

Abstract
DNA sequence variants influence gene expression and cellular phenotypes. In this study, we focused on natural variation in the gene encoding the histone demethylase, KDM4C, which promotes transcriptional activation by removing the repressive histone mark, H3K9me3, from its target genes. We uncovered cis-acting variants that contribute to extensive individual differences in KDM4C expression. We also identified the target genes of KDM4C and demonstrated that variation in KDM4C expression leads to differences in the growth of normal and some cancer cells. Together, our results from genetic mapping and molecular analysis provide an example of how genetic variation affects epigenetic regulation of gene expression and cellular phenotype.
There are extensive individual differences in the expression levels of human genes (Cheung et al. 2003), and there is a genetic basis to this variation (Cheung et al. 2003; Monks et al. 2004; Grundberg et al. 2012). This allows us to treat gene expression levels as quantitative traits in genetic analysis to identify the regulatory variants. By combining quantitative trait mapping with molecular studies, we uncover mechanisms that regulate expression levels of genes without a priori knowledge of the regulatory pathways. Results from such quantitative trait mapping have revealed the locations and identities of regulatory variants (Morley et al. 2004; Cheung et al. 2005; Stranger et al. 2005; Schadt et al. 2008; for reviews, see Pastinen and Hudson 2004; Gibson and Weir 2005; Li and Burmeister 2005; Stranger and Dermitzakis 2005). These variants are either proximal to the target genes, likely acting in cis, or on another chromosome, acting in trans. Some of these regulatory variants have been found to affect disease susceptibility, including asthma and obesity (Moffatt et al. 2007; Emilsson et al. 2008). However, the mechanisms by which these regulatory variants affect gene expression and cellular phenotypes remain largely unknown. While the cis-acting variants have been characterized molecularly, few of the polymorphic trans-acting regulators have been studied.
In parallel to the development of these genetics of gene expression studies, there are many advances in uncovering the epigenetic regulation of gene expression. International efforts such as The ENCODE Project Consortium (The ENCODE Project Consortium 2007, 2012) have created genome-wide maps of chromatin modifications. In addition, many of the enzymes that mediate post-translational modification of histones, either by adding or by removing covalent modifications, have been identified (for reviews, see Greer and Shi 2012; Kooistra and Helin 2012). Even though it is known that histone modifications affect gene expression and play important roles in development and diseases, many aspects remain to be uncovered. The target genes of many histone modifiers are unknown, and individual differences in the activities of these chromatin modifiers are largely unexplored. This information is critical for understanding the functions of each histone modifier and how polymorphic forms can affect their roles in disease development.
In this study, we focused on KDM4C, a polymorphic gene expression regulator of a few hundred genes. We concentrated on KDM4C because as a histone modifying enzyme, it provides us an opportunity to study the genetics of epigenetic regulation of gene expression. With data from a genetics of gene expression study (Cheung et al. 2010), we identified chromosome 9p24, which includes KDM4C, as a region that influences the expression of many genes; this encouraged us to pursue KDM4C's role in gene regulation. Genetic and epigenetic regulations have mostly been studied in parallel; by studying polymorphisms in a histone demethylase, we assessed the genetic and epigenetic influence on gene expression jointly. KDM4C is a member of the Jumonji family of demethylases; it activates genes by removing methyl groups from the repressive histone mark, trimethylated lysine 9 of histone H3 (H3K9me3) (Cloos et al. 2006; Whetstine et al. 2006; Loh et al. 2007; Wissmann et al. 2007; Ishimura et al. 2009; Lu et al. 2012). Prior to our study, MDM2 was one of the few characterized target genes of KDM4C (Ishimura et al. 2009). Results from this study identified cis-acting sequence variants that influence KDM4C expression and a few hundred of its target genes. We showed that KDM4C activates its targets through binding to their promoters and demethylating trimethylated to dimethylated H3K9 marks. Individual differences in KDM4C expression affect its abundance and activity; thus individuals with high KDM4C expression have more dimethylated H3K9 marks at the target gene promoters than those with lower KDM4C expression. In normal and some cancer cells, KDM4C influences cell growth through activating target genes that participate in mitogenic signaling, regulation of the cell cycle, and translation. Our findings increase the number of known target genes of KDM4C, which allowed us to understand how this histone demethylase regulates cell proliferation.
Results
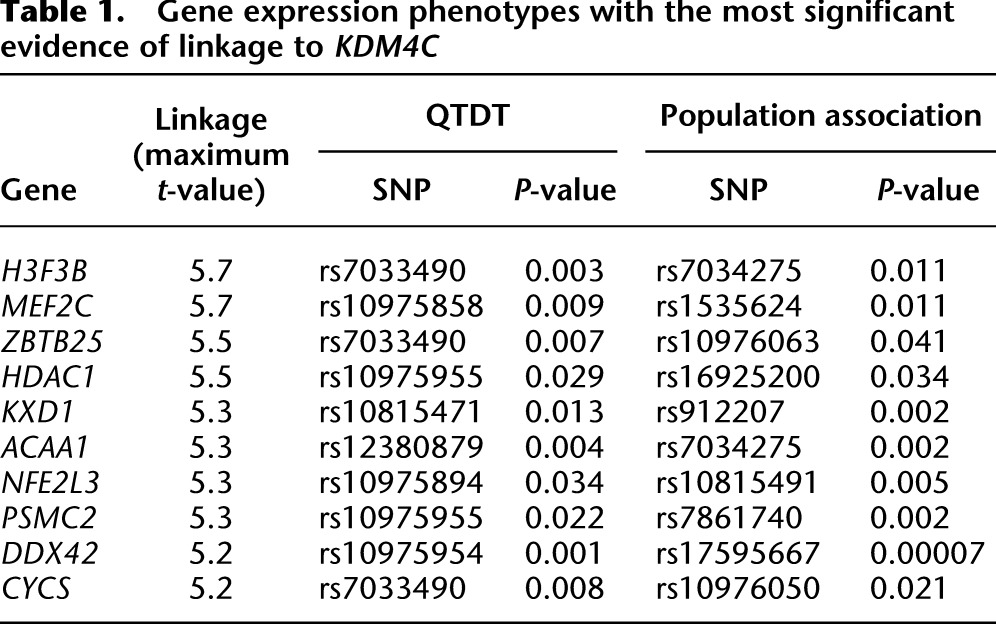
We focused on a regulatory region of gene expression in human B cells on chromosome 9, which includes the KDM4C locus (Cheung et al. 2010). Four genome-wide linkage results of gene expression levels that showed significant linkage to chromosome 9p24 are shown in Figure 1A. At a significance cutoff of t > 4 (corresponding to a logarithm of odds [lod] score of ∼3.4 and a genome-wide corrected significance level of 0.05), 41 gene expression phenotypes showed linkage to this region (Supplemental Fig. S1). The 10 phenotypes with the most significant linkage to chromosome 9p24 are listed in Table 1, and the remaining results are in Supplemental Table S1. These expression phenotypes included several histone variants or chromatin modifiers such as H3F3B (t = 5.7), H2AFV (t = 4.8), and HDAC1 (t = 5.5). At less stringent cutoffs of t > 3 and t > 2, there were 189 and 391 gene expression phenotypes with linkage to chromosome 9p24 (Supplemental Table S1). To follow up the linkage results, we carried out family-based association analyses using the quantitative transmission disequilibrium test (QTDT) (Abecasis et al. 2000). Chromosome 9p24 is not a very gene-rich region; even so, for most of the expression phenotypes, the chromosome 9p24 linkage peaks contain more than 10 genes (see Methods). We performed association analyses with SNPs in or near the expressed genes under the linkage peaks. The results confirmed that the expression levels of 366 out of the 391 expression phenotypes were significantly linked and associated with sequence variants within and around KDM4C (P < 0.05, nominal) (Supplemental Table S1). We then carried out population association analysis on 60 unrelated individuals. The results showed that 376 expression phenotypes are significantly associated with sequence variants of KDM4C in these individuals (P < 0.05, nominal) (Supplemental Table S1). In total, 353 of the 391 expression phenotypes were significantly associated with sequence variants in KDM4C by both family- and population-based association analyses. The linkage and association results from four phenotypes are shown in Figure 1B; those for the 41 phenotypes with the most significant linkage to chromosome 9 are shown in Supplemental Figure S1. The most significant evidence of association for the majority of the phenotypes (>85%) was found in KDM4C (Fig. 1B; Supplemental Fig. 1); thus, the linkage signals for most of the phenotypes can be explained by polymorphisms in KDM4C. However, the expression levels of most genes are influenced by more than one regulator. It has been noted that regulatory elements and genes tend to cluster (Christians and Senger 2007; Hasstedt et al. 2013); thus, it is possible that the chromosome 9 linkage region includes other regulators that influence gene expression. Our genetic results (in particular the significant association data from just 60 individuals) suggest that KDM4C is a key regulator of the expression levels of these genes. For the rest of this study, we focused on KDM4C in molecular studies to study the functional impact of sequence variants in KDM4C and to better understand how individual differences in histone modification affect gene expression.
