

Abstract
Sequence-specific nucleases like TALENs and the CRISPR/Cas9 system have greatly expanded the genome editing possibilities in model organisms such as zebrafish. Both systems have recently been used to create knock-out alleles with great efficiency, and TALENs have also been successfully employed in knock-in of DNA cassettes at defined loci via homologous recombination (HR). Here we report CRISPR/Cas9-mediated knock-in of DNA cassettes into the zebrafish genome at a very high rate by homology-independent double-strand break (DSB) repair pathways. After co-injection of a donor plasmid with a short guide RNA (sgRNA) and Cas9 nuclease mRNA, concurrent cleavage of donor plasmid DNA and the selected chromosomal integration site resulted in efficient targeted integration of donor DNA. We successfully employed this approach to convert eGFP into Gal4 transgenic lines, and the same plasmids and sgRNAs can be applied in any species where eGFP lines were generated as part of enhancer and gene trap screens. In addition, we show the possibility of easily targeting DNA integration at endogenous loci, thus greatly facilitating the creation of reporter and loss-of-function alleles. Due to its simplicity, flexibility, and very high efficiency, our method greatly expands the repertoire for genome editing in zebrafish and can be readily adapted to many other organisms.
Methods of genome engineering are becoming increasingly powerful owing to breakthroughs in the design of artificial nucleases that induce site-specific double-strand breaks (DSBs) in the genome (Gaj et al. 2013). These DSBs, as was shown nearly 20 years ago using the homing endonuclease ISceI, efficiently stimulate homologous recombination (HR) with a gene targeting vector in cultured cells and plants (Jasin 1996). Several types of artificial nucleases can now be designed to make the initial DSB that induces modification of a sequence of interest. Among these, zinc finger and TALE nucleases (TALENs) are fusions of artificial DNA binding domains—arrays of zinc fingers and TALE effector repeats, respectively—to the endonuclease domain of the FokI restriction enzyme. The latter is only active as a dimer and therefore needs to be recruited to the target sequence by fusion to two separate zinc finger or TALE domains binding complementary sequences separated by a short DNA spacer.
More recently, novel RNA-guided nucleases (RGNs) have been developed based on the CRISPR/Cas9 mechanism of bacterial defense against exogenous DNA (Jinek et al. 2012). A short guide RNA (sgRNA) complexed to Streptococcus pyogenes Cas9 endonuclease binds to its complementary DNA target sequence and leads to specific DNA cleavage by Cas9. By changing the 20-bp sgRNA sequence, one can redirect the Cas9 nuclease to predetermined chromosomal target sites (Cho et al. 2013; Cong et al. 2013; Hwang et al. 2013b; Mali et al. 2013).
Important pioneer studies using zinc finger nucleases (ZFNs) have demonstrated the potential of artificial sequence-specific nucleases in the genome engineering of many experimental systems. TALE and CRISPR/Cas9 nucleases have emerged as powerful alternatives that are much easier to engineer. While sequence-specific TALE nucleases can be readily assembled from TALE repeats specific to each nucleotide (Cermak et al. 2011; Huang et al. 2011; Sander et al. 2011), sgRNAs for the CRISPR/Cas9 system can be easily generated by cloning of target-specific oligonucleotides into sgRNA expression vectors. The constraints of the sequences that can be targeted are minimal since TALE nucleases can be assembled to target TN48-54A sequences (Miller et al. 2011) and sgRNA (G/A)(G/A)N18-NGG sequences (Hwang et al. 2013b). Importantly, both systems have been shown to be active in a very high proportion of cases, although efficiencies may vary considerably (Reyon et al. 2012; Hwang et al. 2013b).
The use of sequence-specific TALENs or RGNs based on the CRISPR/Cas9 system allows specific gene disruption in many organisms not previously amenable to forward genetic analyses, for instance, in common experimental models such as the rat or the zebrafish (Huang et al. 2011; Sander et al. 2011; Tesson et al. 2011; Hwang et al. 2013b). Gene inactivation results from small insertions or deletions (indels) introduced during the repair of cleaved DNA by nonhomologous end joining (NHEJ), causing frameshifts and premature stop codons.
However, a broader range of DNA sequence modifications is highly desirable for many purposes such as locus-specific insertion of reporter genes or tagging of open reading frames. Since their first application, both systems have been used for the targeted insertion of short DNA sequences. By co-injection of single-stranded oligonucleotides bearing sequences flanking the cleaved target, site-specific DNA integration was recently demonstrated in mouse and zebrafish (Bedell et al. 2012; Chang et al. 2013; Hwang et al. 2013a; Wang et al. 2013; Wefers et al. 2013). Inducing DSBs with TALENs or RGNs at two sites on a chromosome can be used to trigger chromosomal deletions and inversions in cultured cells and zebrafish (Carlson et al. 2012; Gupta et al. 2013; Lim et al. 2013; Xiao et al. 2013). Artificial nucleases can also stimulate highly precise sequence modification by HR, but the efficiency is generally low. For example, using extremely active TALEN pairs that were able to induce indel mutations at rates up to 98%, Zu et al. could show gene targeting by HR in zebrafish with efficiencies at ∼1.5% (Zu et al. 2013). Linearized donors with >800-bp perfect homology flanking the TALEN target site served as a template for gene targeting by HR and allowed integration of inserts up to 1 kb. In living organisms, low efficiency limits the widespread application of gene targeting by HR because screening a large number of animals may be required to isolate founders carrying the mutation of interest. Here we report highly efficient CRISPR/Cas9-mediated knock-in of >5.7-kb-long DNA cassettes into the zebrafish genome based on homology-independent DSB repair. We show that, due to its flexibility and high efficiency, our method considerably expands the practical possibilities of genome engineering in model organisms.
Results
It was recently shown that zinc finger nucleases and TALENs can drive targeted integration of DNA cassettes in cultured cells (Cristea et al. 2013; Maresca et al. 2013) via homology-independent DSB repair. Although the design strategy slightly differed between the two studies, they both showed that if a donor plasmid is cleaved in transfected cells, it is frequently integrated at a site concomitantly targeted by zinc finger or TALE nucleases. We were interested in testing this approach in a model organism—the zebrafish—as a potential alternative to gene targeting by homologous recombination. Due to its easier design compared to ZFNs and TALE nucleases, we decided to first utilize the CRISPR/Cas9 system to introduce targeted DSBs.
Targeted knock-in of KalTA4 into the Tg(neurod:eGFP) locus
We chose a neurod:eGFP transgene (Obholzer et al. 2008) that is broadly expressed in the central nervous system during embryonic development as the target integration site. The eGFP transgene allows the direct visualization of target gene disruption and should not compromise survival upon loss of gene function.
In our donor plasmid, we inserted the target sequences for two sgRNAs specific to eGFP (hereafter referred to as “bait” sequence) followed by the coding sequence of an improved version of the transcriptional transactivator Gal4 (KalTA4) (Distel et al. 2009). This reading frame was preceded by an E2A peptide linker for multicistronic expression (Fig. 1A; Szymczak et al. 2004). When the donor plasmid was co-injected into an eGFP transgenic line with sgRNAs/Cas9 mRNA, concurrent cleavage of the genomic eGFP locus and bait plasmid sequence occurred. As NHEJ was shown to be highly active in early zebrafish development (Hagmann et al. 1998; Dai et al. 2010; Liu et al. 2012), we speculated that it would trigger integration of the donor plasmid into the opened chromosomal locus through nonspecific ligation of cleaved DNA ends.
Figure 1.

CRISPR/Cas9-mediated knock-in of KalTA4 into the Tg(neurod:eGFP) transgenic line. (A) A schematic of the donor plasmid consisting of an N-terminal eGFPbait with two sgRNA target sites (in orange, PAM sequence in blue). After co-injection of the donor with Cas9 mRNA and one eGFP sgRNA, insertion at the eGFP locus occurs. In-frame fusion of the E2A-KalTA4-pA cassette results in a multicistronic mRNA after successful integration at the eGFP locus. Due to the E2A sequence, the N-terminal eGFP peptide is cleaved from the KalTA4 protein by cotranslational ribosomal skipping. (B) A 6-dpf Tg(neurod:eGFP) × Tg(UAS:RFP, cry1:eGFP) embryo showing a switch from eGFP- to RFP-expressing cells upon injection of the donor plasmid together with sgRNA eGFP 1 and Cas9 mRNA. Successful in-frame knock-in of the donor plasmid into the eGFP open reading frame results in KalTA4 expression. Consecutively, KalTA4 binds to UAS:RFP and triggers RFP expression, leading to the eGFP to RFP switch. Scale bar, 300 μm. Tg(UAS:RFP, cry1:eGFP) transgenic fish express eGFP in the lens (driven by the crystalline promoter cry1:eGFP), thus allowing UAS:RFP transgenic fish to be identified by expression of eGFP in their lens (since without transactivation by KalTA4, no RFP is expressed from this transgene). (C) No RFP-expressing cells could be observed in Tg(neurod:eGFP) × Tg(UAS:RFP, cry1:eGFP) embryos injected with the donor plasmid and Cas9 mRNA but without sgRNA eGFP 1. Scale bar, 300 μm. (D) A representative gel of PCR products obtained from the founder fish shown in B, demonstrating targeted knock-in of the donor plasmid at the eGFP locus. PCR primers were placed flanking the neurod:eGFP locus and outward directed in the donor plasmid. Positions of PCR primers and the resulting fragment nomenclature are shown in A. (E) Sequence analysis at the 5′ and 3′ junctions of five representative targeted integration events. (Orange) sgRNA binding site, (red) base pair changes or insertions. The PAM sequence NGG required for cleavage by Cas9 (Jinek et al. 2012) is shown in blue. Note that only the Δ6 integration events correspond to in-frame insertions of the E2A-KalTA4 sequence. Due to three possible frames and two integration directions, only 16.6% of integration events will result in RFP expression.
