cells and organisms. These nucleases cleave chromosomal DNA in a targeted manner, producing site-specific DNA double-strand breaks (DSBs), the repair of which via homologous recombination (HR) or nonhomologous end-joining (NHEJ) leads to gene disruption (Santiago et al. 2008; Sung et al. 2013), addition (Moehle et al. 2007), correction (Urnov et al. 2005), and targeted chromosomal rearrangements (Brunet et al. 2009; Lee et al. 2010, 2012). Programmable nuclease-based genome editing is now widely used for making gene knockout/knock-in animals and plants as well as genome-modified cell lines.
RGENs are derived from the type II CRISPR (clusters of regularly interspaced palindromic repeats)/Cas (CRISPR-associated) system, an adaptive immune response in bacteria and archaea (Wiedenheft et al. 2012). Cas9, the protein component derived from Streptococcus pyogenes, forms an active nuclease when complexed with transactivating CRISPR RNA (tracrRNA) and CRISPR RNA (crRNA) (Jinek et al. 2012), which are transcribed from the CRISPR sequence encoded in the bacterial genome. This ribonucleoprotein protects host cells from invading phages or plasmids by recognizing and cleaving the DNA sequence corresponding to the crRNA sequence. Recently, we and others have exploited this system to induce site-specific DSBs, thereby modifying genomes in a targeted manner in cells and organisms (Chang et al. 2013; Cho et al. 2013a,b; Cong et al. 2013; Ding et al. 2013; Gratz et al. 2013; Hwang et al. 2013; Jiang et al. 2013; Jinek et al. 2013; Mali et al. 2013b; Shen et al. 2013; Wang et al. 2013).
The specificity of an RGEN is determined by the 20-base pair (bp) sequence in crRNA and the NGG trinucleotide (known as the protospacer-adjacent motif [PAM]) that is recognized by Cas9. Thus, an RGEN recognizes and cleaves a 22-bp target sequence, which can be shown as 5′-X20NGG-3′, where X20 corresponds to the crRNA sequence, and N is any base. Essential portions of crRNA and tracrRNA can be linked to make single-guide RNA (sgRNA). Although the 22-bp specificities of RGENs might be sufficient to ensure targeted mutagenesis without off-target effects in higher eukaryotic cells, a few careful studies duly raised concerns that RGENs might cause collateral damage in the genome. Thus, Cas9 complexed with a crRNA that contains a one-base mismatch with the target sequence can cleave the target DNA, albeit less efficiently, both in vitro (Jinek et al. 2012) and in mammalian (Cong et al. 2013) and bacterial cells (Jiang et al. 2013). RGENs tolerate mismatches especially in the 5′ upstream region, but not in the seed region of 6–11 nt that is immediately upstream of the PAM sequence. Very recently, three groups independently showed that RGENs indeed induce off-target mutations, even at sites that differ by 5 nt from on-target sites in human cells (Fu et al. 2013; Hsu et al. 2013; Pattanayak et al. 2013).
We previously addressed this issue by using the T7 endonuclease I (T7E1) assay and showed that RGENs do not induce mutations at potential off-target sites that are highly homologous to on-target sites in human cells (Cho et al. 2013a); but due to its poor sensitivity, this assay cannot detect off-target mutations that occur at frequencies <1% (Kim et al. 2009). Here, we address this critical issue more thoroughly by using deep sequencing, which can measure off-target mutations that occur at frequencies ranging from 0.01% to 0.1%. In addition, we isolated clonal populations of cells that contained RGEN-induced mutations and investigated whether these mutant cells harbor any off-target mutations in the exome. Our results show that off-target effects of RGENs can be reduced in human cells by choosing unique target sequences and modifying the structure of crRNA or sgRNA. In addition, we report that paired Cas9 nickases that generate two single-strand breaks (SSBs) or nicks on different DNA strands are highly specific in human cells, avoiding off-target mutations without sacrificing genome-editing efficiency.
Results
Analysis of off-target effects of two RGENs at homologous sites
First, we searched for potential off-target sites in the human genome that might be recognized by two RGENs specific for the C4BPB and CCR5 genes (Supplemental Table 1). We chose (1) those sites (CCR5, Off1 to Off14 and C4BPB, Off1 to Off6 in Fig. 1) whose sequences are conserved in the seed region but contain 3–8-base mismatches in the 5′ end; and (2) those sites (CCR5, Off15 to Off28 and C4BPB, Off7 to Off10) whose entire 22-bp sequences are most similar to the target sequence and differ by up to 3 nt from the target sequence. We measured the frequencies of small insertions/deletions (indels) at the on-target site and putative off-target sites using deep sequencing (Fig. 1A,B). The two RGENs induced indels at frequencies of 75% (C4BPB) and 60% (CCR5) at corresponding on-target sites in K562 cells. In sharp contrast, no indels were detected at any measurable frequency (∼0.01%) at any of the putative off-target sites we examined. Note that the CCR5 RGEN did not induce any indels at two sites (Off15 and Off16) that each contained a one-base mismatch in the 11-bp seed region and another one-base mismatch in the upstream region. The Off17 site contained a single-base mismatch in the 20-bp sequence corresponding to the crRNA and another single-base mismatch in the PAM. No indels were detected at this site, suggesting that PAM recognition by Cas9 is also critical.
Figure 1.