Figure 1.

Chromosome 9p24 that includes KDM4C is a gene expression regulatory region. (A) Linkage plots for four gene expression phenotypes that show significant linkage to chromosome 9p24. (B) Results from QTDT and population association with SNPs in expressed genes under the linkage peaks for expression levels of four genes. Expressed genes are depicted in green; genes that are in the region but not expressed in B cells are shown in black. KDM4C is shown in red, and the KDM4C gene region is boxed.
Table 1.
Gene expression phenotypes with the most significant evidence of linkage to KDM4C
KDM4C is cis-regulated
Histone demethylases influence gene expression by removing specific methylation marks from histones and therefore altering chromatin structure (Shi et al. 2004; Lee et al. 2005). Trimethylation of lysine 9 on histone H3 (H3K9me3) is associated with transcriptional repression (Rea et al. 2000; Bannister et al. 2001; Lachner et al. 2001). KDM4C demethylates H3K9me3 and promotes gene activation (Loh et al. 2007; Wissmann et al. 2007; Ishimura et al. 2009). Here, we studied its expression and regulatory function. First, we measured its gene and protein expression levels in B cells. Because KDM4C was not represented on the microarray used in the original genetic mapping, we measured KDM4C gene expression in cultured B cells from 24 unrelated individuals by quantitative RT-PCR. We found that among individuals, KDM4C gene expression varied by as much as 5.5-fold (Fig. 2A). To ensure that this is not specific to cultured B cells, we also measured KDM4C expression in primary B cells, and a similar extent of individual variation was observed (Fig. 2A). This variability is also seen at the protein level; individuals with high gene expression had high KDM4C protein expression (Fig. 2B). The gene and protein expression levels of KDM4C are significantly correlated (P < 0.05) (Fig. 2C).
Figure 2.

Cis-regulation of KDM4C. (A) KDM4C expression levels in cultured B cells of 24 unrelated individuals (open circles) and primary B cells of nine unrelated individuals (filled circles) are shown relative to the individual with the lowest expression, as measured by qRT-PCR. (B) Western blot analysis of KDM4C protein expression with cultured B cells from three individuals with the lowest and three individuals with the highest KDM4C gene expression (n = 3). (C) Scatter plot of KDM4C gene and protein expression in the cultured B cells from 24 individuals in A; significant correlation in transcript and protein expression was found (R2 = 0.39, P < 0.05). (D) KDM4C expression in B cells is shown by their genotype at SNP rs7868863 (P = 0.006, allelic association). The SNP position is indicated.
To determine whether the individual variability in KDM4C gene expression is cis-regulated, we tested single nucleotide polymorphisms (SNPs) in and near KDM4C for association with its expression level. An intronic SNP, rs7868863, near the 3′ end of the KDM4C gene, showed the most significant allelic association (P = 0.006) with KDM4C expression (Fig. 2D). A block of 12 nearby SNPs spanning 106 kb also showed significant association (P < 0.01). To validate this finding, we compared our results to those from other genetics of gene expression studies (http://eqtl.uchicago.edu) and found that others also showed cis-regulation of KDM4C expression. To confirm the cis-regulation of KDM4C experimentally, we looked for evidence of differential allelic expression by measuring the expression of the two allelic forms of KDM4C in 15 individuals heterozygous for an A/C SNP, rs913590, in the 3′ UTR of KDM4C (∼12 kb from rs7868863, and the A-allele of rs7868863 and the C-allele of rs913590 are on the same haplotype). We found that the expression levels of the C-bearing KDM4C transcripts were significantly higher than the A-bearing transcripts (P = 0.001, t-test). The average allelic ratio (C/C + A) is 0.63 compared with the equal allelic expression of 0.5. Consistent with these results, association analysis of this SNP with KDM4C expression showed that the C-allele is associated with higher expression of KDM4C (P = 0.04).
Next, to explore the regulatory potential of the 3′ region of KDM4C that contains sequence variants associated with KDM4C expression, we looked for evidence of enhancer activity. With data from the ENCODE Consortium, we found that a 3.5-kb region showed hypersensitivity to DNase I treatment, consistent with open chromatin. This region is enriched for features of active enhancers (Visel et al. 2009; Creyghton et al. 2010), including EP300 protein, H3K27 acetylation, and H3K4 monomethylation (Fig. 3A). Additionally, the DNA sequences in this region are conserved from platypus to human (Fig. 3B). In order to determine if this region has regulatory function, we used reporter assays and found enhancer activity from a minimal region of 290 bases. Promoter constructs with the enhancer showed 26-fold higher luciferase activity (Fig. 3C) than the promoter alone. Together these results revealed that KDM4C expression is cis-regulated, most likely by an enhancer in the 3′ region.
Figure 3.

The 3′ regulatory region of KDM4C shows enhancer activity. (A) The black bar above the KDM4C gene schematic represents the region containing SNPs with significant association to KDM4C expression; the position of SNP rs7868863 is marked. (Bottom) The putative enhancer region is indicated; ENCODE data from EBV-transformed B cells of GM12878 show enrichment of EP300, H3K27 acetylation, H3K4 monomethylation, and DNase I hypersensitivity. Sequence conservation, from the UCSC Vertebrate Multiz Alignment tract, is also displayed. The gray bar shows the location of the 290-bp fragment used in the luciferase reporter assay. (B) Sequence alignment is shown for a 120-bp DNA segment of the region used in the luciferase reporter assay. Sequences that are identical across the represented species are shown in red and are marked by a star below the alignment. (C) Relative luciferase activity in primary fibroblasts transfected with the reporter containing the putative enhancer region compared with control. Assays were performed in primary fibroblasts from three individuals, in duplicate. Error bars, SEM.