After integration of the donor plasmid resulting in in-frame insertions of the E2A-KalTA4 cDNA (Fig. 1A), former eGFP positive cells were expected to express KalTA4. The simple loss of eGFP expression demonstrates gene disruption by the CRISPR/Cas9 system. In order to visualize integration events of the donor plasmid, we performed injections in embryos also carrying an UAS:RFP transgene [Tg(neurod:eGFP) × Tg(UAS:RFP, cry1:eGFP)] (Fig. 1B,C). If KalTA4 is inserted in-frame at the neurod:eGFP locus (which happens theoretically in 16.6% of integration events given three different frames and two insertion directions of the donor plasmid), the expressed KalTA4 will transactivate RFP expression by binding to the UAS sequence and triggering RFP transcription.
We designed two different sgRNAs targeting the eGFPbait sequence and estimated their efficiency at inducing indel mutations. For this purpose we pooled ten eGFP transgenic embryos after injection of sgRNAs and Cas9 mRNA, isolated genomic DNA, performed locus-specific PCR amplification on the eGFP locus, and estimated the rate of mutations by sequencing individual PCR clones. While sgRNA eGFP 1 was able to induce indel mutations at a rate of 66% (10/15 clones carrying mutations) (Table 1; Supplemental Table 1), the rate for sgRNA eGFP 2 was significantly lower (20%, 3/15 clones carrying mutations).
Table 1.
Knock-in efficiencies at the eGFP and the kif5aa locus
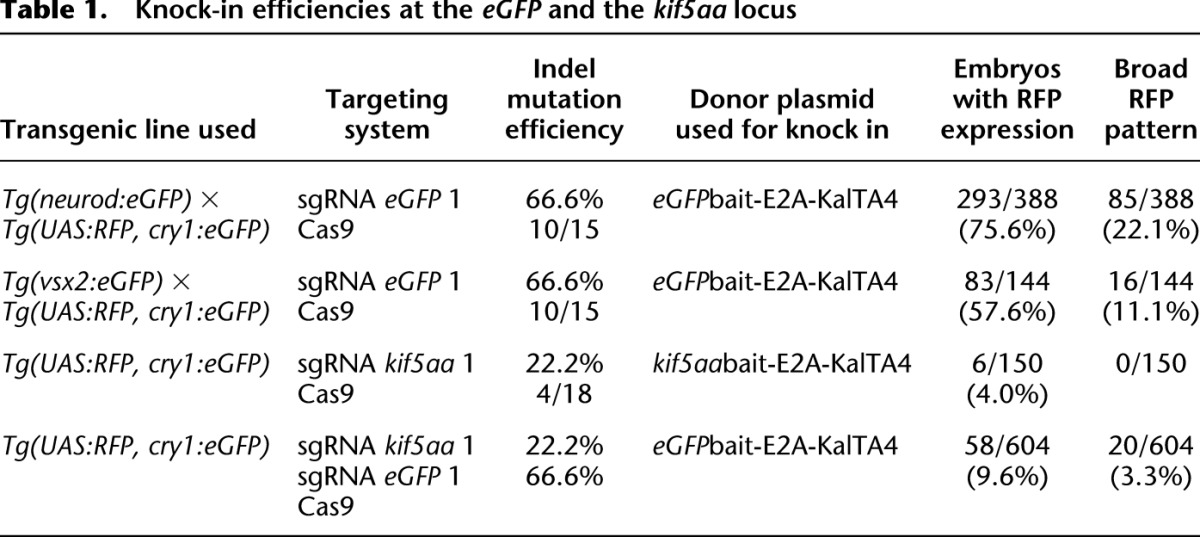
Using sgRNA eGFP 1 and co-injecting it with our eGFPbait-E2A-KalTA4 donor and Cas9 mRNA into Tg(neurod:eGFP) × Tg(UAS:RFP, cry1:eGFP) embryos, we observed RFP-positive cells within the neurod pattern in >75% (293/388) of injected embryos (Table 1). In about 22% (85/388) of injected embryos, RFP-positive cells were largely recapitulating neurod:eGFP expression (Supplemental Fig. 1; Table 1). In such embryos, RFP expression could be simultaneously detected in the brain and caudal neural tube, indicating integration events had likely occurred during the earliest stages of development.
In all confocal images acquired, we never observed co-expression of eGFP and RFP in the same cell. In about 80% of embryos (303/388), eGFP expression was strongly reduced compared to uninjected controls (Supplemental Fig. 1), indicating disruption of the eGFP open reading frame. RFP expression was more often observed in embryos that lost large parts of their neurod:eGFP expression, arguing for higher activity of the CRISPR/Cas9 system in these embryos. Within the group of RFP-positive embryos, <3% (9/388) showed RFP-expressing cells outside the neurod:eGFP expression domain (in muscle or skin cells). To further check for potential off-target integration of the donor plasmid, we performed injections in Tg(UAS:RFP, cry1:eGFP) embryos without the neurod:eGFP target locus. Within these embryos, we could only rarely observe some red muscle or skin cells in 1/300 (0.3%) embryos, arguing for a very low frequency of off-target integration events leading to expression of a functional KalTA4. In Tg(neurod:eGFP) × Tg(UAS:RFP, cry1:eGFP) embryos injected with eGFPbait-E2A-KalTA4 donor DNA and Cas9 mRNA but no sgRNA, we did not detect any RFP-expressing cells (0/243) (Fig. 1C). This indicates that the sgRNA is necessary to trigger integration of the donor plasmid.
After injection of the donor plasmid with the RGNs, successful targeted knock-in events were verified by PCR amplification (Fig. 1D) using integration site- and donor-specific primers (Fig. 1A). Subsequent analysis of the junction sequences revealed indel events typical for DSB repair by classical NHEJ and alternative end-joining mechanisms (Fig. 1E; Supplemental Table 1; Dai et al. 2010; Liu et al. 2012). Analyzing all junction sequences (between target locus and knocked-in donors) obtained in the course of this study, 50% exhibited small deletions (24/48 sequences) and 33% small insertions (16/48), while 17% (8/48) corresponded to ligation of nonmodified DNA sequences from the targeted locus and plasmid (perfect repair).
In a further set of experiments, we made use of the second sgRNA specific for eGFP, sgRNA eGFP 2, and again found phenotypic and molecular evidence for targeted DNA integration (Supplemental Fig. 2). The number of successfully converted embryos (22/149), however, was much lower (15% vs. 76% with sgRNA eGFP 1), consistent with a reduced efficiency of this sgRNA (20%) at directing site-specific indel mutations in the eGFP ORF compared to sgRNA eGFP 1 (66%).
Comparison to co-injection of linearized donor plasmid
We wanted to test whether co-injected linearized donor plasmids would be integrated at the genomic locus cleaved by the CRISPR/Cas9 system. We therefore linearized our donor plasmid prior to injection in vitro with a restriction enzyme, cutting just upstream of the E2A-KalTA4 sequence (close to the sgRNA eGFP 1 binding site). When we co-injected linearized eGFPbait-E2A-KalTA4 donor DNA with sgRNA eGFP 1 and Cas9 mRNA into one-cell stage embryos of the Tg(neurod:eGFP) × Tg(UAS:RFP, cry1:eGFP) cross, we observed an increased death rate compared to when co-injecting circular plasmid (35% vs. 15%, respectively) (Supplemental Fig. 3C). Frequency of in-frame integration events as scored by RFP expression was much lower (11% vs. 76% with circular plasmid) and observed in a sparse manner (Supplemental Fig. 3A,E). Altogether, this experiment demonstrates that co-injection of a circular plasmid that is cleaved concurrently with the endogenous target locus is less toxic and more efficient in triggering plasmid integration at the desired locus.
Targeted knock-in of KalTA4 into the Tg(vsx2:eGFP) transgenic line
We next sought to confirm the efficiency of our approach using a second eGFP transgenic line [Tg(vsx2:eGFP)] (Kimura et al. 2006) integrated at a different genomic locus and with a more restricted expression pattern. Vsx2:eGFP drives eGFP expression in the zebrafish embryonic retina and hindbrain cells in 2-dpf-old embryos (Fig. 2A). The eGFPbait-E2A-KalTA4 donor plasmid was co-injected with sgRNA eGFP 1 and fish embryos examined at 2 dpf. As shown in Figure 2B, conversion of the eGFP to the KalTA4 transgene could be directly visualized by the appearance of red fluorescent cells in the retina in the Tg(vsx2:eGFP) × Tg(UAS:RFP, cry1:eGFP) genetic background. Cells in the hindbrain also switched from eGFP to RFP expression (Fig. 2C). Efficiency of targeted DNA integration was estimated to range around 60% (83/144 embryos) (Table 1), based on the green to red fluorescence conversion. Eleven percent of embryos (16/144) thereby showed a broad expression pattern, with red cells spread over the whole retina (∼5% of retinal cells) (Fig. 2B) and the hindbrain (Fig. 2C). PCR and sequence analysis further confirmed that targeted DNA integration had taken place, and indel mutations typical of homology-independent repair pathways such as NHEJ were detected at junction sequences (Fig. 2D).