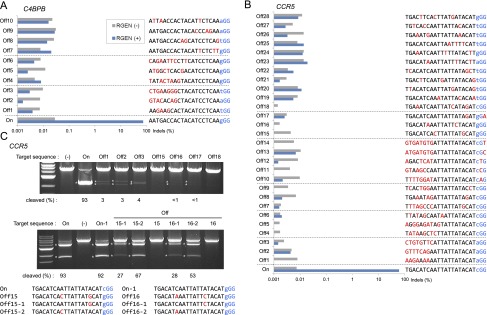
Mutation frequencies at on-target and potential off-target sites of the C4BPB- and CCR5-specific RGENs in K562 cells. (A,B) Cells were transfected with crRNA, tracrRNA, and the Cas9 plasmid or the Cas9 plasmid alone (negative control). PCR amplicons that span the on-target site and potential off-target sites were subjected to deep sequencing. Sequences that contained indels around the expected cleavage site were considered to be RGEN-induced mutations. Mismatched bases are shown in red. The PAM sequence is shown in blue. (C) In vitro cleavage assay of on-target or potential off-target sequences by the CCR5-specific RGEN. Plasmids that contain putative off-target (upper) or hybrid (middle) sequences were digested with the recombinant Cas9 protein complexed with crRNA and tracrRNA. Asterisks indicate cleaved DNA bands. (Bottom) DNA sequences of the on-target, off-target, and hybrid sites.
We also tested these two RGENs in another human cell line, HeLa (Supplemental Fig. 1A,B), and found that these RGENs, again, discriminated on-target sites from their most likely off-target sites by a factor of >10,000-fold. It is worth noting that off-target mutations were not detected in either K562 or HeLa cells at the C4BPB Off1 site, whose three mismatches are all located in the upstream region. Taken together, these results suggest that the 5′ upstream region outside the seed region in the target sequence also contributes to the specificity of RGEN cleavage, and that RGENs cannot efficiently cleave chromosomal DNA with mismatches of two or more nucleotides, at least one of which occurs in the seed region.
We then examined whether RGENs can differentially cleave target and off-target DNA sequences in vitro. We incubated the recombinant Cas9 protein/crRNA/tracrRNA complex with plasmids that contained either the CCR5 on-target sequence or off-target sequences. Interestingly, the CCR5-specific RGEN displayed a low level (<1%) of cleavage activity toward plasmids that contained the off-target sequences (Fig. 1C), suggesting that RGENs function less discriminatingly in vitro than they do in cells under our experimental conditions. We also constructed and tested plasmids that contained hybrid sequences with a single-base mismatch (Off15-1, 15-2, 16-1, and 16-2 in Fig. 1C). These hybrid sequences were cleaved much more efficiently by the ribonucleoprotein than were the off-target sequences with two-base mismatches (Off15 and Off16). This result suggests that RGENs can distinguish on-target sites from off-target sites with two-base mismatches but not those with a single-base mismatch.
Analysis of off-target effects of additional RGENs
To investigate off-target effects of RGENs further, we tested nine additional RGENs that target the CCR5 gene. CCR5 is highly homologous to CCR2: Both genes encode G-protein-coupled chemokine receptors. We chose these additional sites so that the RGENs would have off-target sites in the CCR2 gene that harbor one- or two-base mismatches. We measured and compared mutation frequencies of these RGENs at the CCR5 on-target and CCR2 off-target sites using deep sequencing. Three RGENs (#1, 2, and 3 in Fig. 2) did not induce any off-target mutations at the CCR2 sites that carried two-base mismatches: Indel frequencies were not much different from those obtained with the empty vector control. One RGEN (#4) induced off-target mutations at the CCR2 site that carried a one-base mismatch in the seed region and another one-base mismatch in the upstream region at a frequency of 1.8%, 29-fold lower than the on-target mutation frequency.
Figure 2.

Mutation frequencies at CCR5 on-target sites and CCR2 off-target sites. K562 cells were transfected with RGENs that target nine different sites. PCR amplicons that span the on-target and off-target sites were subjected to deep sequencing.
We found, however, that RGENs cannot efficiently discriminate on-target sites from off-target sites that differ by a single base. Thus, all four RGENs (#5–#8) induced indels at frequencies that ranged from 1.6% to 43% at off-target sites that carried a single-base mismatch either in the seed region or the upstream region. Two RGENs (#5 and #8) were still able to discriminate on-target sites from off-target sites with a single-base mismatch by a factor of 7.2- to 42-fold. One RGEN (#9) had an off-target site with a single-base mismatch in the PAM. This RGEN discriminated the on-target site from the off-target site by 490-fold, confirming, again, the importance of PAM recognition by Cas9. These RGENs did not induce any indels at several potential off-target sites identified in other loci (Supplemental Table 2). Taken together, these results suggest that RGENs can distinguish on-target sites from off-target sites that differ by at least two bases, and that one could avoid or minimize off-target effects of RGENs by choosing unique target sites that do not have homologous sequences elsewhere in the genome.
Exome sequencing analysis of RGEN-induced mutant clones
Next, we performed exome sequencing to identify RGEN-induced off-target mutations, if any, on a genomic scale. Using limiting dilution, we isolated clonal populations of CCR5- or C4BPB-disrupted cells that contained RGEN-induced indels (Supplemental Fig. 2). Of 37 or 47 clones, 29 (78%) or 35 (74%) had mutations in the CCR5 or C4BPB genes, respectively. These high mutation frequencies were comparable to those observed with deep sequencing (Fig. 1A,B). Genomic DNA isolated from one C4BPB-disrupted clone, two CCR5-disrupted clones, and wild-type K562 cells was subjected to exome sequencing. Analysis of the exome sequence data revealed a total of 308 indels present in the four clones, including wild-type cells, compared to the reference genome—255 in CCR5-disrupted clone #1, 246 in CCR5 clone #2, 233 in the C4BPB clone, and 253 in the wild-type clone (the numbers are comparable in these clones) (Supplemental Fig. 3A). Among these, 190 indels were present in all four clones, including wild-type cells, and 74 indels were shared in at least two clones. These indels were excluded from further analysis. Ten indels were found only in the wild-type clone, which suggests that the coverage of exome sequencing was not 100%. Likewise, 8, 9, and 17 indels were found only in the C4BPB clone, CCR5 clone #1, and CCR5 clone #2, respectively (Supplemental Table 3). We excluded 13 indels reported in a public database (dbSNP build 130, NCBI), because they are unlikely to result from RGEN-mediated DNA cleavages. We also used Sanger and deep sequencing to show that most of these “clone-specific” indels are present even in wild-type cells. We then searched for putative off-target sequences around the remaining indels in the reference genome (Supplemental Table 4). Other than the on-target mutations induced by the RGENs in these clones, none of these indels were associated with potential off-target sites (Supplemental Table 3; Methods). It is most likely that the indels we found in gene-disrupted clones were the result of incomplete coverage of exome sequencing (Clark et al. 2011). Exons are captured before sequencing, but this process covers only <90% of the human exome. More than 10% of exons missed in wild-type cells can be captured in mutant clones. To rule out the possibility that the remaining sites are cleaved by off-target RGEN activity (although their sequences are quite different from the on-target sequences), we incubated the RGENs with PCR amplicons that contain the potential off-target sites in vitro. No cleavage was detected (Supplemental Fig. 3B), suggesting that these sites are not off-target sites. We note, however, that exome sequencing analysis is limited by high false negative results (Karakoc et al. 2012): Off-target indels present in clones might have been missed.