KDM4C knockdown affects target gene expression
Next, we carried out molecular studies to follow-up the genetic mapping results that pointed to the role of KDM4C as a gene expression regulator. RNA interference was used to knockdown KDM4C expression in cultured B cells and in primary fibroblasts. First, we carried out transient knockdown of KDM4C in cultured B cells. KDM4C expression decreased by 60% at the transcript level and by ∼50% at the protein level (Fig. 4A). To assess the effect of KDM4C knockdown on target gene expression, we first studied 15 target genes with significant evidence of linkage and association to KDM4C; these genes were chosen because their expression levels were most significantly linked to the region of chromosome 9 where the KDM4C locus resides and because of our interests in some of these genes such as HRAS and SQSTM1 that encode an oncogene and a protein in the signaling pathway that activates NFKB, respectively. The results showed that the expression of 12 genes, including ZBTB25, RNMT, H3F3B, and MAP3K5, decreased significantly (P < 0.05) following KDM4C knockdown (Fig. 4B). Given that genes have different stability and combinatorial regulation, we do not expect to see changes in the expressions of all the target genes when examining one time-point following KDM4C knockdown. Seeing that the majority of the genes were affected, we examined a larger set of KDM4C target genes. We established stable knockdown of KDM4C in primary fibroblasts by expressing shRNA constructs targeting KDM4C or a control shRNA. Skin fibroblasts provide us primary cells to examine the relationship between KDM4C and its target genes. Gene knockdown was confirmed by Western blot (Fig. 4C). All four shRNA constructs were equally efficient at decreasing KDM4C expression. By quantitative reverse transcription PCR (qRT-PCR), 13 of the 15 target genes showed a significant (P < 0.05) decrease in expression following stable knockdown of KDM4C in primary fibroblasts (Fig. 4D), indicating that KDM4C regulates a common set of genes in B cells and primary fibroblasts. We then used microarrays to examine expression changes in a larger set of genes following KDM4C knockdown. Of the 391 genes whose expression levels showed evidence of linkage to the KDM4C locus in B cells, 340 were expressed in fibroblasts. Following knockdown of KDM4C in primary fibroblasts, the expression levels of 132 of the 340 target genes decreased by ≥20% (Fig. 4E; Supplemental Table S2). Eighty-nine target genes increased expression by ≥20% (Fig. 4E; Supplemental Table S2). Even though KDM4C is a positive regulator of gene expression, not all the target genes are expected to be its direct targets, and feedback regulation is also expected; thus, not all the target genes would decrease in expression following KDM4C knockdown. Together, these results confirm the target genes identified in our genetic study and show that many of the KDM4C target genes identified in B cells are also targets of KDM4C in fibroblasts.
Figure 4.

Molecular validation of KDM4C target genes in B cells and primary fibroblasts. (A) B cells from four individuals were treated with siRNA targeting KDM4C or a control siRNA. (Top) qRT-PCR data confirming KDM4C depletion at the transcript level; (bottom) a representative Western blot. (B) Gene expression changes in B cells from four individuals for 15 target genes, measured by qRT-PCR, following KDM4C knockdown. RPS6 is the control gene. Error bars, SEM ([*] P < 0.05, t-test). (C) Western blot of cells transfected with shRNA constructs targeting KDM4C or a control shRNA confirms knockdown of KDM4C in primary skin fibroblasts. (D) Gene expression changes of 15 target genes in primary fibroblasts by qRT-PCR following stable knockdown of KDM4C using the four shRNA constructs from C. RPS6 is the control gene. Ctr indicates control. Error bars, SEM ([*] P < 0.05, t-test). (E) Gene expression changes in 340 target genes measured by microarrays following KDM4C knockdown with sh1 (two biological replicates, in duplicate). Data from two additional shRNA constructs showed highly similar results (P < 0.001) (Supplemental Fig. S2B,C).
KMD4C binds to target genes, demethylates H3K9me3, and increases gene expression
We next investigated the mechanism by which KDM4C affects the expression levels of its target genes. We carried out chromatin immunoprecipitation (ChIP) assays to assess if KDM4C binds to its target genes. We also measured the relative abundance of tri- and dimethylated H3K9.
First, we carried out ChIP assays in human B cells with antibodies against KDM4C. The results showed that there was significant enrichment of KDM4C binding to target gene promoters. Results for eight target genes are shown in Supplemental Figure 3. For example, KDM4C binding was fivefold and eightfold greater at the promoters of ZBTB25 and RNMT, respectively, than at the promoter of the nontarget control RPS6. Next, since we are interested in individual variation of KDM4C, we asked if differences in KDM4C expression affect the binding of KDM4C to its target genes. We found that more KDM4C bound to the target genes in cells from individuals with higher KDM4C expression levels than those with lower KDM4C expression levels (Fig. 5A). For example, in KDM4C immunoprecipitates, sequences from the promoter of ZBTB25 were 2.4-fold and those of RNMT were 5.6-fold more enriched in cells from individuals with high versus low KDM4C expression. Together, these results show that KDM4C binds to its target genes, and KDM4C expression influences the amount of KDM4C bound to its target genes.
Figure 5.

KDM4C expression influences target gene expression levels. (A) Chromatin immunoprecipitation (ChIP) for KDM4C at target gene promoters in B cells from three individuals with high and three individuals with low KDM4C expression. The negative controls are the promoter of RPS6 and an intronic region. (B) The ratio of H3K9me2 to H3K9me3 at the eight target genes in A is shown for individuals with high KDM4C expression compared with low KDM4C expression (n = 3 per group). (C) Target gene expression levels in seven individuals with high KDM4C expression compared with seven individuals with low KDM4C expression, by qRT-PCR. RPS6 is used as the control gene. P < 0.05 for all target genes except RRP7A (t-test). Error bars, SEM.
Given that KDM4C removes methyl groups from H3K9me3 to produce H3K9me2 (and, to a lesser extent, H3K9me1), we next measured the relative abundance of H3K9me2 and H3K9me3 at the target genes. Individuals with high KDM4C expression had higher ratios of H3K9me2 to H3K9me3 at its target genes than did individuals with low KDM4C expression (P < 0.005, t-test) (Fig. 5B). For example, at ZBTB25, this ratio is 2.1-fold and at RNMT, it is 1.6 higher in individuals with high KDM4C expression than individuals with low KDM4C expression. The difference in H3K9me2 level is specific to the KDM4C target genes since the global levels of methylated H3K9 were not significantly different among individuals with high and low KDM4C expression (Supplemental Fig. S4). These results show that KDM4C demethylates trimethylated H3K9 at its target genes and that the efficiency of the demethylase activity is influenced by KDM4C expression.
Next, we examined if differences in KDM4C demethylase activity affect the expression levels of its target genes. Removal of H3K9me3, a repressive mark, to its dimethylated form promotes gene activation. We compared the expression levels of KDM4C target genes in individuals with high and low KDM4C expression and found that individuals with higher KDM4C expression have significantly higher target gene expression levels than did those with lower KDM4C expression. For example, ZBTB25 and RNMT expression levels were 2.5- and 2.1-fold higher in individuals with high KDM4C than in individuals with low KDM4C, respectively (P < 0.05, t-test) (Fig. 5C). Thus, our results show that differences in KDM4C expression affect its demethylase activity and, subsequently, its target gene expression.
Target genes of KDM4C influence cell growth
Having found that KDM4C influences the expression of its target genes, we next assessed if this regulatory role affects cellular phenotypes. We studied the functions of the target genes and found that they are enriched for proto-oncogenes (Pc = 0.02, Benjamini), genes involved in programmed cell death (Pc = 0.003, Benjamini), and cell cycle regulation (P = 0.006). An examination of these target genes suggests that KDM4C regulates cell growth through various pathways. For instance, KDM4C induces HRAS and MYC, which are potent activators of cell growth, and CDC5L, which promotes progression from G2 to M phase in the cell cycle (Bernstein and Coughlin 1998). KDM4C also promotes cell growth by repressing cell cycle inhibitors, such as CCNG2 (Horne et al. 1997). In addition, KDM4C induces BIRC5 and MEF2C, which inhibit apoptosis (Tamm et al. 1998; Wang et al. 2005), and induces RNMT and several aminoacyl tRNA synthetases that promote growth by regulating the translation of cell cycle genes (Cowling 2010).
To test if KDM4C affects cell growth, we measured several parameters. Growth curve analysis (Fig. 6A) showed that B-cell counts from individuals with high KDM4C expression increase faster than those from individuals with low KDM4C expression (P = 3 × 10−6, ANOVA). We also compared relative BrdU incorporation in individuals with high and low KDM4C expression. On average, individuals with high KDM4C expression had twofold more BrdU incorporation compared with individuals with low KDM4C expression (Fig. 6B). To assess the extent that KDM4C contributes to individual differences in cell proliferation, we regressed BrdU incorporation on KDM4C expression levels. The results showed that KDM4C expression level explains ∼25% of individual differences in cell proliferation (Fig. 6C). We next compared the proportion of B cells in different phases of the cell cycle between individuals with high and low KDM4C expression. The results showed that KDM4C expression correlates with cell cycle progression; with higher KDM4C expression, fewer cells (6.5 ± 0.01%) were arrested in G0/G1 and more cells (5.0 ± 0.3%) were in the S phase, G2, and M phases of the cell cycle (Fig. 6D).