Figure 2.

CRISPR/Cas9-mediated knock-in of KalTA4 into the Tg(vsx2:eGFP) transgenic line. (A) Tg(vsx2:eGFP) shows eGFP expression in retina progenitor cells and the hindbrain region in 2dpf transgenic embryos. Scale bar, 100 μm. (B) eGFP to KalTA4 conversion in retina progenitor cells of Tg(vsx2:eGFP) × Tg(UAS:RFP, cry1:eGFP) embryos as revealed by RFP expression. The same donor plasmid and sgRNA eGFP 1 as in Figure 1 were used. Scale bar, 50 μm. (C) eGFP to KalTA4 conversion was seen as well in the developing hindbrain. Zoom-in of region indicated in A. Scale bar, 50 μm. (D) Using PCR, the targeted integration events could be verified. Sequence analysis of the 5′ junction and the 3′ junction. (E) F1 embryo (from founder A) with stable expression of the Tg(vsx2:eGFPbait-E2A-KalTA4, UAS:RFP) transgene activating RFP expression from UAS:RFP in the retina. Scale bar, 300 μm. (F) List of 5′ junctions of alleles identified in stable transgenic founders. Within 12 screened potential founder fish, six alleles could be detected, whereas four founders showed in-frame integration of the transgene. (Orange) sgRNA binding site; (blue) PAM sequence NGG.
As we used cry1:eGFP (resulting in eGFP expression in the lens) as a transgenesis marker for the UAS:RFP transgene in the Tg(UAS:RFP, cry1:eGFP) line, we offered a further potential target site for eGFP-specific sgRNAs. In a few cases, we could observe RFP expression in the lens of the Tg(UAS:RFP, cry1:eGFP) transgenic fish (Supplemental Fig. 4). This event likely reflects the insertion of the KalTA4 DNA cassette into the cry1:eGFP transgene and was rarely detected (8/388 [2%] of injected embryos), owing to the extremely restricted expression pattern of the cry1 promoter.
Targeted knock-in at the Tg(pou4f3:mGFP) locus
Subsequently, to test our method with a different target gene while still benefiting from the visual read-out of the GFP-to-KalTA4 switch, we targeted a transgene encoding an older, noncodon optimized version of GFP present in the Tg(pou4f3:mGFP) transgenic line (Xiao et al. 2005). We designed a sgRNA specific to the noncodon optimized GFP coding sequence and generated a new matching bait sequence for our E2A-KalTA4 donor plasmid. Co-injection with Cas9 mRNA into the Tg(pou4f3:mGFP) × Tg(UAS:RFP, cry1:eGFP) cross led to the GFP-to-KalTA4 switch (Supplemental Fig. 5A,B), and targeted DNA integration was confirmed at the DNA level by PCR and DNA sequence analysis of the junctions at the integration site (Supplemental Fig. 5C,D). The previous experiments show that we can successfully target eGFP and GFP transgenes and convert them to KalTA4 expression.
Targeted knock-in at the zebrafish kif5aa locus
To further extend the validity of CRISPR/Cas9-mediated knock-in on an endogenous target gene, we chose to target integration of KalTA4 cDNA to the kinesin family member 5Aa (kif5aa, ENSEMBL ID: ENSDARG00000005470.9) locus. Using in situ hybridization, we detected mRNA expression of this gene from 24 h post-fertilization onward in the spinal cord (Fig. 3A), consistent with a recently published expression pattern (Campbell and Marlow 2013). At 3 dpf, kif5aa is broadly expressed in the brain, while BAC transgenesis using the medaka (Oryzias latipes) ortholog showed additional kif5aa transcription in the spinal cord and motoneurons at later stages of development (Kawasaki et al. 2012). We first designed a sgRNA specific to kif5aa, whose efficiency at inducing indel mutations was determined to range around 22% (4/18) (Supplemental Table 1). Furthermore, we replaced the eGFPbait sequence in the previously described KalTA4 targeting vector with a bait sequence for kif5aa (Fig. 3D). Successful integration of KalTA4 was revealed by RFP expression after co-injection of the kif5aabait-E2A-KalTA4 donor vector, sgRNA kif5aa 1, and Cas9 mRNA into Tg(UAS:RFP, cry1:eGFP) embryos. RFP-positive cells could be detected in 4% (6/150) of injected embryos within the endogenous kif5aa expression domain (Fig. 3B,C; Table 1), while the remaining 96% of embryos did not show any RFP expression. We observed RFP-expressing cells in the spinal cord, hindbrain, cerebellum, and motoneurons. Insertion in the kif5aa locus was confirmed by PCR and subsequent sequence analysis (Fig. 3E). In contrast to experiments on the two eGFP transgenes, however, we did not observe embryos with extensive red fluorescent labeling, indicating that knock-in efficiency was lower (76% of RFP-positive cells when using the eGFP knock-in set vs. 4% when using the kif5aa knock-in set) when using a less efficient sgRNA (66% of indel mutations for sgRNA eGFP 1 vs. 22% for sgRNA kif5aa 1).
Figure 3.

CRISPR/Cas-mediated knock-in of KalTA4 into the kif5aa locus. (A) Kif5aa expression in zebrafish embryos revealed by in situ hybridization. Dorsal (A′) and lateral (A′′) views of 24-hpf embryos and dorsal view of 3-dpf embryo head and trunk region (A′′′) showing kif5aa expression in various brain regions and the spinal cord. (B,C) Representative confocal pictures of a Tg(UAS:RFP, cry1:eGFP) embryo showing RFP expression in the brain and spinal cord upon injection of the kif5aa bait donor plasmid together with sgRNA kif5aa 1 and Cas9 mRNA. Lateral view of the spinal cord (B′,C′), dorsal view of the head and trunk region (B′′,C′′), and high magnification of the spinal cord region (B′′′,C′′′) showing RFP expression in motoneurons. Scale bar, 50 μm. (sc) Spinal cord, (cb) cerebellum, (hb) hindbrain, (mn) motoneuron (cf. the GFP expression in the kif5aa BAC transgenic line reported by Kawasaki et al. [2012]). (D) A schematic of the used donor plasmid consisting of an N-terminal kif5aa bait with the sgRNA target site. The same E2A-KalTA4-pA cassette as in Figure 1A was used. (E) Sequence analysis at the 5′ and 3′ junctions of representative targeted integration events after PCR-based amplification. Binding sites of primers used for amplification are shown in D. (Orange) sgRNA binding site; (blue) PAM sequence NGG; (red) integrated additional base pairs. Note that the sgRNA is targeting the minus strand.
Combination of multiple sgRNAs to increase knock-in efficiency
To overcome this reduced efficiency and demonstrate the flexibility of the CRISPR/Cas9 system for targeted knock-in, we co-injected Cas9 mRNA, the eGFPbait-E2A-KalTA4 donor plasmid, and the more efficient sgRNA eGFP 1 together with sgRNA kif5aa 1 (Fig. 4A). While sgRNA eGFP 1 guides Cas9 nuclease activity to cut the donor plasmid in the eGFPbait sequence, sgRNA kif5aa 1 is used to target the endogenous target locus. By more efficient cutting of the donor plasmid (66% vs. 22% indel rates for sgRNA eGFP 1 and sgRNA kif5aa 1, respectively), more linearized donor is expected to be present for integration.
Figure 4.

CRISPR/Cas-mediated knock-in of KalTA4 into the kif5aa locus using the eGFPbait donor plasmid. (A) For integration of the E2A-KalTA4-pA cassette into the kif5aa locus, we used the eGFPbait donor plasmid in combination with two different sgRNAs. While sgRNA kif5aa 1 guides cleavage to the endogenous kif5aa locus, sgRNA eGFP 1 is employed for cleavage of the donor plasmid. (B,C) Representative confocal pictures of Tg(UAS:RFP, cry1:eGFP) 2-dpf embryos showing RFP expression in various brain regions and the spinal cord. Dorsal view (B′,C′) of the brain region and lateral view of an entire embryo (B′′,C′′) showing RFP expression in the whole length of the spinal cord and in the midbrain. Scale bar (B′,C′): 50 μm, (B′′,C′′): 200 μm. (dc) Diencephalon, (cb) cerebellum, (ot) optic tectum, (hb) hindbrain, (mb) midbrain, (sc) spinal cord. (D) Sequence analysis at the 5′ junction of representative targeted integration events after PCR-based amplification. Binding sites of primers used for amplification are shown in A. (Black) kif5aa locus; (blue) NGG PAM sequences for sgRNA kif5aa 1 and sgRNA eGFP 1; (green) parts of the eGFP bait sequence; (red) integrated additional base pairs. Note that, in this case, due to the frame difference between the kif5aa and eGFP genes, only +2 or −1 indels will produce functional fusion protein.
Indeed, we observed a 2.5-fold increase in integration of the DNA cassette at the specific kif5aa locus (9.6% [58/604] vs. 4% [6/150]) (Table 1). Furthermore, 3.3% (20/604) of the injected embryos now exhibited a broad RFP expression in the entire kif5aa expression domain (Fig. 4B,C). Successful integration events were confirmed by PCR and subsequent sequence analysis (Fig. 4D). These results indicate that, when only low-efficiency sgRNAs are available to target the chromosomal sequence of interest, as in the case of kif5aa, the integration frequency can be significantly improved by the co-injection of a more efficient sgRNA for in vivo cleavage of the donor vector. In addition, this experiment demonstrated that our knock-in strategy is independent from any sequence homology between the target locus and the bait sequence in the donor plasmid.