Guide RNA structure
Recently, three groups reported that RGENs had off-target effects in human cells (Fu et al. 2013; Hsu et al. 2013; Pattanayak et al. 2013). To our surprise, RGENs induced mutations efficiently at off-target sites that differ by 3–5 nt from on-target sites. We noticed, however, that there were several differences between our RGENs and those used by others. First, we used dualRNA, which is crRNA plus tracrRNA, rather than single-guide RNA (sgRNA) that is composed of essential portions of crRNA and tracrRNA. Second, we transfected K562 cells (but not HeLa cells) with synthetic crRNA rather than plasmids encoding crRNA. HeLa cells were transfected with crRNA-encoding plasmids. Other groups used sgRNA-encoding plasmids. Third, our guide RNA had two additional guanine nucleotides at the 5′ end, which are required for efficient transcription by T7 polymerase in vitro. No such additional nucleotides were included in the sgRNA used by others. Thus, the RNA sequence of our guide RNA can be shown as 5′-GGX20, whereas 5′-GX19, in which X20 or GX19 corresponds to the 20-bp target sequence, represents the sequence used by others. The first guanine nucleotide is required for transcription by RNA polymerase in cells. To test whether off-target RGEN effects can be attributed to these differences, we chose four RGENs that induced off-target mutations in human cells at high frequencies (Fu et al. 2013). First, we compared our method of using in vitro transcribed dualRNA with the method of transfecting sgRNA-encoding plasmids in K562 cells and measured mutation frequencies at the on-target and off-target sites via the T7E1 assay. Three RGENs showed comparable mutation frequencies at on-target and off-target sites regardless of the composition of guide RNA. Interestingly, one RGEN (VEGFA site 1) did not induce indels at one validated off-target site, which differs by 3 nt from the on-target site (termed OT1-11) (Supplemental Fig. 4), when synthetic dualRNA was used. However, the synthetic dualRNA did not discriminate the other validated off-target site (OT1-3), which differs by 2 nt from the on-target site.
Next, we tested whether the addition of two guanine nucleotides at the 5′ end of sgRNA could make RGENs more specific by comparing 5′-GGX20 (or 5′-GGGX19) sgRNA with 5′-GX19 sgRNA. Four GX19 sgRNAs complexed with Cas9 induced indels equally efficiently at on-target and off-target sites, tolerating up to 4 nucleotide mismatches. In sharp contrast, GGX20 sgRNAs discriminated off-target sites effectively. In fact, the T7E1 assay barely detected RGEN-induced indels at six of the seven validated off-target sites when we used the four GGX20 sgRNAs (Fig. 3). We noticed, however, that two GGX20 sgRNAs (VEGFA sites 1 and 3) were less active at on-target sites than were the corresponding GX19 sgRNAs. These results show that the extra nucleotides at the 5′ end can affect mutation frequencies at on-target and off-target sites, perhaps by altering guide RNA stability, concentration, or secondary structure. Our results suggest that three factors—the use of synthetic guide RNA rather than guide RNA-encoding plasmids, dualRNA rather than sgRNA, and GGX20 sgRNA rather than GX19 sgRNA—have cumulative effects on the discrimination of off-target sites.
Figure 3.

Comparison of guide RNA structure. Mutation frequencies of the RGENs reported in Fu et al. (2013) were measured at on-target and off-target sites using the T7E1 assay. K562 cells were cotransfected with the Cas9-encoding plasmid and the plasmid encoding GX19 sgRNA or GGX20 sgRNA. Off-target sites (OT1-3, etc.) are labeled as in Fu et al. (2013).
Paired Cas9 nickases
Previously, we showed that two zinc finger (ZF) nickases that generate two adjacent SSBs or nicks on different DNA strands, which give rise to a composite DSB, enable targeted mutagenesis efficiently in human cells (Kim et al. 2012). In principle, SSBs cannot be repaired by error-prone NHEJ but still trigger high fidelity homology-directed repair (HDR) or base excision repair; but nickase-induced targeted mutagenesis via HDR is much less efficient than is nuclease-induced mutagenesis (Ramirez et al. 2012; Wang et al. 2012). We reasoned that paired Cas9 nickases would produce composite DSBs, which trigger DNA repair via NHEJ or HDR, leading to efficient mutagenesis (Fig. 4A). Furthermore, paired nickases would double the specificity of Cas9-based genome editing.
Figure 4.

In vitro DNA cleavage by Cas9 nickases. (A) Schematic overview of the Cas9 nuclease and the paired Cas9 nickase. The PAM sequences and cleavage sites are shown in red. (B) Target sites in the human AAVS1 locus. The position of each target site is shown in red. (C) Schematic overview of DNA cleavage reactions. FAM dyes (shown in green) were linked to both 5′ ends of the DNA substrate. (D) DSBs and SSBs analyzed using fluorescent capillary electrophoresis. Fluorescently labeled DNA substrates were incubated with Cas9 nucleases or nickases before electrophoresis.