Figure 6.

KDM4C expression is associated with cell growth in B cells. (A) Growth curve of B cells from five individuals with high or low KDM4C expression (P = 3 × 10−6, ANOVA). (B) BrdU incorporation in B cells from six individuals with high or low KDM4C expression, measured in triplicate ([*] P < 0.05, t-test). (C) BrdU incorporation is regressed on KDM4C expression level in B cells from 22 individuals, measured in triplicate. Error bars, SEM. (D) Proportion of cells at different stages of the cell cycle in individuals with high or low KDM4C expression. Error bars, SEM.
To confirm that the relationship between KDM4C and cell growth is not merely correlative, we overexpressed KDM4C in skin fibroblasts by transfection, which increased KDM4C expression by fivefold (Fig. 7A). The expression levels of KDM4C target genes, including MYC, MAP3K5, NCK1, and RNMT, increased following KDM4C overexpression (Fig. 7B). The cells that overexpress KDM4C grew significantly faster than control cells (P = 0.003, ANOVA) (Fig. 7C). BrdU incorporation assays showed that overexpression of KDM4C significantly increases cell proliferation (1.5-fold, P = 0.007, t-test) (Fig. 7D). Cell cycle analysis showed that KDM4C overexpression led to a decrease in the percentage of cells in G0/G1 (−28.0 ± 2.1%) and an increase in the percentage of cells between the S phase, G2, and M phases of the cell cycle (27.1 ± 1.3%) (Fig. 7E). Together, these results demonstrate that KDM4C promotes cell growth through regulation of its target genes.
Figure 7.

KDM4C overexpression accelerates cell growth. (A) Overexpression of KDM4C in primary fibroblasts was confirmed by qRT-PCR and Western blot relative to control cells. (B) Target gene expression changes following KDM4C overexpression in primary fibroblasts (RPS6 is used as a control). (C) Cell growth following KDM4C overexpression was measured by growth curve analysis (measured in quadruplicate; P = 0.003, ANOVA). (D) BrdU incorporation was measured in triplicate relative to control cells ([*] P < 0.05, t-test). (E) Changes in the proportion of cells at different stages of the cell cycle following KDM4C overexpression. Ctr indicates control cells. Error bars, SEM.
KMD4C promotes growth of cancer cells
Next, we asked if KDM4C contributes to the growth of normal cells whether it also plays a role in cancer cells. We measured KDM4C expression levels in 289 tumor and 68 matched normal samples from 18 different tissues. The results showed that in 10 of the 18 tissues, the average KDM4C expression levels were higher in the tumors than in normal samples. In seven tissues, KDM4C expression was at least 20% higher in cancer than in matching normal samples (Fig. 8A). In the pancreatic tissues, KDM4C expression was significantly higher in the cancer cells than the matching controls (P = 0.04, t-test). Amplification of the chromosomal region 9p24, which includes KDM4C, has been observed in a variety of cancers, including pancreatic tumors (Cloos et al. 2006; Rui et al. 2010; Zhang et al. 2011). We next used the Gene Expression Database of Normal and Tumor Tissues (GENT) (Shin et al. 2011) to validate the increased KDM4C expression in cancer. Data from GENT show that KDM4C expression is higher in six of the seven cancer types that we found to show at least 20% higher KDM4C expression than their matching normal controls (Fig. 8B). In addition, in a recent paper, Whetstine and colleagues (Whetstine et al. 2006), using sequencing data from TCGA, also noted higher KDM4C expression and the amplification of the KDM4C locus in cancers (Black et al. 2013). Increased KDM4C expression can affect cellular function; the highly expressed KDM4C in tumors may work cooperatively with other amplified genes to promote cell survival, as has been reported in lymphoma (Rui et al. 2010). Yamamoto and colleagues showed that KDM4C expression is necessary for colonsphere formation in colon cancer cells through interaction with beta catenin (Yamamoto et al. 2013). Even though independently our samples and those in the GENT database and TCGA are small, the fact that they showed uniformly that KDM4C expression is higher in some tumors than in normal cells suggests that this lysine demethylase is overexpressed in these cancers.
Figure 8.

KDM4C expression level in cancer cells. (A) Average KDM4C expression level in cancer tissues relative to matching normal tissues, by qRT-PCR. (B) Ratios of KDM4C expression levels in cancers and the matching controls in seven tissues by our qRT-PCR compared with data from the GENT database of microarray data. (C) KDM4C expression in HT-29 cells expressing shRNA targeting KDM4C or a control shRNA, by qRT-PCR (top) and by Western blot (bottom). (D) ChIP for KDM4C at two target genes, MYC and RNMT, following KDM4C knockdown in HT-29 cells. (E) The ratio of H3K9me2 to H3K9me3 levels at promoters of MYC and RNMT, as determined by ChIP in HT-29 cells following KDM4C knockdown relative to controls. (F) Expression changes in KDM4C target genes following KDM4C knockdown in HT-29 cells (PDIA6 is the control gene). (G) Growth curve analysis (measured in quadruplicate; P = 7 × 10−8, ANOVA), BrdU incorporation (H; measured in triplicate) and changes in the proportion of cells at different stages of the cell cycle (I) are shown for HT-29 cells with and without KDM4C knockdown. Ctr indicates control cells ([*] P < 0.05, t-test). Error bars, SEM.
Next, we examined experimentally if KDM4C overexpression influences cancer cell growth. If high levels of KDM4C expression contribute to cell growth in cancer cells, then lowering KDM4C expression may slow the proliferation of cancer cells. To test this hypothesis, we knocked down KDM4C in HT-29 colorectal cancer cells (Fig. 8C). ChIPs of KDM4C showed that KDM4C binding was reduced at the promoters of MYC and RNMT in shKDM4C-treated cells compared with controls (Fig. 8D). This led to more H3K9me3 and less H3K9me2 at promoters of these genes (Fig. 8E). The lower KDM4C expression also resulted in a lower expression of the growth promoting target genes such as MYC (Fig. 8F).
Next, we examined if KDM4C knockdown affects tumor cell growth. We found that silencing of KDM4C in HT-29 colorectal cancer cells led to a significant attenuation of growth by growth curve analysis (P = 7 × 10−8, ANOVA) (Fig. 8G) and BrdU incorporation (P = 0.03, t-test) (Fig. 8H). By flow cytometry, we found more cells were in the G0/G1 stages of the cell cycle following knockdown of KDM4C compared with control cells (9.7 ± 4.0%) and fewer cells progressed into the later stages of the cell cycle (S/G2/M; −9.0 ± 3.1%) (Fig. 8I). These results show that in addition to normal cells, KDM4C regulates growth in some cancer cells.
Discussion
In summary, normal variation in human gene expression allowed us to uncover cis-regulation of KDM4C expression and to identify a few hundred target genes of KDM4C. Although prior to our study, it was known that KDM4C is a histone demethylase, little was known about its cellular impact, since only a few of its target genes had been identified. Here, we uncovered DNA sequence variants of KDM4C that explain the extensive individual differences in transcript and protein expression of the gene itself, and showed that they are associated with differences in the demethylase activity of KDM4C on genes such as MYC and RNMT, which in turn affect cell growth. Fibroblasts and B cells from normal individuals with high KDM4C grow faster than those from individuals with lower KDM4C levels. We then extended the analysis to cancer and found 20% higher KDM4C expression in seven different cancer types relative to matched normal tissues. To show that the relationship between KDM4C expression level and cell growth is not merely correlative, we overexpressed KDM4C in dermal fibroblasts from normal individuals and observed the expected changes in cell growth. In addition, we were able to attenuate cell proliferation in colorectal cancer cells by knockdown of KDM4C.