Homology-independent knock-in with TALE nucleases
Because the current design of the CRISPR/Cas9 system allows one to target statistically one sequence every 32 bp, in specific cases it may be necessary to use TALENs to target DSBs at specific loci (Hwang et al. 2013b). Therefore, we wanted to test the compatibility of our knock-in method with TALE nucleases in zebrafish. We designed a TALEN pair targeting the kif5aa locus. As previously described for our sgRNAs, we estimated the TALEN efficiency at inducing indel mutations by PCR amplification on genomic DNA from a pool of ten injected embryos and subsequent sequence analysis of individual PCR clones. Thereby, this TALEN pair showed an efficiency of 60% (6/10 clones carrying mutations) at inducing indel mutations (Supplemental Table 1). For the visualization of integration events, we designed a plasmid donor with a kif5aabait sequence followed by an UAS:eGFP cassette (Supplemental Fig. 6A). This DNA reporter construct shows eGFP expression independently from the direction and the frame of its insertion, allowing an easy assessment of integration events. Injections of the donor plasmid together with the kif5aa TALEN mRNAs were performed into the double transgenic line Tg(UAS-mcherry) × Et(-1.5hsp70l:Gal4-VP16)s1013t (Scott et al. 2007) that expresses Gal4 and mcherry in the central nervous system and the notochord. This approach can be used without any prior knowledge of the target gene expression pattern and allows an efficient preselection of potential founders with targeted integration. More than 30% of injected embryos showed correct eGFP expression in the notochord compared to controls (injection without TALEN mRNAs or injection of TALEN mRNAs plus donor with scrambled bait sequence) (Supplemental Fig. 6B) showing no eGFP signal. Integration events were verified by PCR and sequence analysis (Supplemental Fig. 6C,D). In a few cases, eGFP fluorescence could also be detected in muscle cells in control embryos, which may correspond to rare random DNA integration or persistence of plasmid DNA at later developmental stages.
Germline transmission of knocked-in transgenes
To investigate the transmission of knocked-in donor plasmids through the germline to the next generation, we raised embryos of the Tg(neurod:eGFP) transgenic line that were injected with the eGFPbait-E2A-KalTA4 donor plasmid together with sgRNA eGFP 1 and Cas9 mRNA. This allowed an unbiased determination of the germline transmission rate without prior selection for positive integration events. Potential founder fish were out-crossed to Tg(UAS:RFP, cry1:eGFP) embryos and screened for RFP expression. We could detect germline transmission of in-frame knock-in events in three out of 29 (10.3%) F0 fish (Fig. 5B; Table 2). The degree of transmission of the knocked-in transgene to the next generation thereby ranged from 1.2% (3/244) to 34.2% (93/272) in F1 progeny (Supplemental Table 2). If no RFP expression was observed in at least 50 embryos, these were pooled and analyzed by PCR for out-of frame insertion of the targeting vector not resulting in expression of a functional KalTA4. In six further founders, we detected forward insertion of the KalTA4 transgene by PCR, and sequence analysis confirmed out-of-frame insertion into the eGFP locus (Fig. 5C; see Fig. 5D for a list of sequenced 5′ junctions). This argues for a germline transmission rate of forward integrated donors of 31% (9/29 tested founders) (Table 2).
Figure 5.

Analysis of stable germline transmission of the Tg(neurod:eGFPbait-E2A-KalTA4) transgene. (A) Schematic depicting the Southern blot design to detect KalTA4 transgene integration. The neurod locus-specific probe 1 detects a 2.7-kb fragment after HindIII digest in the wild-type allele. The transgenic BAC neurod:eGFP locus is digested into a 2.6-kb fragment and, in the case of a partial digest in the BAC backbone, into a 4.4-kb fragment. After insertion of the KalTA4 cassette, a 6.6-kb fragment is detected. (B) Brightfield and fluorescent images of a transgenic Tg(neurod:eGFPbait-E2A-KalTA4) embryo at 2 dpf. (C) Screening for transgene integration by PCR in eight potential founders. Two show the expected fragment size (478 bp) (cf. Fig. 1A for primer positions and amplicon size). Note that the amplicon of founder B is slightly larger, as confirmed by sequencing and shown in D. (D) Sequences of 5′ junction sites of alleles identified in stable transgenic founders. Out of 11 founders showing stable transgene integration and transmission, five had an in-frame integration of the transgene. (Orange) sgRNA binding site; (blue) PAM sequence NGG; (red) integrated additional base pairs. (E) Analysis of the stable founder C for site-specific transgene integration by Southern blot analysis. As controls, wild-type and Tg(neurod:eGFP) embryos were used. Compare the schematic shown in A for expected fragment sizes. The 2.7-kb wild-type neurod fragment can be seen in all three samples (white arrow). The Tg(neurod:eGFP) sample shows a further fragment at 2.6 kb with greater intensity (black arrow) consistent with multiple insertions of the BAC construct. A shorter exposure is shown below to better distinguish the two separate bands. A further fragment at 4.4 kb is visible (asterisk), probably arising from incomplete digest of the neurod:GFP BAC trangene. In founder C, the neurod:eGFP band is no longer visible—instead, a fragment at 6.6 kb corresponding to the integration of KalTA4 into the eGFP sequence is detected.
Table 2.
Rate of germline transmission of KalTA4 knock-in into the eGFP locus
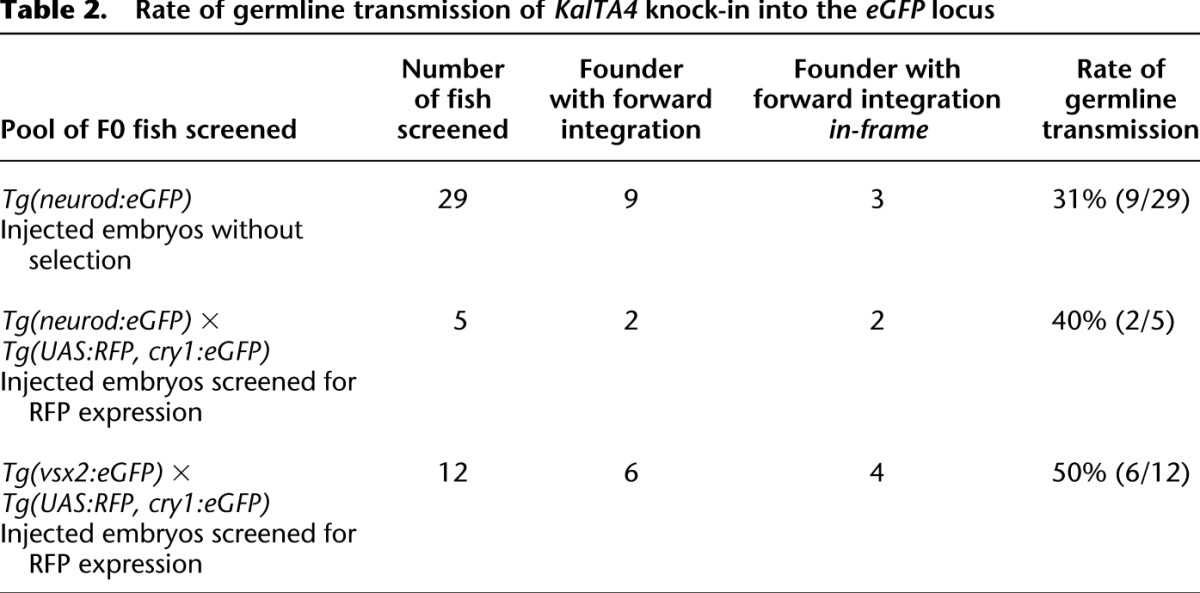
Taking advantage of the visual readout of integration, we also selectively raised Tg(neurod:eGFP) × Tg(UAS:RFP, cry1:eGFP) embryos injected with the eGFPbait-E2A-KalTA4 donor plasmid together with sgRNA eGFP 1 that showed expression of RFP in parts of the neurod:eGFP expression domain. Within the pool of RFP-selected embryos, we found germline transmission of two in-frame knock-in events in five founders screened (40%, 2/5) (Table 2). This argues for an enrichment of in-frame integration by selection for RFP expression in F0 fish as expected.
Similarly, for the Tg(vsx2:eGFP) transgene, we could identify transmission through the germline at a comparable rate of 50% (6/12) in RFP-selected Tg(vsx2:eGFP) × Tg(UAS:RFP, cry1:eGFP) embryos (Table 2), with 33% (4/12) showing in-frame integration (Fig. 2E; see Fig. 2F for a list of sequenced 5′ junctions).
The identical expression pattern of RFP and eGFP clearly argues for the insertion of the eGFPbait-E2A-KalTA4 transgene into the eGFP locus as confirmed by PCR analysis.