We first tested several Cas9 nucleases and nickases designed to target sites in the AAVS1 locus (Fig. 4B) in vitro via fluorescent capillary electrophoresis. Unlike Cas9 nucleases that cleaved both strands of DNA substrates, Cas9 nickases composed of guide RNA and a mutant form of Cas9 in which a catalytic aspartate residue is changed to an alanine (D10A Cas9) cleaved only one strand, producing site-specific nicks (Fig. 4C,D). Interestingly, however, some nickases (AS1, AS2, AS3, and S6 in Fig. 5A) induced indels at target sites in human cells, suggesting that nicks can be converted to DSBs, albeit inefficiently, in vivo. Paired Cas9 nickases producing two adjacent nicks on different DNA strands yielded indels at frequencies that ranged from 14% to 91%, comparable to the effects of paired nucleases (Fig. 5A). In line with two recent reports (Mali et al. 2013a; Ran et al. 2013), the repair of two nicks that would produce 5′ overhangs led to the formation of indels much more frequently than those producing 3′ overhangs at three genomic loci (Fig. 5A; Supplemental Fig. 5). In addition, paired nickases enabled targeted genome editing via homology-directed repair more efficiently than did single nickases (Supplemental Fig. 6).
Figure 5.
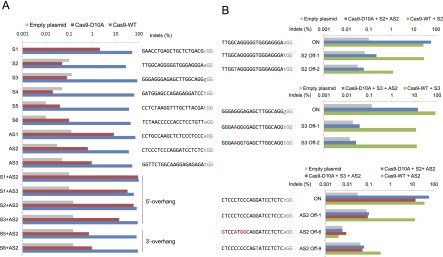
Comparison of Cas9 nuclease and nickase behavior. (A) On-target mutation frequencies associated with Cas9 nucleases (WT), nickases (D10A), and paired nickases. Paired nickases that would produce 5′ overhangs or 3′ overhangs are indicated. (B) Analysis of off-target effects of Cas9 nucleases and paired nickases. A total of seven potential off-target sites for three sgRNAs were analyzed.
We next measured mutation frequencies of paired nickases and nucleases at off-target sites using deep sequencing. Cas9 nucleases complexed with three sgRNAs induced off-target mutations at six sites that differ by 1 or 2 nt from their corresponding on-target sites with frequencies that ranged from 0.5% to 10% (Fig. 5B). In contrast, paired Cas9 nickases did not produce indels above the detection limit of 0.1% at any of the six off-target sites. The S2 Off-1 site that differs by a single nucleotide at the first position in the PAM (i.e., N in NGG) from its on-target site can be considered as another on-target site. As expected, the Cas9 nuclease complexed with the S2 sgRNA was equally efficient at this site and the on-target site. In sharp contrast, D10A Cas9 complexed with the S2 and AS2 sgRNAs discriminated this site from the on-target site by a factor of 270-fold. This paired nickase also discriminated the AS2 off-target sites (Off-1 and Off-9 in Fig. 5B) from the on-target site by factors of 160-fold and 990-fold, respectively.
Chromosomal DNA splicing induced by paired Cas9 nickases
We and others have shown previously that two concurrent DSBs produced by engineered nucleases, such as ZFNs and TALENs, can promote large deletions of the intervening chromosomal segments (Carlson et al. 2012). We tested whether two SSBs induced by paired Cas9 nickases can also produce deletions in human cells. We used PCR to detect deletion events and found that seven paired nickases induced deletions of up to 1.1-kbp chromosomal segments as efficiently as paired Cas9 nucleases did (Fig. 6A,B). DNA sequences of the PCR products confirmed the deletion events (Fig. 6C). Interestingly, the sgRNA-matching sequence remained intact in two of seven deletion-specific PCR amplicons (underlined in Fig. 6C). In contrast, Cas9 nuclease pairs did not produce sequences that contained intact target sites. This finding suggests that two distant nicks were not converted to two separate DSBs to promote deletions of the intervening chromosomal segment. In addition, it is unlikely that two nicks separated by more than 100 bp can produce a composite DSB with large overhangs under physiological conditions because the melting temperature is very high. We propose that two distant nicks are repaired by strand displacement in a head-to-head direction, resulting in the formation of a DSB in the middle, whose repair via NHEJ causes small deletions (Fig. 6D). Because the two target sites remain intact during this process, nickases can induce SSBs again, triggering the cycle repeatedly until the target sites are deleted. This mechanism explains why two offset nicks producing 5′ overhangs but not those producing 3′ overhangs induced indels efficiently at three loci.
Figure 6.
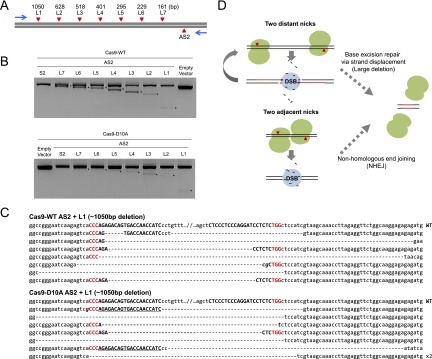
DNA splicing induced by paired Cas9 nickases. (A) The target sites of paired nickases in the human AAVS1 locus. The distances between the AS2 site and each of the other sites are shown. Arrows indicate PCR primers. (B) Genomic deletions detected using PCR. Asterisks indicate deletion-specific PCR products. (C) DNA sequences of deletion-specific PCR products obtained using AS2 and L1 sgRNAs. Target site PAM sequences are shown in red, and sgRNA-matching sequences are shown in capital letters. Intact sgRNA-matching sequences are underlined. (D) A schematic model of paired Cas9 nickase-mediated chromosomal deletions. Newly synthesized DNA strands are shown in red.
We then investigated whether Cas9 nucleases and nickases can induce unwanted chromosomal translocations that result from NHEJ repair of on-target and off-target DNA cleavages (Fig. 7A). We were able to detect translocations induced by Cas9 nucleases using PCR (Fig. 7B,C). No such PCR products were amplified using genomic DNA isolated from cells transfected with the plasmids encoding the AS2 + S3 Cas9 nickase pair. This result is in line with the fact that both AS2 and S3 nickases, unlike their corresponding nucleases, did not produce indels at off-target sites (Fig. 5B).