These results are important for elucidating the function of KDM4C, but they also show the role of genetic variation in functional studies and the relationship between genetic and epigenetic regulations. Individual variation is often viewed as an unwanted complication in mechanistic studies. Here, by taking advantage of individual differences in KDM4C expression, we determined the genetic basis of this variation and identified its target genes and downstream effect on cellular phenotype. Furthermore, since KDM4C is a chromatin modifier, our results showed that epigenetic regulation of gene expression can be influenced by DNA sequence polymorphisms, thus demonstrating that genetic and epigenetic regulations are not distinct pathways of gene regulation.
Genetic variation generates phenotypic differences that enable mechanistic studies. However, to conduct these studies, genetic and phenotypic data are needed from a large number of individuals. Advances in technologies have facilitated such data collection. Here, the knowledge of the genetic regulation of KDM4C broadened our understanding of how it regulates other genes and cellular functions in normal and cancer cells. To date, most studies have focused on sequence variants that act in cis to regulate gene expression. Extension of our approach will complement on-going studies of cis-regulation to identify the impact of regulatory variants that act in trans to affect gene regulation and phenotypes. Together the results will provide a more comprehensive view of the mechanistic basis of how sequence variants affect gene regulation and ultimately disease susceptibility.
Methods
Linkage analysis and association studies
Linkage analysis was performed as described previously (Cheung et al. 2010). The data were from members of 45 three-generation CEPH-Utah families (CEPH 1328, 1330, 1331, 1332, 1333, 1334, 1340, 1341, 1344, 1345, 1346, 1347, 1349, 1350, 1353, 1354, 1356, 1357, 1358, 1362, 1375, 1400, 1408, 1413, 1416, 1418, 1420, 1421, 1423, 1424, 1444, 1447, 1451, 1454, 1456, 1458, 1459, 1463, 1477, 1582, 13281, 13291, 13292, 13293, 13294). Individuals are United States residents of northern and western European descent. For linkage analysis, only the offspring were used. On average, there were eight sibs per family. In this study, we focused on a region on chromosome 9p24 where a few hundred expression phenotypes showed significant evidence of linkage. This region includes the histone demethylase, KDM4C, gene locus (chr9: 6,710,863–7,175,648). For each gene expression phenotype, the expressed genes within the linkage peak (t > 2) were defined as those with an RPKM > 1 in B cells, as previously described (Cheung et al. 2010). Supplemental Table S5 lists the number of expressed genes within the linkage peaks on chromosome 9p24 for the 391 gene expression phenotypes. QTDT analyses of SNPs within or near the expressed genes under each linkage peak (t > 2) were performed using the orthogonal (ao) model and variance component options (wega) (Abecasis et al. 2000). The regions and the number of SNPs tested in association studies for each of the expressed genes are listed in Supplemental Table S6. Then, we followed up all the significant QTDT results (expressed genes for which a SNP had a P < 0.05) by population association analyses with SNPs within or near genes that were significant by QTDT analysis. The population association analyses were carried out using expression data from B cells of 60 unrelated individuals of northern and western European descent. The QTDT and population association results for phenotypes with significant linkage of t > 4 are plotted in Supplemental Figure S1; the most significant P-values in KDM4C are listed in Supplemental Table S1.
Cell culture and mRNA analysis
Cultured B cells from unrelated individuals of northern and western European descent from the CEPH-Utah collections (GM12056, GM11839, GM06994, GM12891, GM07034, GM12004, GM07022, GM07345, GM12813, GM12155, GM11992, GM12145, GM12750, GM12717, GM12874, GM12264, GM07056, GM12005, GM12144, GM12872, GM06993, GM12815, GM12003, and GM06986) were cultured in RPMI 1640 with 15% fetal bovine serum, 2 mM L-glutamine, and 100 units/mL penicillin-streptomycin. Primary B cells were obtained from the University of Pennsylvania Human Immunology Core Facility. HT-29 cells were purchased from ATCC and cultured in RPMI 1640 as above, except supplemented with 10% fetal bovine serum. Primary dermal fibroblasts were isolated from neonatal foreskin (University of Pennsylvania Skin Disease Research Center Core) and cultured in MEM supplemented with 10% fetal bovine serum, 2 mM L-glutamine, and 100 units/mL penicillin–streptomycin. For gene expression and protein analyses, B cells were suspended at 5 × 105 cells/mL in culture media, and RNA and protein were extracted 24 h later. HT-29 cells and primary fibroblasts were grown to 70% confluence and then lysed for RNA or protein analysis. Following RNA extraction, cDNA was generated using the TaqMan Reverse Transcription kit (Invitrogen) and primed with oligo dT. Gene expression levels were determined by qRT-PCR on a 7900 HT PCR machine (Applied Biosystems) using Power SYBR Green dye (Applied Biosystems) to detect products. For primer sequences, see Supplemental Table 4. Single gene products were confirmed by dissociation curve analysis and by visualization on agarose gel. Relative gene expression levels were analyzed using the ΔΔCt method: Ct values were normalized to the housekeeping gene, NDUFA4, and then expressed as the fold difference relative to the individual with the lowest KDM4C expression (GM12056).
Protein analysis
For Western blot analysis, cells were lysed in 1× lysis buffer (20 mM Tris-HCl at pH 7.5, 150 mM NaCl, 1 mM Na2EDTA, 1 mM EGTA, 1% Triton X-100; Cell Signaling) supplemented with 1× complete protease inhibitors (Roche). Twenty to 50 μg of total protein was incubated overnight at 4°C with the following antibodies: 5 μg KDM4C antibody (A300-885A, Bethyl Laboratories), 5 μg hnRNPL antibody (ab6106, Abcam), 2 μg beta-tubulin (Millipore), or 2 μg TBP (ab818, Abcam). KDM4C protein levels were quantified using ImageJ software normalized to hnRNPL protein levels and are shown as the fold difference relative to the individual with the lowest expression.
Association analysis of expression level of KDM4C
To test for cis-association with the expression level of KDM4C, we carried out population association analysis by linear regression. KDM4C expression level in B cells from the 24 unrelated individuals, as a dependent variable, was regressed on SNP genotypes (coded 0, 1, 2). We tested SNPs within the KDM4C gene, ±50 kb (chr9: 6,660,863–7,215,648).
Differential allelic expression
Allele-specific real-time PCR was performed in heterozygous individuals using a TaqMan assay for SNP rs913590 (C_1416486_10, Life Technologies). Allele-specific gene expression was measured in 15 individuals (GM06993, GM06994, GM07056, GM07345, GM12005, GM12144, GM12145, GM12264, GM12750, GM12813, GM12815, GM12874, GM12891, GM07034, GM12717) by real-time PCR on an 7900 HT real-time PCR machine (Applied Biosystems) using 5 ng of cDNA. A standard curve (linear regression line) was generated for each SNP by mixing gDNAs from two homozygous individuals, one of each genotype, at ratios of 8:1, 4:1, 2:1, 1:1, 1:2, 1:4, and 1:8. The TaqMan assay contains probes specific for each allele of rs913590, which are labeled with the fluorescent dye FAM (C-allele) or VIC (A-allele). Each sample was run in triplicate according to the manufacturer's instructions. Fluorescence signal was read after the final cycle and the prerun fluorescence signal subtracted as background using SDS 2.4 software (Applied Biosystems). The log of FAM mean Ct/VIC mean Ct values were plotted against the log of the gDNA ratio. The linear graph (correlation coefficients > 0.95) was used to calculate the corresponding allele-specific gene expression. Significance was determined by t-test.