To further confirm locus-specific knock-in events, we performed Southern blot analysis. With a probe hybridizing to the neurod locus flanking sequence (Fig. 5A), we could detect a specific band of the expected size (6.6 kb) for insertion of the donor plasmid into the neurod:eGFP locus in the progeny of founder C (Fig. 5E, black arrow). Furthermore, the probe detected a 2.7-kb band in the wild-type zebrafish embryos corresponding to the endogenous neurod locus (white arrowhead), present also in all other samples as expected. In the transgenic animals used for our knock-in experiments, this band was accompanied by a smaller 2.6-kb band corresponding to the neurod:eGFP BAC transgene (black arrowhead), as well as an additional weaker band of 4.4 kb (asterisk), that likely corresponded to a partially digested fragment (see Fig. 5A for a graphic explanation). The signals corresponding to the transgenic locus were much more intense than the wild-type one, consistent with the presence of multiple transgene copies in the Tg(neurod:eGFP) line (Fig. 5E, inset), which is frequently observed in classical BAC transgenesis used to generate this line (Obholzer et al. 2008). In the knock-in animals derived from founder C, the bands corresponding to the neurod:eGFP transgene were no longer detected and were replaced by the 6.6-kb band resulting from the KalTA4 integration.
To examine if multiple copies of donor plasmid were integrated, we performed PCR analysis on DNA of founder progeny. We used five different primer combinations to detect 5′ and 3′ junctions of integrations at the target locus and potential head-to-head, tail-to-tail, or head-to-tail plasmid concatemers (Supplemental Fig. 8A). We detected head-to-tail concatemer formation as well as potential single-copy integration in different stable lines, as shown in Supplemental Figure 8B. These results were further confirmed using a KalTA4 transgene-specific probe in Southern blot analysis (Supplemental Fig. 8C,D).
This indicates that, in our approach, similar to ISceI-mediated transgenesis (Thermes et al. 2002), small concatemers can integrate at the target locus. For most applications this should not create any inconvenience, but given the high number of founders generated, single-copy integration can be identified if needed as shown here (Supplemental Fig. 8).
Analysis of potential off-target indel mutations and integration events
As it was recently shown that CRISPR/Cas9 nucleases show a high frequency of off-target mutagenesis in human cells (Fu et al. 2013), we analyzed off-target indel mutations or integrations in our approach. Using the fuzznuc program from the EMBOSS bioinformatics suite, we identified no potential off-target binding sites of sgRNA eGFP 1 in the zebrafish genome (Zv9 assembly) with up to three mismatches. Two sequences showed four mismatches and a conserved PAM sequence (5′-NGG) and 19 sequences five mismatches and a conserved PAM sequence (5′-NGG) compared to the original sgRNA sequence. Of these, 14 were annotated as part of a gene in the UCSC database and were selected for further examination (Supplemental Table 3). Eleven could be amplified and checked for mutations by T7 endonucle ase I digestion in pools of Tg(neurod:eGFP) × Tg(UAS:RFP, cry1:eGFP) embryos with and without injection of sgRNA eGFP 1, Cas9 mRNA, and the eGFPbait-E2A-KalTA4 donor plasmid.
As expected, we could detect T7E1-mediated cleavage at the neurod:eGFP locus in the pool of injected embryos (Supplemental Fig. 9). In contrast, no mutations could be detected at eight of the 11 potential off-target loci tested. For off#7 we saw the same T7E1 activity in controls as in injected embryos, and we determined by sequencing of PCR products that this was caused by a polymorphism in the Tg(neurod:eGFP) × Tg(UAS:RFP, cry1:eGFP) genetic background (16:43707701–43707722: TGTTTATTTTTTGTTTTTTTA → TG- - -A- - - - -TGTTTTTTTA). At two loci (off#1, off#8), we detected T7E1-mediated cleavage that was more prominent in injected embryos compared to controls (Supplemental Fig. 9). By direct sequencing of PCR clones from these loci, we did not detect any indel mutations at the potential off-target site off#8 in 33 clones (0/33 clones carrying mutations), arguing for a cleavage frequency <3% at this locus. For off#1, we sequenced 34 clones. Thus we detected the presence of a polymorphic microsatellite region with various alleles within our amplicon (1:40240770–40240783: GTGTGTGTGTGT) that would lead to fragment sizes, after T7E1 cleavage, of around 140 bp + 250 bp. Furthermore, we detected no indel mutations at the potential off-target site (0/34 clones carrying mutations). Also at this locus, the cleavage frequency of sgRNA eGFP 1/Cas9 must be <3%.
To check for knock-in of our donor plasmid at the two off-target sites, off#1 and off#8, we looked for plasmid insertion by PCR at these two locations in injected embryos and could not detect any evidence for off-target insertion (Supplemental Fig. 10). Similarly, when analyzing the progeny of one founder fish [Tg(neurod:eGFPbait-E2A-KalTA4)—founder H], no integration of our donor plasmid at these two potential off-target genomic locations could be observed, consistent with our Southern blot data.
Discussion
In the experiments described here, we showed for the first time in an in vivo model that CRISPR/Cas9-mediated DSBs can be used to efficiently knock-in donor plasmids at predetermined target sites. We were able to knock-in donors as large as 5.7 kb compared to up to 1 kb when gene targeting was performed by HR in zebrafish (Zu et al. 2013).
In previous cell culture studies, Cristea et al. (2013) showed that including a short DNA sequence bearing the nuclease target sequence onto a plasmid (that we call bait sequence) was sufficient for targeted integration at the nuclease chromosomal target sequence by homology-independent pathways of DSB repair upon cotransfection of plasmid and nuclease expression vectors. In contrast, Maresca et al. (2013) reported a different design where further cleavage of the integrated plasmid was prevented due to the specific utilization of nucleases with FokI mutants that only heterodimerize. In our case, using CRISPR/Cas9, we showed that both designs were efficient, since recleaving of the integrated sequence is possible in the case of the KalTA4 insertion into the eGFP locus but impossible after the insertion of the same DNA cassette into the kif5aa locus, as shown in Figure 4. In the first case, upon integration and end-joining in the absence of indels, we expect to re-create a complete sgRNA target sequence necessary for Cas9 activity, while in the second case, a hybrid sequence between the endogenous gene and the GFP bait sequence will be generated and no longer be recognized by the sgRNAs. In our study, we have not examined which homology-independent mechanisms are mediating DNA integration. Further studies would be necessary to determine if classical NHEJ or alternative end-joining pathways are involved. Nevertheless, in agreement with previous studies in cell culture systems (Maresca et al. 2013), classical NHEJ is the most likely mechanism involved.
In order to test our knock-in method we chose to target eGFP transgenes, and we have shown that our eGFPbait-E2A-KalTA4 construct can be directly applied to efficiently convert any eGFP into a KalTA4 transgenic line. Given the wealth of eGFP enhancer and gene trap lines previously generated in zebrafish (Kawakami et al. 2004; Parinov et al. 2004; Ellingsen et al. 2005), this offers new possibilities for deeper analysis of the marked cell types by tissue-specific expression of various UAS-driven constructs. The same approach, using the same target plasmid and sgRNA, can also be used in other species, such as Drosophila, where large collections of eGFP transgenic lines exist and CRISPR/Cas9 has been shown to work (Gratz et al. 2013).
Previously, when performing a knock-in by HR, Zu et al. (2013) showed germline transmission in zebrafish at rates of 1.5%, using highly efficient TALEN pairs (up to 98% indel rates). In our case, the most efficient nuclease, sgRNA eGFP 1/Cas9, had an indel mutation rate of 66%. Nevertheless, we observed germline transmission rates for the neurod:eGFP locus up to 31%. Even just taking in-frame integrations into account, with 10.3%, the rate of functional targeting of the locus was still higher. Taking advantage of positive selection, as done when screening for RFP-positive founders, we could increase this rate up to 40%. This high rate of in-frame founders after selection held true for a second locus, Tg(vsx2:eGFP), with four in-frame insertion events in 12 screened founder fish (4/12, 33%). Therefore, it seems that knock-in events by homology-independent DSB repair mechanisms are more frequent and lead to higher rates of germline transmission than HR-mediated events. This is in line with previous studies that showed that NHEJ, the major homology-independent mechanism of DSB repair, is at least 10-fold more active than HR during early zebrafish development (Hagmann et al. 1998; Dai et al. 2010; Liu et al. 2012).
Importantly, when targeting the kif5aa locus, we found that integration efficiency was considerably increased by using a combination of the kif5aa-specific sgRNA kif5aa 1 and sgRNA eGFP 1, with its corresponding eGFP DNA donor (Fig. 4). This strategy can be easily applied to any gene of interest without designing locus-specific donor plasmids. Our efficient sgRNA 1 for eGFP seems to direct only a very low degree of off-target nuclease activity, and no integration of the donor vector at predicted off-target sites could be detected. Therefore, sgRNA eGFP 1 together with its donor plasmid can be used to efficiently insert KalTA4 at any genomic locus targeted by a site-specific sgRNA, even of modest efficacy. Furthermore, KalTA4 can be easily replaced with reporter genes such as GFP to generate fluorescent fusion proteins, or other heterologous transcription factors such as TetR or LexA.
Our strategy, due to its simplicity and high efficiency, may become a new standard to generate mutant alleles that can be readily visualized and screened for in different transgenic backgrounds. This has the advantage of creating reporter lines at the same time (as compared to BAC recombineering), as we demonstrated for the kif5aa locus. The possibility to select for integration events already in the F0 will greatly reduce the number of animals to raise and screen to obtain mutants, so far blindly selected by PCR. In addition, the simplicity of the DNA target vector preparation will offer an easier alternative to BAC transgenesis. In fact, as bait sequences are of small size, they can be generated easily by PCR or oligonucleotide cloning, and no long homology stretches between donor and target site are required.