Figure 7.
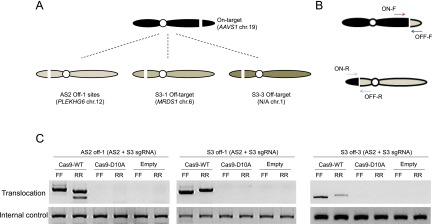
Paired Cas9 nickases do not induce translocations. (A) Schematic overview of chromosomal translocations between the on-target and off-target sites. (B) PCR amplification to detect chromosomal translocations. (C) Translocations induced by Cas9 nucleases but not by the nickase pair.
Discussion
Applications of engineered nucleases in research, biotechnology, and medicine are hampered by their off-target effects. Both ZFNs and TALENs can induce mutations at off-target sites that are highly homologous to on-target sites (Gabriel et al. 2011; Mussolino et al. 2011; Pattanayak et al. 2011). Too much off-target DNA cleavage causes cellular toxicity (Kim et al. 2009). Furthermore, the repair of off-target DSBs gives rise to unwanted chromosomal rearrangements such as deletions, inversions, and translocations (Brunet et al. 2009; Lee et al. 2010; Lee et al. 2012; Kim et al. 2013a). The possibility that unknown off-target mutations may cause activation of oncogenes or inactivation of tumor suppressor genes is of particular concern to researchers interested in using engineered nucleases in gene or cell therapy.
RGENs are a new member in the growing family of programmable nucleases but are distinguished from the other members, ZFNs and TALENs, in several respects. First, RGENs function as monomers, whereas ZFNs and TALENs work in pairs to cleave DNA. Use of an obligatory heterodimeric FokI nuclease domain can make ZFNs and TALENs even more specific (Miller et al. 2007; Szczepek et al. 2007). Second, the specificity of these FokI-based nucleases can be enhanced by increasing the number of zinc finger or TALE modules (Reyon et al. 2012). ZFNs and TALENs that recognize DNA sequences longer than 30-bp are now routinely used. In contrast, the specificity of RGENs is fixed to 22 bp. Third, zinc fingers (Bae et al. 2003) and TALEs (Boch et al. 2009) are derived from transcription factors that function in higher eukaryotic organisms, whereas RGENs are derived from prokaryotes whose genomes are ∼1000-fold smaller than those of higher eukarytotes. The specificity of RGENs might be limited to the small size of prokaryotic genomes. These differences suggest that RGENs may have more severe off-target effects than do ZFNs and TALENs.
Unlike ZFNs and TALENs, whose DNA-targeting specificity is determined solely by protein–DNA interactions that are often context-dependent and unpredictable, RGENs recognize target sites mostly by the simple Watson-Crick base-pairing rules. For example, ZFNs induce off-target mutations at cryptic sites that cannot be predicted based on sequence homology alone (Gabriel et al. 2011). It is unlikely, however, that RGENs cleave off-target DNA sequences whose recognition does not follow the base-pairing rules. Thus, off-target sites of RGENs can be more reliably predicted by sequence homology alone than are those of ZFNs or TALENs. The question is then how many mismatches RGENs can tolerate to trigger off-target DNA cleavage.
In this study, we showed that RGENs do indeed induce off-target mutations at sites with a single-base mismatch. We found, however, that RGENs efficiently discriminate on-target sites from off-target sites that differ by only two bases. Furthermore, exome sequencing showed that no off-target mutations were present in four clonal populations of mutant cells. Our results suggest that one could avoid or minimize off-target effects of RGENs by choosing unique target sites that do not have any homologous sequences elsewhere in the genome, a strategy we had used to avoid off-target effects of TALENs (Kim et al. 2013a). We also found that the structure and composition of guide RNA can be modified to reduce off-target mutations. We cannot rule out the possibility that the 11 RGENs we created in this study induce off-target mutations at sites not examined here, which could be revealed by deep sequencing at other less-homologous candidate sites or by whole genome sequencing. In addition, in vitro selection of cleavage sites (Pattanayak et al. 2011) or the IDLV capture approach (Gabriel et al. 2011) used for the identification of ZFN off-target sites may reveal cryptic sites cleaved by RGENs, although it is difficult to imagine that RGENs recognize off-target sites that are not complementary to crRNA sequences at an appreciable level. It is also worth noting that RGENs cleave DNA much less discriminatingly in vitro than they do in cells (Fig. 1C), limiting the in vitro selection method.
Since our initial submission, two groups reported that paired Cas9 nickases can reduce off-target activity in human cell lines (Mali et al. 2013a; Ran et al. 2013). Here, we also showed that paired Cas9 nickases do not induce indels at off-target sites at which their corresponding nucleases induce mutations. Furthermore, we showed that paired Cas9 nickases can induce large deletions of up to 1-kbp chromosomal segments in human cells and proposed a plausible mechanism that is supported by mutant DNA sequences. Importantly, unlike nucleases, paired nickases do not promote unwanted translocations associated with off-target DNA cleavages. In principle, paired nickases double the specificity of Cas9-mediated mutagenesis and will broaden the utility of RNA-guided enzymes in applications that require precise genome editing such as gene and cell therapy. One caveat to this approach is that two highly active sgRNAs are needed to make an efficient nickase pair, limiting targetable sites. As shown in this and other studies, not all sgRNAs are equally active. When single clones rather than populations of cells are used for further studies or applications, the choice of guide RNAs that represent unique sequences in the genome and the use of optimized guide RNAs would suffice to avoid off-target mutations associated with Cas9 nucleases. We propose that both Cas9 nucleases and paired nickases are powerful options that will facilitate precision genome editing in cells and organisms.