Enhancer identification and enhancer reporter assay
IGV2.2 software (Broad Institute) (Robinson et al. 2011; Thorvaldsdottir et al. 2012) was used to visualize the following ENCODE data sets (The ENCODE Project Consortium 2012): GM12878 EP300 SC584 standard signal from ENCODE/SYDH, GM12878 H3K27 acetylation ChIP-seq signal from ENCODE/BROAD, GM12878 H3K4 monomethylation ChIP-seq signal from ENCODE/BROAD, and GM12878 DNase-seq signal from ENCODE/DUKE within the genomic region (hg18; chr9: 7,096,769–7,203,459). This region contains SNPs that were significantly associated with KDM4C expression level. Sequence conservation from the UCSC Vertebrate Multiz Alignment (28 species) genome browser tract is also displayed. A 290-bp fragment of the putative enhancer region (hg18; chr9: 7,108,126–7,108,415) was cloned upstream of a pCLuc Mini-TK2 luciferase reporter vector with a minimal reporter (New England Biolabs) using XhoI and KpnI restriction endonucleases (New England Biolabs). Primary fibroblasts were transfected with 0.5 μg of either the enhancer_pCLuc Mini-TK2 or the empty pCLuc Mini-TK2 vector and with 20 ng of control luciferase vector (pCMV GLuc control vector; New England Biolabs). Cells from three individuals were transfected in duplicate with Lipofectamine LTX transfection reagent (Invitrogen) for 12 h; luciferase activity was assayed in the cell culture media at 24 h using the BioLux Cypridina luciferase (CLuc) and BioLux Gaussia luciferase (GLuc) assay kits (New England Biolabs); and luminescence was read on a veritas microplate luminometer (Turner Biosystems). The CLuc/GLuc ratio was determined, and the fold difference was reported for the enhancer containing reporter relative to the empty vector. Significance was determined by t-test.
RNA interference and KDM4C overexpression
Cultured B cells from four individuals (GM12813, GM12144, GM11992, and GM12712) were transiently transfected with Silencer Select siRNA (Applied Biosystems) targeting KDM4C (s225931) or a control siRNA (control no 1) by electroporation using the Neon electroporation system (Invitrogen) with the following settings: 1350 V, 30 msec, and one pulse. RNA and protein were harvested 24 h after electroporation in order to assess knockdown efficiency and gene expression changes. Significance was determined by t-test. Primary fibroblasts were transfected with four shRNA constructs targeting KDM4C or a control shRNA (SureSilencing shRNA plasmids; SABiosciences) using Lipofectamine LTX transfection reagent (Invitrogen). HT-29 cells were transfected as above except with a cocktail of four shRNA constructs (sh1–4) (see Supplemental Fig. S2A) or the control shRNA. Stable clones were selected with 1 μg/mL puromycin (EMD) and then maintained at 0.5 μg/mL puromycin in complete culture media. RNA and protein were harvested 24 h after electroporation in order to assess knockdown efficiency and gene expression changes. Significance was determined by t-test. The KDM4C open reading frame (NM_015061.3) was cloned into the pcDNA3.1D/V5-His-Topo expression vector using the pcDNA3.1 Directional TOPO expression kit (Invitrogen) (for primer sequences, see Supplemental Table S4) and transfected into primary fibroblasts using Lipofectamine LTX transfection reagent (Invitrogen) and 1 μg of vector. Stable clones were selected with 200 μg/mL geneticin (Gibco) and then maintained at 100 μg/mL geneticin in complete culture media. RNA and protein were harvested in order to assess knockdown efficiency and gene expression changes. Significance was determined by t-test.
Microarray analysis
RNA was extracted from two primary fibroblast clones selected for stable expression of the control shRNA and five clones expressing shRNA constructs targeting KDM4C (two expressing shKDM4C1, two expressing shKDM4C2, one expressing shKDM4C3) (see Supplemental Fig. S2A). RNA was hybridized to the Gene Chip Human Genome U133A Plus 2.0 array (∼38,500 genes; Affymetrix), in duplicate. Expression intensities were scaled to 500 using the global scaling method implemented in the Expression Console software from Affymetrix and log2 transformed. Genes were considered if they were called as “present” in the control samples or in at least 3/5 of the knockdown samples. Three hundred forty of the 391 target genes identified in B cells were present in primary fibroblasts, and the average gene expression differences were determined for each shRNA construct in the KDM4C knockdown samples relative to controls. Gene expression differences were determined for each shRNA construct relative to the control shRNA. Effects on gene expression are highly similar between the different shRNAs (Supplemental Fig. S2B,C).
Gene ontology analysis
The 132 genes that decreased and 89 genes that increased by ≥20% following KDM4C knockdown in primary fibroblasts were analyzed by the DAVID Functional Annotation Tool (Dennis et al. 2003), compared with a background of the 11,509 genes expressed in primary fibroblasts.
Chromatin immunoprecipitation
ChIP was performed as previously described (Ernst et al. 2011), using ∼1 × 107 B cells for each immunoprecipitation. KDM4C immunoprecipitations were performed with 10 μg of antibody against KDM4C (Bethyl Laboratories) or a rabbit IgG control antibody (Millipore) per reaction in B cells from three individuals with high KDM4C expression (GM12003, GM12144, GM12815) and three individuals with low KDM4C expression (GM12891, GM11839, GM07022). An individual with high and an individual with low KDM4C expression were paired into three sets, and ChIPs for each set were performed together. Immunoprecipitations for histone modification levels were also carried out in these individuals. ChIP for histone modification level was performed with the following antibodies: 4 μg trimethylated H3K9 (17-625; Millipore), 4 μg dimethylated H3K9 (39683; Active Motif), and 2 μg total H3 (Abcam). For each ChIP, precipitated DNA was resuspended in 80 μL TE, and 2 μL was used for each qPCR reaction. Sequence enrichment was determined by qPCR relative to input and was performed in triplicate. Primer sequences are listed in Supplemental Table S4: The same primer sequences were used for KDM4C and histone ChIP-qPCR experiments. When determining the fold difference in the H3K9me2-to-H3K9me3 ratio between individuals with high and low KDM4C expression, the H3K9me2-to-H3K9me3 ratio at a target gene was calculated for both individuals in a set. For the eight target genes (CLTA, TRA2B, ZBTB25, MEF2C, RRP7A, H3F3B, RNMT, and MYC), we compared by t-test the H3K9me2/3 ratio for individuals with high versus low KDM4C expression. ChIP was also performed in HT-29 control or shKDM4C-treated cells as described above in B cells, except that the following antibody concentrations were used: 15 μg of antibody against KDM4C, 15 μg of rabbit IgG control antibody, 8 μg trimethylated H3K9, 8 μg dimethylated H3K9, 8 μg monomethylated H3K9, and 2 μg total H3. ChIPs were performed in duplicate.
Histone extractions and determination of histone modification levels among individuals
B cells from four individuals with low KDM4C expression (GM11839, GM06994, GM12891, GM07034) and four individuals with high KDM4C expression (GM07056, GM12144, GM12815, GM12003) were washed in PBS and suspended in Triton lysis buffer (PBS with 0.5% Triton X 100 [v/v], 1× complete protease inhibitors [Roche], and 5 mM sodium butyrate) at 1 × 107 cells/mL and lysed for 10 min on ice. Cell nuclei were collected by centrifugation and resuspended in 250 μL of 0.2 M HCl and incubated overnight at 4°C to extract histones. The supernatant was collected, and 10–20 μg of protein were analyzed by Western blot with the following antibodies: 2 μg trimethylated H3K9 (17-625; Millipore), 2 μg dimethylated H3K9 (39683; Active Motif), 5 μg monomethylated H3K9 (39681; Active Motif), or 0.5 μg total H3 (Abcam). Quantitative Western blot detection was carried out using secondary antibodies coupled to fluorophores (Licor), and the fluorescent signal was detected on an Odyssey fluorescent scanner (Licor). A representative Western blot is shown in greyscale in Supplemental Figure S4. No significant difference in fluorescent signal was detected among individuals with high and low KDM4C expression for any of the methylation states of H3K9, by t-test. Histone extractions from B cells were repeated, and the same results were obtained.