However, in contrast to gene targeting by HR, which allows for precise, predetermined transgene insertion sites, knock-in events mediated by homology-independent mechanisms have to be selected for appropriate in-frame insertions. In our case, this did not seem to be a major limitation due to the high knock-in rate. In many cases, choosing target sequences within introns and employing splice acceptor sites in the donor plasmid will avoid problems due to imprecise end-joining, and it could even further increase the number of functional insertions. As a great advantage, CRISPR/Cas9 allows the simultaneous targeting of several sequences (Cong et al. 2013; Wang et al. 2013) and may also be used for gene replacement by targeting sequences upstream of and downstream from a given locus at the same time.
Methods
Fish lines and husbandry
For this study, the Tg(neurod:eGFP) (Obholzer et al. 2008), Tg(vsx2:eGFP) (Kimura et al. 2006), Tg(pou4f3:mGFP) (Xiao et al. 2005), Tg(UAS:mCherry) × Et(1.5hsp70l:Gal4-VP16)s1013t16 (Scott et al. 2007), and Tg(UAS:RFP, cry1:eGFP) transgenic lines were used. Breeding and raising of zebrafish followed standard protocols.
Molecular cloning
The UAS:RFP/cry1:eGFP construct was cloned combining the cry1:eGFP fragment (Balciunas et al. 2004) with an 14×UAS sequence upstream of RFP (Koster and Fraser 2001) in a vector containing Tol2 sites (Kawakami et al. 2000). The eGFPbait-E2A-KalTA4 donor plasmid was generated by forward insertion of a PCR-amplified eGFP fragment into the pCRII-TOPO (TOPO TA Cloning Kit Dual Promoter, Invitrogen) vector. Primers used were (5′ to 3′) eGFP_fwd: ATAGTGGTACCATGGTGAGCAAGGGCGAGGAGC, eGFP_rev: GTAGCGGCTGAAGCACTGCACGC. The E2A-KalTA4-pA fragment was generated by fusion of individual PCR products using Phusion High-Fidelity DNA Polymerase (Thermo Scientific); E2A was amplified with the primers (5′ to 3′) E2A_fwd: TGCAGATATCCAGGAGGAGGACAGTGTACTAATTATGCTC, E2A_rev: TTCCTCCTCCGGGACCTGGGTTGCTC from a previously generated E2A sequence (Szymczak et al. 2004). KalTA4-pA was amplified with (5′ to 3′) KalTA4_fwd: CCCAGGTCCCGGAGGAGGAAAACTGCTC, KalTA4_rev: CATGCTCGAGTCCACTAGTTCTAGAGCG, using the 4 × Kaloop vector as template (Distel et al. 2009). Subsequently, both fragments were fused, amplified, and inserted into pCRII-TOPO-eGFPbait with EcoRV and XhoI. The GFPbait-E2A-KalTA4-pA donor plasmid was generated by forward insertion of a PCR-amplified GFP fragment into the pCRII-TOPO vector. Primers used (5′ to 3′) were GFP_fwd: ATGAGTAAAGGAGAAGAAC, GFP_rev: TCCGTATGTTGCATCACC. The E2A-KalTA4 fragment was transferred by an EcoRV and XhoI digest from the eGFPbait-E2A-KalTA4 donor plasmid. The kif5aa bait sequence was amplified from genomic zebrafish (TL genotype) DNA using the following primers (5′ to 3′): Kif5aa_fwd: TCTTCAACCACATCTTCTCC, Kif5aa_rev: TACCTTGATGTGGAACTCCAG, and inserted into the pCRII-TOPO vector. The E2A-KalTA4-pA fragment was transferred by an EcoRV and XhoI digest from the eGFPbait-E2A-KalTA4 donor plasmid. To generate the kif5aabait-UAS:eGFP-pA vector, an 4 × UAS:eGFP-pA fragment (Akitake et al. 2011) was excised by XhoI/SpeI digestion and inserted into the XhoI/XbaI-digested kif5aa bait vector. All constructs were verified by sequencing.
TALEN and sgRNA generation
TALENs were assembled by a method derived from Huang et al. (2011). For each TALEN subunit, the fragment containing the 16 RVD segment was obtained from single-unit plasmids kindly provided by Bo Zhang (Peking University, China). The assembled TALE repeats were subcloned in a pCS2 vector containing appropriate Δ152 Nter TALE, +63 Cter TALE, and FokI cDNA sequences with the appropriate half-TALE repeat (derived from the original pCS2 vector [Huang et al. 2011]). Sequences of encoded TALEN proteins are listed in Supplemental Table 4. sgRNAs guide sequences (listed in Supplemental Table 4) were cloned into the DR274 (Addgene ref 42250) plasmid vector for synthesis of sgRNA by T7 RNA polymerase as recommended (Hwang et al. 2013b).
Production of sgRNAs, Cas9 mRNA, and TALEN mRNAs
sgRNAs and Cas9 mRNA were generated as described previously (Hwang et al. 2013b). TALEN expression vectors were linearized by NotI digestion. Capped RNAs were synthesized using the mMESSAGE mMACHINE SP6 Kit (Life Technologies) and purified using the NucleoSpin RNA II Kit (Macherey-Nagel).
Injection of zebrafish embryos
TALEN mRNAs or sgRNA/Cas9 mRNA were co-injected into one-cell stage zebrafish embryos with fresh Qiagen midiprep (Qiagen) purified donor DNA. Each embryo was injected with 1 nl of solution containing ∼75 ng/μl of each TALEN mRNA or ∼7 ng/μl of sgRNA and ∼150 ng/μl Cas9 mRNA together with ∼7 ng/μl of donor plasmid. When two sgRNAs were co-injected, 7 ng/μl of each sgRNA were used. On the next day, injected embryos were inspected under a stereomicroscope. Only embryos that developed normally were assayed. Fluorescent protein expression was monitored over consecutive days. Genomic DNA was extracted from either single embryos or pools of embryos (as indicated) and then used for PCR, mapping, and DNA sequencing experiments as described below.
Insertion mapping
For insertion mapping, the primers used are listed in Supplemental Table 5. Genomic DNA was extracted following standard protocols. PCR was performed using Phusion High-Fidelity DNA Polymerase (Thermo Scientific). For sequence analysis of PCR products, PCR amplicons were tailed using Taq Polymerase (Life Technologies), cloned into the pCRII-TOPO (TOPO TA Cloning Kit Dual Promoter, Life Technologies) vector, and sent for sequencing. Mutant alleles were identified by comparison to the wild-type unmodified sequence. Mapping products were compared to the theoretical fusion products of cutting sites.
Detection of germline transmission
Potential founder fish were out-crossed to the Tg(UAS:RFP, cry1:eGFP) transgenic line. Fluorescent protein expression was monitored over the following days of development and the rate of mosaicism of germline transmission determined for RFP-positive in-frame founders. If no RFP signal was detected in at least 50 embryos, embryos were pooled, and genomic DNA was extracted and screened for locus-specific transgene integration by PCR. Subsequently PCR amplicons were sequenced.
Immunohistochemistry
Zebrafish larvae were processed for immunohistochemistry using standard protocols. Briefly, 4-dpf larvae were fixed in 4% paraformaldehyde (PFA; w/v, pH 7.4) overnight at 4°C, equilibrated in 30% sucrose (w/v) in phosphate-buffered saline (PBS) overnight at 4°C, and embedded in Tissue-Tek O.C.T. Compound (Sakura Finetech). Blocks were then frozen at −80°C on dry ice. Embedded larvae were sectioned horizontally on a cryostat (Leica Instruments,). The 12-μm sections were collected on Superfrost Plus slides (Fisher Scientific), air dried for 30 min–2 h, and rehydrated in PBS. Sections were incubated with blocking reagent containing 10% (v/v) normal goat serum (Jackson ImmunoResearch Laboratories) and 0.1% Tween-20 (v/v; Sigma) in PBS (pH 7.4) for 1 h at room temperature. Slides were left overnight in primary antibody diluted in blocking solution at 4°C in a humidified chamber. The following day, sections were washed three times in PBS/0.1% Tween-20 and then incubated for 2 h in a blocking solution containing Alexa fluorophore-conjugated secondary antibody diluted 1:500 (Invitrogen Molecular Probes) with DAPI nuclear marker (Sigma), washed three times in PBS/0.1% Tween-20, and mounted in Fluoromount (Sigma). Slides were air-dried in the dark from 4 h to overnight. Images were acquired using a Zeiss LSM 710 confocal microscope (Zeiss). Primary antibody used and concentrations: anti-GFP antibody (GeneTex), 1:1000; anti-RFP antibody (Evrogen), 1:400.
In situ hybridization
In situ hybridization was performed on 24-hpf- and 3-dpf-old embryos (TL) as described (Di Donato et al. 2013). For generation of a kif5aa specific antisense-probe, the following primers were used (5′ to 3′): Kif5aa-is-fwd: AGCATCGTCTACTCGACGGGGTTTT, Kif5aa-is-rev: GCTGCTCCCGTCTTACTGACCTTCT.
Microscopy
For low magnification imaging, a Leica MZ FLIII stereomicroscope (Leica) equipped with a Leica DFC310FX digital camera (Leica) was used. Confocal microscopy was performed using a Zeiss LSM 710 confocal microscope (Zeiss) and a 40× or 25× water immersion or 10× objective. Z volumes were acquired with a 1- to 3-μm resolution and images processed using Adobe Photoshop and Adobe Illustrator software. Three-dimensional reconstructions of Z-volumes were done using Imaris.