Methods
Cell culture and transfection
K562 (ATCC, CCL-243) cells were grown in RPMI-1640 with 10% FBS and a penicillin/streptomycin mix (100 units/mL and 100 μg/mL, respectively). 2 × 106 K562 cells were transfected with 20 μg of Cas9-encoding plasmid using the 4D-Nucleofector, SF Cell Line 4D-Nucleofector X Kit, Program FF-120 (Lonza) according to the manufacturer's protocol. After 24 h, 60 μg and 120 μg of in vitro transcribed crRNA and tracrRNA, respectively, were transfected into 1 × 106 K562 cells. Cells were collected 48 h after RNA transfection and genomic DNA were isolated. Under these conditions, RGENs showed the highest mutation frequencies.
HeLa cells (ATCC, CCL-2) were maintained in Dulbecco's modified Eagle's medium (DMEM) supplemented with 100 units/mL penicillin, 100 μg/mL streptomycin, 0.1 mM nonessential amino acids, and 10% FBS. 0.8 × 105 HeLa cells were transfected with Cas9-encoding plasmid (0.5 μg) and a tracrRNA /crRNA expression plasmid (0.5 μg) using Lipofectamine 2000 (Invitrogen) according to the manufacturer's protocol.
Plasmids
The Cas9-coding plasmid used in this study was described previously (Cho et al. 2013a). In this vector, the Cas9 protein is expressed under the control of the CMV promoter and is fused to a peptide tag (NH2-GGSGPPKKKRKVYPYDVPDYA-COOH) containing the HA epitope and a nuclear localization signal (NLS) at the C terminus. Plasmids that encode tracrRNA, crRNA, and sgRNA contained RNA sequences under the control of the U6 promoter. The plasmid encoding D10A Cas9 was made by site-directed mutagenesis.
Cas9 purification and in vitro DNA cleavage assay
The pET plasmid that encodes His-tagged Cas9 was transformed into BL21(DE3). The expression of Cas9 and D10A Cas9 was induced using 0.5 mM IPTG for 4 h at 25°C. The Cas9 protein was purified using Ni-NTA agarose resin (Qiagen) and dialyzed against 20 mM HEPES (pH 7.5), 150 mM KCl, 1 mM DTT, and 10% glycerol. More than 90% of the plasmid (350 ng) that contained the CCR5 on-target sequence was cleaved within 1 h with 33 ng Cas9 protein, 10 ng tracrRNA, and 5 ng crRNA. In this study, we incubated 350 ng target or off-target plasmid DNA or 200 ng PCR amplicons with 1700 ng Cas9 protein, 500 ng tracrRNA, and 250 ng crRNA in a reaction volume of 20 μL in NEB buffer 3 for 1 h at 37°C.
Fluorescent capillary electrophoresis
Substrate DNA for in vitro cleavage reactions was prepared by PCR using 5′-FAM labeled primers. The PCR products were purified and used for in vitro DNA cleavage assays. The reaction products were analyzed using fluorescent capillary electrophoresis.
RNA preparation
RNA was in vitro transcribed through run-off reactions by T7 RNA polymerase. Templates for crRNA were generated by annealing two complementary oligonuceotides (Supplemental Table 5). Transcribed RNA was resolved on an 8% denaturing urea-PAGE gel. RNA was recovered in nuclease-free water followed by phenol:chloroform extraction, chloroform extraction, and ethanol precipitation. Purified RNA was quantified by spectrometry.
Targeted deep sequencing
The on-target and potential off-target regions were amplified using Phusion polymerase (New England Biolabs) and used for library construction. Equal amounts of the PCR amplicons were subjected to paired-end read sequencing using Illumina MiSeq at Bio Medical Laboratories. Insertions or deletions located around the RGEN cleavage site (3 bp upstream of the PAM) were considered as a signature of NHEJ-mediated mutagenesis.
Exome sequencing
RGEN-induced mutant clones were isolated by limiting dilution of populations of K562 cells transfected with the Cas9 plasmid (20 μg), tracrRNA (120 μg), and crRNA (60 μg). Genomic DNA from each clone was isolated using a G-DEXIIc kit (iNtRON Biotechnology). Exome-captured libraries were constructed using a TrueSeq Exome Enrichment kit (Illumina). PCR enriched libraries were subjected to Illumina HiSeq 2000 at Bio Medical Laboratories. Exome sequence data were analyzed essentially as described (Ousterout et al. 2013). Raw reads in FASTQ format were aligned to the NCBI36/Hg18 reference genome using BWA-0.5.9. PCR duplicates were removed using SAMtools-0.1.16. Single nucleotide variants and indels were called using GATK-1.4. We then verified variants in several ways: (1) Because engineered nucleases rarely induce substitutions via error-prone NHEJ (Kim et al. 2013b), we excluded any single nucleotide variations; (2) shared indels found in at least two clones were also excluded, except those shared in the CCR5 #1 and CCR5 #2 clones; (3) naturally occurring common indels were excluded by comparing to dbSNP build 130; (4) we used Sanger and deep sequencing to exclude indels present in wild-type K562 cells; and (5) we developed an algorithm to search for putative off-target sequences around the remaining indels in the reference genome. To quantify the similarity or homology between the target sequence and query sequences, we considered a fixed ungapped alignment between these sequences. Thus, we compared the 20-bp target sequence (excluding the PAM) with a query sequence that spans from 23-bp upstream to 23-bp downstream from the position of each indel and counted the number (S) of matched bases. Because the probability that one base of the query sequence would match the corresponding base of the targeted sequence is 1/4, the P-value is given as
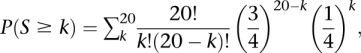 |
where k is the number of matched bases.
Data access
The deep sequencing data have been submitted to the DNA Data Bank of Japan (DDBJ Sequence Read Archive; http://trace.ddbj.nig.ac.jp/dra/index_e.html) under accession number DRA001195.
Competing interest statement
S.W.C., S.K., and J.-S.K. have filed a patent application based on this work.
Acknowledgments
J.-S.K. is supported by the National Research Foundation of Korea (2013000718).
Footnotes
[Supplemental material is available for this article.]
Article published online before print. Article, supplemental material, and publication date are at http://www.genome.org/cgi/doi/10.1101/gr.162339.113.