Cell growth assays
BrdU incorporation was determined using the cell proliferation ELISA, BrdU (colorimetric, 11647229001; Roche). BrdU incorporation was measured in B cells from 22 individuals in triplicate. Cells were seeded at 5 × 105 cells/mL and incubated with BrdU for 20 h. These data were reanalyzed for six individuals with high KDM4C expression (GM12815, GM12144, GM07056, GM12264, GM12750, and GM12003) and six individuals with low KDM4C expression (GM07345, GM11839, GM07034, GM07022, GM12006, and GM12891). BrdU incorporation was analyzed in HT-29 cells in triplicate following KDM4C knockdown versus control cells and in primary fibroblasts overexpressing KDM4C versus control cells, in triplicate. Cells were seeded at 2 × 105 cells/mL and incubated with BrdU for 20 h. Significance was determined by t-test. BrdU incorporation was also repeated at a later date with the same result. Growth curve analysis was performed in B cells from five individuals with high KDM4C expression (GM12005, GM12144, GM12815, GM07056, and GM12750) and five individuals with low KDM4C expression (GM12891, GM11839, GM07022, GM12006, and GM07034), in HT-29 cells following KDM4C knockdown compared with control cells, and primary fibroblasts overexpressing KDM4C compared with control cells. On day 0, 2 × 105 cells/mL was plated in culture media in triplicate for immortalized B cells or 1 × 105 cells/mL was plated in culture media in at least triplicate for HT-29 cells and primary fibroblasts. Cell number was determined on subsequent days by counting cells on a cellometer (Nexcelom). B-cell data were analyzed for each individual in triplicate, and then the mean was taken for individuals with high or low KDM4C expression. The significance was determined by ANOVA. Growth curve analysis was also repeated at a later date for individuals with high and low KDM4C expression; the results were highly similar.
Cancer tissue sample mRNA analysis
KDM4C expression level was assayed on a TissueScan disease tissue qPCR cDNA array (Origene) and normalized to the housekeeping gene NDUFA4. Expression levels were measured in duplicate. KDM4C expression levels were then analyzed as the fold difference relative to the sample with the lowest expression for each tissue. The mean of KDM4C expression in cancer samples was compared with the mean of KDM4C expression in the matched normal samples for each tissue type. In total, 68 normal samples and 289 cancer samples were studied (Supplemental Table S3).
Cancer tissue expression analysis from GENT
The Gene Expression in Normal and Tumor Tissues Database (Shin et al. 2011) is a collection of data microarray experiments. Fifteen of the 18 tissues that we investigated by qRT-PCR were represented in this database. Only experiments with both normal and cancer samples were included in our analysis. The average KDM4C expression level in cancer was compared with the average KDM4C expression level in normal samples for each experiment. Data from 1311 normal samples and 2793 cancer samples were studied (Supplemental Table S3).
Cell cycle analysis
Cells from an individual with high KDM4C expression (GM12750) and an individual with low KDM4C expression (GM12004) were resuspended at 5 × 105 cells/mL, and 2 million cells were harvested 24 h later. Cells were washed in cold PBS and then fixed in 70% EtOH for at least 12 h at −20°C. Cells were then washed in cold PBS and resuspended in 300 μL of propidium iodide staining solution (0.1% [v/v] Triton X-100 [Roche], 0.2 mg/mL RNase A [Roche], and 40 μg/mL propidium iodide [Sigma] in PBS). Resuspended cells were stained for 15 min at 37°C. Flow cytometry analysis was performed on an LSRFortessa (BD Biosciences), and the proportion of cells in the G0/G1, S, and G2/M phases of the cell cycle were modeled using FlowJo 7.6.5 software (TreeStar), ±SEM. Cell cycle analysis was also performed for HT-29 cells treated with shKDM4C and control cells, as described above. Primary fibroblasts overexpressing KDM4C and control cells were resuspended at 2 × 105 cells/mL and harvested 24 h later for cell cycle analysis, as described above.
Statistical analysis
Statistical analysis was performed using two-sided methodologies with a significance level of P < 0.05. Continuous variables were described according to group classification (KDM4C expression level) by mean and compared across groups by t-test. Growth analysis was analyzed by ANOVA: two factor with replication.
Data access
Microarray data have been submitted to the NCBI Gene Expression Omnibus (GEO; http://www.ncbi.nlm.nih.gov/geo/) under accession number GSE41040.
Acknowledgments
We thank Michael Morley and Zhengwei Zhu for help with data analysis; The Children's Hospital of Philadelphia Flow Cytometry Core for assistance with cell cycle analysis; Drs. Marisa Bartolomei, Brian Gregory, Kristen Lynch, Joshua Plotkin, and Zhaolan Zhou for discussion of the work; and members of the Cheung laboratory for suggestions and comments on the manuscript. This work is funded by grants from the National Institutes of Health and support from the Howard Hughes Medical Institute.
Footnotes
[Supplemental material is available for this article.]
Article published online before print. Article, supplemental material, and publication date are at http://www.genome.org/cgi/doi/10.1101/gr.156141.113.
References
- Abecasis GR, Cardon LR, Cookson WO 2000. A general test of association for quantitative traits in nuclear families. Am J Hum Genet 66: 279–292 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bannister AJ, Zegerman P, Partridge JF, Miska EA, Thomas JO, Allshire RC, Kouzarides T 2001. Selective recognition of methylated lysine 9 on histone H3 by the HP1 chromo domain. Nature 410: 120–124 [DOI] [PubMed] [Google Scholar]
- Bernstein HS, Coughlin SR 1998. A mammalian homolog of fission yeast Cdc5 regulates G2 progression and mitotic entry. J Biol Chem 273: 4666–4671 [DOI] [PubMed] [Google Scholar]
- Black JC, Manning AL, Van Rechem C, Kim J, Ladd B, Cho J, Pineda CM, Murphy N, Daniels DL, Montagna C, et al. 2013. KDM4A lysine demethylases induces site-specific copy gain and rereplication of regions amplified in tumors. Cell 154: 541–555 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheung VG, Conlin LK, Weber TM, Arcaro M, Jen KY, Morley M, Spielman RS 2003. Natural variation in human gene expression assessed in lymphoblastoid cells. Nat Genet 33: 422–425 [DOI] [PubMed] [Google Scholar]
- Cheung VG, Spielman RS, Ewens KG, Weber TM, Morley M, Burdick JT 2005. Mapping determinants of human gene expression by regional and genome-wide association. Nature 437: 1365–1369 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheung VG, Nayak RR, Wang IX, Elwyn S, Cousins SM, Morley M, Spielman RS 2010. Polymorphic cis- and trans-regulation of human gene expression. PLoS Biol 8: e1000480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christians JK, Senger LK 2007. Fine mapping dissects pleiotrophic growth quantitative trait locus in linked loci. Mamm Genome 18: 240–245 [DOI] [PubMed] [Google Scholar]
- Cloos PA, Christensen J, Agger K, Maiolica A, Rappsilber J, Antal T, Hansen KH, Helin K 2006. The putative oncogene GASC1 demethylates tri- and dimethylated lysine 9 on histone H3. Nature 442: 307–311 [DOI] [PubMed] [Google Scholar]
- Cowling VH 2010. Enhanced mRNA cap methylation increases cyclin D1 expression and promotes cell transformation. Oncogene 29: 930–936 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Creyghton MP, Cheng AW, Welstead GG, Kooistra T, Carey BW, Steine EJ, Hanna J, Lodato MA, Frampton GM, Sharp PA, et al. 2010. Histone H3K27ac separates active from poised enhancers and predicts developmental state. Proc Natl Acad Sci 107: 21931–21936 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dennis G Jr, Sherman BT, Hosack DA, Yang J, Gao W, Lane HC, Lempicki RA 2003. DAVID: Database for Annotation, Visualization, and Integrated Discovery. Genome Biol 4: 3. [PubMed] [Google Scholar]