Genomic DNA extraction for Southern blot analysis
Genomic DNA was isolated from pools of 20–50 out-crossed embryos harvested 5 dpf. Samples were digested for 1 h at 55°C in 0.5mL lysis buffer (10 mM Tris, pH 8.0, 10 mM NaCl, 10 mM EDTA, and 2% SDS) with proteinase K (0.17 mg/mL, Roche Diagnostics) and centrifuged for 10 min at 14,000 rpm. The supernatant was transferred to a phase lock gel tube (Dutscher), 0.5 mL of phenol/chloroform (Life Technologies) added, briefly mixed and centrifuged for 10 min at 14,000 rpm. One milliliter of 100% ethanol and 10% of 3 M sodium acetate, pH 6.0 were added to the supernatant and centrifuged for 30 min at 14,000 rpm at 4°C. The pellet was washed with 70% ethanol, dried, and resuspended in 100 μL H20.
Southern blot analysis
Genomic DNA (3–5 μg) was digested overnight with 50 units of HindIII (New England Biolabs, High Fidelity) restriction enzyme. The digested genomic DNA was separated by standard gel electrophoresis on a 1% agarose gel in 1× TAE buffer. Transfer of DNA was done overnight by upward capillarity transfer in 10× SSC to a Hybond N+ membrane (Amersham Biosciences). The membrane was UV cross-linked using a UV cross-linker (Fisher Biotech). A neurod locus-specific probe (565 bp, probe 1) and an E2A-KalTA4-specific probe (491 bp, probe 2) were amplified using the PCR DIG Probe Synthesis Kit (Roche), according to the manufacturer's protocol. Probes were amplified starting from genomic wild-type DNA (AB) or the eGFPbait-E2A-KalTA4 plasmid as templates, respectively. Fwd primer probe 1 (5′ to 3′): CAACACACCCTAGGTATGTGATCTG, Rev primer probe 1 (5′ to 3′): GTGATAAGTACGTTCTCACAAGTTC. Fwd primer probe 2 (5′ to 3′): CAGTGTACTAATTATGCTCTC, Rev primer probe 2 (5′ to 3′): CTCTGTCCCTTGTTAGAAGACTC. Hybridization was done overnight at 68°C, and for detection, the CDP-Star Kit (Roche) was used according to the manufacturer's instructions.
Insertion mapping and concatemer detection
For insertion mapping and concatemer detection, the primers used are listed in Supplemental Table 5. PCR was performed using Phusion High-Fidelity DNA Polymerase (Thermo Scientific). For sequence analysis of PCR products, PCR amplicons were tailed using Taq Polymerase (Life Technologies), cloned into the pCRII-TOPO (TOPO TA Cloning Kit Dual Promoter, Life Technologies) vector, and sequenced. Mutant alleles were identified by comparison to the wild-type unmodified sequence. Mapping products were compared to the theoretical fusion products of cutting sites.
Identification of off-target sites and T7E1 assay
Potential off-targets of sgRNA eGFP 1 (GGCGAGGGCGATGCCACCTACGG) in the Danio rerio Zv9 assembly were identified using fuzznuc from the EMBOSS suite, and no off-targets bearing up to three mismatches were detected. Out of 21 sequences with up to five mismatches, 14 were annotated as part of genes in the UCSC database (Supplemental Table 3). For amplification of these loci and the neurod:eGFP locus, primers listed in Supplemental Table 6 were used.
Genomic DNA was isolated from pools of 25 5-dpf embryos of the Tg(Tg(neurod:eGFP)) × Tg(UAS:RFP, cry1:eGFP) cross with and without injection of the eGFPbait-E2A-KalTA4 donor plasmid together with sgRNA eGFP 1 and Cas9. PCR was performed using Phusion Polymerase (New England Biolabs) following the manufacturer's protocol. Five microliters of unpurified PCR product + 5 μL of NEBuffer 2 (2×) (New England Biolabs) were melted and annealed (95°C for 5 min, 95°C to 25°C at −0.5°C/30 sec, and 4°C for 15 min) to form heteroduplex DNA. The annealed DNA was treated (or untreated) with 0.75 units of T7 endonuclease 1 (New England Biolabs) for 20 min at 37°C and run on a 2.4% agarose gel after stopping the reaction by adding 10 μL of Proteinase K (0.4 mg/μL) in 50% sucrose. To check for frequency of indel mutations at the off-target sites off#1 and off#8, PCR amplicons were tailed using Taq Polymerase (Life Technologies), cloned into the pCRII-TOPO (TOPO TA Cloning Kit Dual Promoter, Life Technologies) vector and sent for sequencing. Mutant alleles were identified by comparison to the wild-type unmodified sequence. For detection of the polymorphism at off#7, multiple PCR clones were sent for sequencing and alleles compared. For insertion mapping at the two off-target sites, the primers listed in Supplemental Table 6 were used.
Data access
Sequences of the primers are listed in the Methods and Supplemental Tables 5 and 6. The target sites of the sgRNAs and TALENs are listed in Supplemental Table 4. The TALEN RVD sequences are provided in Supplemental Table 4.
Acknowledgments
We thank A. Shkumatava, H. Baier, and L. Poggi for the Tg(neurod:eGFP), Tg(pou4f3:mGFP), and Tg(vsx2:eGFP) lines, respectively. We thank I. Rentero Rebollo and A. Shkumatava for critical reading of the manuscript. We also thank D. Balciunas for the UAS:RFP plasmid, C. Giovannangeli and members of the Del Bene laboratory for general discussion and comments, and M. Charpentier for the generation of the kif5aa TALEN plasmids and excellent technical help. We thank the members of the Developmental Biology Curie imaging facility (PICT-IBiSA@BDD, UMR3215/U934) for their help and advice with confocal microscopy. The Del Bene laboratory “Neural Circuits Development” is part of the Laboratoire d'Excellence (LABEX) entitled DEEP (ANR -11-LABX-0044), and of the École des Neurosciences de Paris Ile-de-France network. T.O.A. was supported by a Boehringer Ingelheim Fonds Ph.D. fellowship. This work has been supported by an ATIP/AVENIR program starting grant (F.D.B.), ERC-StG #311159 (F.D.B.), ANR TEFOR (J.P.C.), CNRS, INSERM, Institut Curie and Muséum National d'Histoire Naturelle.
Author contributions: T.O.A., J.P.C., and F.D.B. conceived and designed experiments; J.P.C. prepared sgRNA constructs. A.D.C. performed the T7E1 assays. T.O.A. and K.D. built donor constructs, carried out the PCR diagnosis, Southern blotting, and in situ hybridization. T.O.A. performed microinjections and microscopy and F.D.B. prepared the cryosections. T.O.A., J.P.C., and F.D.B. wrote the manuscript.
Footnotes
[Supplemental material is available for this article.]
Article published online before print. Article, supplemental material, and publication date are at http://www.genome.org/cgi/doi/10.1101/gr.161638.113.