Freely available online through the Genome Research Open Access option.
References
- Bae KH, Kwon YD, Shin HC, Hwang MS, Ryu EH, Park KS, Yang HY, Lee DK, Lee Y, Park J, et al. 2003. Human zinc fingers as building blocks in the construction of artificial transcription factors. Nat Biotechnol 21: 275–280 [DOI] [PubMed] [Google Scholar]
- Bibikova M, Beumer K, Trautman JK, Carroll D 2003. Enhancing gene targeting with designed zinc finger nucleases. Science 300: 764. [DOI] [PubMed] [Google Scholar]
- Boch J, Scholze H, Schornack S, Landgraf A, Hahn S, Kay S, Lahaye T, Nickstadt A, Bonas U 2009. Breaking the code of DNA binding specificity of TAL-type III effectors. Science 326: 1509–1512 [DOI] [PubMed] [Google Scholar]
- Brunet E, Simsek D, Tomishima M, DeKelver R, Choi VM, Gregory P, Urnov F, Weinstock DM, Jasin M 2009. Chromosomal translocations induced at specified loci in human stem cells. Proc Natl Acad Sci 106: 10620–10625 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carlson DF, Tan W, Lillico SG, Stverakova D, Proudfoot C, Christian M, Voytas DF, Long CR, Whitelaw CB, Fahrenkrug SC 2012. Efficient TALEN-mediated gene knockout in livestock. Proc Natl Acad Sci 109: 17382–17387 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang N, Sun C, Gao L, Zhu D, Xu X, Zhu X, Xiong JW, Xi JJ 2013. Genome editing with RNA-guided Cas9 nuclease in zebrafish embryos. Cell Res 23: 465–472 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho SW, Kim S, Kim JM, Kim JS 2013a. Targeted genome engineering in human cells with the Cas9 RNA-guided endonuclease. Nat Biotechnol 31: 230–232 [DOI] [PubMed] [Google Scholar]
- Cho SW, Lee J, Carroll D, Kim JS 2013b. Heritable gene knockout in Caenorhabditis elegans by direct injection of Cas9-sgRNA ribonucleoproteins. Genetics doi: 10.1534/genetics.113.155853 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark MJ, Chen R, Lam HY, Karczewski KJ, Euskirchen G, Butte AJ, Snyder M 2011. Performance comparison of exome DNA sequencing technologies. Nat Biotechnol 29: 908–914 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cong L, Ran FA, Cox D, Lin S, Barretto R, Habib N, Hsu PD, Wu X, Jiang W, Marraffini LA, et al. 2013. Multiplex genome engineering using CRISPR/Cas systems. Science 339: 819–823 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding Q, Regan SN, Xia Y, Oostrom LA, Cowan CA, Musunuru K 2013. Enhanced efficiency of human pluripotent stem cell genome editing through replacing TALENs with CRISPRs. Cell Stem Cell 12: 393–394 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu Y, Foden JA, Khayter C, Maeder ML, Reyon D, Joung JK, Sander JD 2013. High-frequency off-target mutagenesis induced by CRISPR-Cas nucleases in human cells. Nat Biotechnol 31: 822–826 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gabriel R, Lombardo A, Arens A, Miller JC, Genovese P, Kaeppel C, Nowrouzi A, Bartholomae CC, Wang J, Friedman G, et al. 2011. An unbiased genome-wide analysis of zinc-finger nuclease specificity. Nat Biotechnol 29: 816–823 [DOI] [PubMed] [Google Scholar]
- Gratz SJ, Cummings AM, Nguyen JN, Hamm DC, Donohue LK, Harrison MM, Wildonger J, O'Connor-Giles KM 2013. Genome engineering of Drosophila with the CRISPR RNA-guided Cas9 nuclease. Genetics doi: 10.1534/genetics.113.152710 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsu PD, Scott DA, Weinstein JA, Ran FA, Konermann S, Agarwala V, Li Y, Fine EJ, Wu X, Shalem O, et al. 2013. DNA targeting specificity of RNA-guided Cas9 nucleases. Nat Biotechnol 31: 827–832 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hwang WY, Fu Y, Reyon D, Maeder ML, Tsai SQ, Sander JD, Peterson RT, Yeh JR, Joung JK 2013. Efficient genome editing in zebrafish using a CRISPR-Cas system. Nat Biotechnol 31: 227–229 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang W, Bikard D, Cox D, Zhang F, Marraffini LA 2013. RNA-guided editing of bacterial genomes using CRISPR-Cas systems. Nat Biotechnol 31: 233–239 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jinek M, Chylinski K, Fonfara I, Hauer M, Doudna JA, Charpentier E 2012. A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science 337: 816–821 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jinek M, East A, Cheng A, Lin S, Ma E, Doudna J 2013. RNA-programmed genome editing in human cells. Elife 2: e00471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karakoc E, Alkan C, O'Roak BJ, Dennis MY, Vives L, Mark K, Rieder MJ, Nickerson DA, Eichler EE 2012. Detection of structural variants and indels within exome data. Nat Methods 9: 176–178 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim HJ, Lee HJ, Kim H, Cho SW, Kim JS 2009. Targeted genome editing in human cells with zinc finger nucleases constructed via modular assembly. Genome Res 19: 1279–1288 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim S, Lee MJ, Kim H, Kang M, Kim JS 2011. Preassembled zinc-finger arrays for rapid construction of ZFNs. Nat Methods 8: 7. [DOI] [PubMed] [Google Scholar]
- Kim E, Kim S, Kim DH, Choi BS, Choi IY, Kim JS 2012. Precision genome engineering with programmable DNA-nicking enzymes. Genome Res 22: 1327–1333 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim Y, Kweon J, Kim A, Chon JK, Yoo JY, Kim HJ, Kim S, Lee C, Jeong E, Chung E, et al. 2013a. A library of TAL effector nucleases spanning the human genome. Nat Biotechnol 31: 251–258 [DOI] [PubMed] [Google Scholar]