References
- Akitake CM, Macurak M, Halpern ME, Goll MG 2011. Transgenerational analysis of transcriptional silencing in zebrafish. Dev Biol 352: 191–201 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balciunas D, Davidson AE, Sivasubbu S, Hermanson SB, Welle Z, Ekker SC 2004. Enhancer trapping in zebrafish using the Sleeping Beauty transposon. BMC Genomics 5: 62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bedell VM, Wang Y, Campbell JM, Poshusta TL, Starker CG, Krug RG, Tan W, Penheiter SG, Ma AC, Leung AYH, et al. 2012. In vivo genome editing using a high-efficiency TALEN system. Nature 491: 114–118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campbell PD, Marlow FL 2013. Temporal and tissue specific gene expression patterns of the zebrafish kinesin-1 heavy chain family, kif5s, during development. Gene Expr Patterns 13: 271–279 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carlson DF, Tan W, Lillico SG, Stverakova D, Proudfoot C, Christian M, Voytas DF, Long CR, Whitelaw CB, Fahrenkrug SC 2012. Efficient TALEN-mediated gene knockout in livestock. Proc Natl Acad Sci 109: 17382–17387 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cermak T, Doyle EL, Christian M, Wang L, Zhang Y, Schmidt C, Baller JA, Somia NV, Bogdanove AJ, Voytas DF 2011. Efficient design and assembly of custom TALEN and other TAL effector-based constructs for DNA targeting. Nucleic Acids Res 39: e82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang N, Sun C, Gao L, Zhu D, Xu X, Zhu X, Xiong JW, Xi JJ 2013. Genome editing with RNA-guided Cas9 nuclease in zebrafish embryos. Cell Res 23: 465–472 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho SW, Kim S, Kim JM, Kim JS 2013. Targeted genome engineering in human cells with the Cas9 RNA-guided endonuclease. Nat Biotechnol 31: 230–232 [DOI] [PubMed] [Google Scholar]
- Cong L, Ran FA, Cox D, Lin S, Barretto R, Habib N, Hsu PD, Wu X, Jiang W, Marraffini LA, et al. 2013. Multiplex genome engineering using CRISPR/Cas systems. Science 339: 819–823 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cristea S, Freyvert Y, Santiago Y, Holmes MC, Urnov FD, Gregory PD, Cost GJ 2013. In vivo cleavage of transgene donors promotes nuclease-mediated targeted integration. Biotechnol Bioeng 110: 871–880 [DOI] [PubMed] [Google Scholar]
- Dai J, Cui X, Zhu Z, Hu W 2010. Non-homologous end joining plays a key role in transgene concatemer formation in transgenic zebrafish embryos. Int J Biol Sci 6: 756–768 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Donato V, Auer TO, Duroure K, Del Bene F 2013. Characterization of the calcium binding protein family in zebrafish. PLoS ONE 8: e53299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Distel M, Wullimann MF, Köster RW 2009. Optimized Gal4 genetics for permanent gene expression mapping in zebrafish. Proc Natl Acad Sci 106: 13365–13370 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ellingsen S, Laplante MA, Konig M, Kikuta H, Furmanek T, Hoivik EA, Becker TS 2005. Large-scale enhancer detection in the zebrafish genome. Development 132: 3799–3811 [DOI] [PubMed] [Google Scholar]
- Fu Y, Foden JA, Khayter C, Maeder ML, Reyon D, Joung JK, Sander JD 2013. High-frequency off-target mutagenesis induced by CRISPR-Cas nucleases in human cells. Nat Biotechnol 31: 822–826 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaj T, Gersbach CA, Barbas CF 3rd 2013. ZFN, TALEN, and CRISPR/Cas-based methods for genome engineering. Trends Biotechnol 7: 397–405 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gratz SJ, Cummings AM, Nguyen JN, Hamm DC, Donohue LK, Harrison MM, Wildonger J, O'Connor-Giles KM 2013. Genome engineering of Drosophila with the CRISPR RNA-guided Cas9 nuclease. Genetics 194: 1029–1035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gupta A, Hall VL, Kok FO, Shin M, McNulty JC, Lawson ND, Wolfe SA 2013. Targeted chromosomal deletions and inversions in zebrafish. Genome Res 23: 1008–1017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hagmann M, Bruggmann R, Xue L, Georgiev O, Schaffner W, Rungger D, Spaniol P, Gerster T 1998. Homologous recombination and DNA-end joining reactions in zygotes and early embryos of zebrafish (Danio rerio) and Drosophila melanogaster. Biol Chem 379: 673–681 [DOI] [PubMed] [Google Scholar]
- Huang P, Xiao A, Zhou M, Zhu Z, Lin S, Zhang B 2011. Heritable gene targeting in zebrafish using customized TALENs. Nat Biotechnol 29: 699–700 [DOI] [PubMed] [Google Scholar]
- Hwang WY, Fu Y, Reyon D, Maeder ML, Kaini P, Sander JD, Joung JK, Peterson RT, Yeh JR 2013a. Heritable and precise zebrafish genome editing using a CRISPR-Cas system. PLoS ONE 8: e68708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hwang WY, Fu Y, Reyon D, Maeder ML, Tsai SQ, Sander JD, Peterson RT, Yeh J-RJ, Joung JK 2013b. Efficient genome editing in zebrafish using a CRISPR-Cas system. Nat Biotechnol 31: 227–229 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jasin M 1996. Genetic manipulation of genomes with rare-cutting endonucleases. Trends Genet 12: 224–228 [DOI] [PubMed] [Google Scholar]
- Jinek M, Chylinski K, Fonfara I, Hauer M, Doudna JA, Charpentier E 2012. A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science 337: 816–821 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawakami K, Shima A, Kawakami N 2000. Identification of a functional transposase of the Tol2 element, an Ac-like element from the Japanese medaka fish, and its transposition in the zebrafish germ lineage. Proc Natl Acad Sci 97: 11403–11408 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawakami K, Takeda H, Kawakami N, Kobayashi M, Matsuda N, Mishina M 2004. A transposon-mediated gene trap approach identifies developmentally regulated genes in zebrafish. Dev Cell 7: 133–144 [DOI] [PubMed] [Google Scholar]
- Kawasaki T, Kurauchi K, Higashihata A, Deguchi T, Ishikawa Y, Yamauchi M, Sasanuma M, Hori H, Tsutsumi M, Wakamatsu Y, et al. 2012. Transgenic medaka fish which mimic the endogenous expression of neuronal kinesin, KIF5A. Brain Res 1480: 12–21 [DOI] [PubMed] [Google Scholar]
- Kimura Y, Okamura Y, Higashijima S 2006. alx, a zebrafish homolog of Chx10, marks ipsilateral descending excitatory interneurons that participate in the regulation of spinal locomotor circuits. J Neurosci 26: 5684–5697 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koster RW, Fraser SE 2001. Tracing transgene expression in living zebrafish embryos. Dev Biol 233: 329–346 [DOI] [PubMed] [Google Scholar]
- Lim S, Wang Y, Yu X, Huang Y, Featherstone MS, Sampath K 2013. A simple strategy for heritable chromosomal deletions in zebrafish via the combinatorial action of targeting nucleases. Genome Biol 14: R69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J, Gong L, Chang C, Liu C, Peng J, Chen J 2012. Development of novel visual-plus quantitative analysis systems for studying DNA double-strand break repairs in zebrafish. J Genet Genomics 39: 489–502 [DOI] [PubMed] [Google Scholar]
- Mali P, Yang L, Esvelt KM, Aach J, Guell M, DiCarlo JE, Norville JE, Church GM 2013. RNA-guided human genome engineering via Cas9. Science 339: 823–826 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maresca M, Lin VG, Guo N, Yang Y 2013. Obligate ligation-gated recombination (ObLiGaRe): Custom-designed nuclease-mediated targeted integration through nonhomologous end joining. Genome Res 23: 539–546 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller JC, Tan S, Qiao G, Barlow KA, Wang J, Xia DF, Meng X, Paschon DE, Leung E, Hinkley SJ, et al. 2011. A TALE nuclease architecture for efficient genome editing. Nat Biotechnol 29: 143–148 [DOI] [PubMed] [Google Scholar]
- Obholzer N, Wolfson S, Trapani JG, Mo W, Nechiporuk A, Busch-Nentwich E, Seiler C, Sidi S, Sollner C, Duncan RN, et al. 2008. Vesicular glutamate transporter 3 is required for synaptic transmission in zebrafish hair cells. J Neurosci 28: 2110–2118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parinov S, Kondrichin I, Korzh V, Emelyanov A 2004. Tol2 transposon-mediated enhancer trap to identify developmentally regulated zebrafish genes in vivo. Dev Dyn 231: 449–459 [DOI] [PubMed] [Google Scholar]
- Reyon D, Tsai SQ, Khayter C, Foden JA, Sander JD, Joung JK 2012. FLASH assembly of TALENs for high-throughput genome editing. Nat Biotechnol 30: 460–465 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sander JD, Cade L, Khayter C, Reyon D, Peterson RT, Joung JK, Yeh JR 2011. Targeted gene disruption in somatic zebrafish cells using engineered TALENs. Nat Biotechnol 29: 697–698 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scott EK, Mason L, Arrenberg AB, Ziv L, Gosse NJ, Xiao T, Chi NC, Asakawa K, Kawakami K, Baier H 2007. Targeting neural circuitry in zebrafish using GAL4 enhancer trapping. Nat Methods 4: 323–326 [DOI] [PubMed] [Google Scholar]
- Szymczak AL, Workman CJ, Wang Y, Vignali KM, Dilioglou S, Vanin EF, Vignali DAA 2004. Correction of multi-gene deficiency in vivo using a single ‘self-cleaving’ 2A peptide-based retroviral vector. Nat Biotechnol 22: 589–594 [DOI] [PubMed] [Google Scholar]
- Tesson L, Usal C, Menoret S, Leung E, Niles BJ, Remy S, Santiago Y, Vincent AI, Meng X, Zhang L, et al. 2011. Knockout rats generated by embryo microinjection of TALENs. Nat Biotechnol 29: 695–696 [DOI] [PubMed] [Google Scholar]
- Thermes V, Grabher C, Ristoratore F, Bourrat F, Choulika A, Wittbrodt J, Joly JS 2002. I-SceI meganuclease mediates highly efficient transgenesis in fish. Mech Dev 118: 91–98 [DOI] [PubMed] [Google Scholar]
- Wang H, Yang H, Shivalila CS, Dawlaty MM, Cheng AW, Zhang F, Jaenisch R 2013. One-step generation of mice carrying mutations in multiple genes by CRISPR/Cas-mediated genome engineering. Cell 153: 910–918 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wefers B, Meyer M, Ortiz O, Hrabe de Angelis M, Hansen J, Wurst W, Kuhn R 2013. Direct production of mouse disease models by embryo microinjection of TALENs and oligodeoxynucleotides. Proc Natl Acad Sci 110: 3782–3787 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao T, Roeser T, Staub W, Baier H 2005. A GFP-based genetic screen reveals mutations that disrupt the architecture of the zebrafish retinotectal projection. Development 132: 2955–2967 [DOI] [PubMed] [Google Scholar]
- Xiao A, Wang Z, Hu Y, Wu Y, Luo Z, Yang Z, Zu Y, Li W, Huang P, Tong X, et al. 2013. Chromosomal deletions and inversions mediated by TALENs and CRISPR/Cas in zebrafish. Nucleic Acids Res 41: e141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zu Y, Tong X, Wang Z, Liu D, Pan R, Li Z, Hu Y, Luo Z, Huang P, Wu Q, et al. 2013. TALEN-mediated precise genome modification by homologous recombination in zebrafish. Nat Methods 10: 329–331 [DOI] [PubMed] [Google Scholar]