- Kim Y, Kweon J, Kim JS 2013b. TALENs and ZFNs are associated with different mutation signatures. Nat Methods 10: 185. [DOI] [PubMed] [Google Scholar]
- Lee HJ, Kim E, Kim JS 2010. Targeted chromosomal deletions in human cells using zinc finger nucleases. Genome Res 20: 81–89 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee HJ, Kweon J, Kim E, Kim S, Kim JS 2012. Targeted chromosomal duplications and inversions in the human genome using zinc finger nucleases. Genome Res 22: 539–548 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mali P, Aach J, Stranges PB, Esvelt KM, Moosburner M, Kosuri S, Yang L, Church GM 2013a. CAS9 transcriptional activators for target specificity screening and paired nickases for cooperative genome engineering. Nat Biotechnol 31: 833–838 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mali P, Yang L, Esvelt KM, Aach J, Guell M, DiCarlo JE, Norville JE, Church GM 2013b. RNA-guided human genome engineering via Cas9. Science 339: 823–826 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller JC, Holmes MC, Wang J, Guschin DY, Lee YL, Rupniewski I, Beausejour CM, Waite AJ, Wang NS, Kim KA, et al. 2007. An improved zinc-finger nuclease architecture for highly specific genome editing. Nat Biotechnol 25: 778–785 [DOI] [PubMed] [Google Scholar]
- Miller JC, Tan S, Qiao G, Barlow KA, Wang J, Xia DF, Meng X, Paschon DE, Leung E, Hinkley SJ, et al. 2011. A TALE nuclease architecture for efficient genome editing. Nat Biotechnol 29: 143–148 [DOI] [PubMed] [Google Scholar]
- Moehle EA, Rock JM, Lee YL, Jouvenot Y, DeKelver RC, Gregory PD, Urnov FD, Holmes MC 2007. Targeted gene addition into a specified location in the human genome using designed zinc finger nucleases. Proc Natl Acad Sci 104: 3055–3060 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mussolino C, Morbitzer R, Lutge F, Dannemann N, Lahaye T, Cathomen T 2011. A novel TALE nuclease scaffold enables high genome editing activity in combination with low toxicity. Nucleic Acids Res 39: 9283–9293 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ousterout DG, Perez-Pinera P, Thakore PI, Kabadi AM, Brown MT, Qin X, Fedrigo O, Mouly V, Tremblay JP, Gersbach CA 2013. Reading frame correction by targeted genome editing restores dystrophin expression in cells from duchenne muscular dystrophy patients. Mol Ther 21: 1718–1726 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pattanayak V, Ramirez CL, Joung JK, Liu DR 2011. Revealing off-target cleavage specificities of zinc-finger nucleases by in vitro selection. Nat Methods 8: 765–770 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pattanayak V, Lin S, Guilinger JP, Ma E, Doudna JA, Liu DR 2013. High-throughput profiling of off-target DNA cleavage reveals RNA-programmed Cas9 nuclease specificity. Nat Biotechnol 31: 839–843 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramirez CL, Certo MT, Mussolino C, Goodwin MJ, Cradick TJ, McCaffrey AP, Cathomen T, Scharenberg AM, Joung JK 2012. Engineered zinc finger nickases induce homology-directed repair with reduced mutagenic effects. Nucleic Acids Res 40: 5560–5568 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ran FA, Hsu PD, Lin CY, Gootenberg JS, Konermann S, Trevino AE, Scott DA, Inoue A, Matoba S, Zhang Y, et al. 2013. Double nicking by RNA-guided CRISPR Cas9 for enhanced genome editing specificity. Cell 154: 1380–1389 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reyon D, Tsai SQ, Khayter C, Foden JA, Sander JD, Joung JK 2012. FLASH assembly of TALENs for high-throughput genome editing. Nat Biotechnol 30: 460–465 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santiago Y, Chan E, Liu PQ, Orlando S, Zhang L, Urnov FD, Holmes MC, Guschin D, Waite A, Miller JC, et al. 2008. Targeted gene knockout in mammalian cells by using engineered zinc-finger nucleases. Proc Natl Acad Sci 105: 5809–5814 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen B, Zhang J, Wu H, Wang J, Ma K, Li Z, Zhang X, Zhang P, Huang X 2013. Generation of gene-modified mice via Cas9/RNA-mediated gene targeting. Cell Res 23: 720–723 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sung YH, Baek IJ, Kim DH, Jeon J, Lee J, Lee K, Jeong D, Kim JS, Lee HW 2013. Knockout mice created by TALEN-mediated gene targeting. Nat Biotechnol 31: 23–24 [DOI] [PubMed] [Google Scholar]
- Szczepek M, Brondani V, Buchel J, Serrano L, Segal DJ, Cathomen T 2007. Structure-based redesign of the dimerization interface reduces the toxicity of zinc-finger nucleases. Nat Biotechnol 25: 786–793 [DOI] [PubMed] [Google Scholar]
- Urnov FD, Miller JC, Lee YL, Beausejour CM, Rock JM, Augustus S, Jamieson AC, Porteus MH, Gregory PD, Holmes MC 2005. Highly efficient endogenous human gene correction using designed zinc-finger nucleases. Nature 435: 646–651 [DOI] [PubMed] [Google Scholar]
- Wang J, Friedman G, Doyon Y, Wang NS, Li CJ, Miller JC, Hua KL, Yan JJ, Babiarz JE, Gregory PD, et al. 2012. Targeted gene addition to a predetermined site in the human genome using a ZFN-based nicking enzyme. Genome Res 22: 1316–1326 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H, Yang H, Shivalila CS, Dawlaty MM, Cheng AW, Zhang F, Jaenisch R 2013. One-step generation of mice carrying mutations in multiple genes by CRISPR/Cas-mediated genome engineering. Cell 153: 910–918 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiedenheft B, Sternberg SH, Doudna JA 2012. RNA-guided genetic silencing systems in bacteria and archaea. Nature 482: 331–338 [DOI] [PubMed] [Google Scholar]