- Emilsson V, Thorleifsson G, Zhang B, Leonardson AS, Zink F, Zhu J, Carlson S, Helgason A, Walters GB, Gunnarsdottir S, et al. 2008. Genetics of gene expression and its effect on disease. Nature 452: 423–428 [DOI] [PubMed] [Google Scholar]
- The ENCODE Project Consortium 2007. Identification and analysis of functional elements in 1% of the human genome by the ENCODE pilot project. Nature 447: 799–816 [DOI] [PMC free article] [PubMed] [Google Scholar]
- The ENCODE Project Consortium 2012. An integrated encyclopedia of DNA elements in the human genome. Nature 489: 57–74 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ernst J, Kheradpour P, Mikkelsen TS, Shoresh N, Ward LD, Epstein CB, Zhang X, Wang L, Issner R, Coyne M, et al. 2011. Mapping and analysis of chromatin state dynamics in nine human cell types. Nature 473: 43–49 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gibson G, Weir B 2005. The quantitative genetics of transcription. Trends Genet 21: 616–623 [DOI] [PubMed] [Google Scholar]
- Greer EL, Shi Y 2012. Histone methylation: A dynamic mark in health, disease and inheritance. Nat Rev Genet 13: 343–357 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grundberg E, Small KS, Hedman AK, Nica AC, Buil A, Keildson S, Bell JT, Yang TP, Meduri E, Barrett A, et al. 2012. Mapping cis- and trans-regulatory effects across multiple tissues in twins. Nat Genet 44: 1084–1089 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hasstedt SJ, Highland HM, Elbein SC, Harris CL, Das SK, the American Diabetes Association GENNID Study Group 2013. Five linkage regions harbor multiple type 2 diabetes genes in the African American subset of the GENNID Study. J Hum Genet 58: 378–383 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horne MC, Donaldson KL, Goolsby GL, Tran D, Mulheisen M, Hell JW, Wahl AF 1997. Cyclin G2 is up-regulated during growth inhibition and B cell antigen receptor-mediated cell cycle arrest. J Biol Chem 272: 12650–12661 [DOI] [PubMed] [Google Scholar]
- Ishimura A, Terashima M, Kimura H, Akagi K, Suzuki Y, Sugano S, Suzuki T 2009. Jmjd2c histone demethylase enhances the expression of Mdm2 oncogene. Biochem Biophys Res Commun 389: 366–371 [DOI] [PubMed] [Google Scholar]
- Kooistra SM, Helin K 2012. Molecular mechanisms and potential functions of histone demethylases. Nat Rev Mol Cell Biol 13: 297–311 [DOI] [PubMed] [Google Scholar]
- Lachner M, O'Carroll D, Rea S, Mechtler K, Jenuwein T 2001. Methylation of histone H3 lysine 9 creates a binding site for HP1 proteins. Nature 410: 116–120 [DOI] [PubMed] [Google Scholar]
- Lee MG, Wynder C, Cooch N, Shiekhattar R 2005. An essential role for CoREST in nucleosomal histone 3 lysine 4 demethylation. Nature 437: 432–435 [DOI] [PubMed] [Google Scholar]
- Li J, Burmeister M 2005. Genetical genomics: Combining genetics with gene expression analysis. Hum Mol Genet 14 (Suppl 2): R163–R1639 [DOI] [PubMed] [Google Scholar]
- Loh YH, Zhang W, Chen X, George J, Ng HH 2007. Jmjd1a and Jmjd2c histone H3 Lys 9 demethylases regulate self-renewal in embryonic stem cells. Genes Dev 21: 2545–2557 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu C, Ward PS, Kapoor GS, Rohle D, Turcan S, Abdel-Wahab O, Edwards CR, Khanin R, Figueroa ME, Melnick A, et al. 2012. IDH mutation impairs histone demethylation and results in a block to cell differentiation. Nature 483: 474–478 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moffatt MF, Kabesch M, Liang L, Dixon AL, Strachan D, Heath S, Depner M, Von Berg A, Bufe A, Rietschel E, et al. 2007. Genetic variants regulating ORMDL3 expression contribute to the risk of childhood asthma. Nature 448: 470–473 [DOI] [PubMed] [Google Scholar]
- Monks SA, Leonardson A, Zhu H, Cundiff P, Pietrusiak P, Edwards S, Phillips JW, Sachs A, Schadt EE 2004. Genetic inheritance of gene expression in human cell lines. Am J Hum Genet 75: 1094–1105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morley M, Molony CM, Weber TM, Devlin JL, Ewens KG, Spielman RS, Cheung VG 2004. Genetic analysis of genome-wide variation in human gene expression. Nature 430: 743–747 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pastinen T, Hudson TJ 2004. Cis-acting regulatory variation in the human genome. Science 306: 647–650 [DOI] [PubMed] [Google Scholar]
- Rea S, Eisenhaber F, O'Carroll D, Strahl BD, Sun ZW, Schmid M, Opravil S, Mechtler K, Ponting CP, Allis CD, et al. 2000. Regulation of chromatin structure by site-specific histone H3 methyltransferases. Nature 406: 593–599 [DOI] [PubMed] [Google Scholar]
- Robinson JT, Thorvaldsdottir H, Winckler W, Guttman M, Lander ES, Getz G, Mesirov JP 2011. Integrative genomics viewer. Nat Biotechnol 29: 24–26 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rui L, Emre NC, Kruhlak MJ, Chung HJ, Steidl C, Slack G, Wright GW, Lenz G, Ngo VN, Shaffer AL, et al. 2010. Cooperative epigenetic modulation by cancer amplicon genes. Cancer Cell 18: 590–605 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schadt EE, Molony C, Chudin E, Hao K, Yang X, Lum PY, Kasarskis A, Zhang B, Wang S, Suver C, et al. 2008. Mapping the genetic architecture of gene expression in human liver. PLoS Biol 6: e107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi Y, Lan F, Matson C, Mulligan P, Whetstine JR, Cole PA, Casero RA, Shi Y 2004. Histone demethylation mediated by the nuclear amine oxidase homolog LSD1. Cell 119: 941–953 [DOI] [PubMed] [Google Scholar]
- Shin G, Kang TW, Yang S, Baek SJ, Jeong YS, Kim SY 2011. GENT: Gene expression database of normal and tumor tissues. Cancer Inform 10: 149–157 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stranger BE, Dermitzakis ET 2005. The genetics of regulatory variation in the human genome. Hum Genomics 2: 126–131 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stranger BE, Forrest MS, Clark AG, Minichiello MJ, Deutsch S, Lyle R, Hunt S, Kahl B, Antonarakis SE, Tavare S, et al. 2005. Genome-wide associations of gene expression variation in humans. PLoS Genet 1: e78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tamm I, Wang Y, Sausville E, Scudiero DA, Vigna N, Oltersdorf T, Reed JC 1998. IAP-family protein survivin inhibits caspase activity and apoptosis induced by Fas (CD95), Bax, caspases, and anticancer drugs. Cancer Res 58: 5315–5320 [PubMed] [Google Scholar]
- Thorvaldsdottir H, Robinson JT, Mesirov JP 2012. Integrative Genomics Viewer (IGV): High-performance genomics data visualization and exploration. Brief Bioinform 14: 178–192 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Visel A, Blow MJ, Li Z, Zhang T, Akiyama JA, Holt A, Plajzer-Frick I, Shoukry M, Wright C, Chen F, et al. 2009. ChIP-seq accurately predicts tissue-specific activity of enhancers. Nature 457: 854–858 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Tang X, Li M, Marshall J, Mao Z 2005. Regulation of neuroprotective activity of myocyte-enhancer factor 2 by cAMP-protein kinase A signaling pathway in neuronal survival. J Biol Chem 280: 16705–16713 [DOI] [PubMed] [Google Scholar]
- Whetstine JR, Nottke A, Lan F, Huarte M, Smolikov S, Chen Z, Spooner E, Li E, Zhang G, Colaiacovo M, et al. 2006. Reversal of histone lysine trimethylation by the JMJD2 family of histone demethylases. Cell 125: 467–481 [DOI] [PubMed] [Google Scholar]
- Wissmann M, Yin N, Muller JM, Greschik H, Fodor BD, Jenuwein T, Vogler C, Schneider R, Gunther T, Buettner R, et al. 2007. Cooperative demethylation by JMJD2C and LSD1 promotes androgen receptor-dependent gene expression. Nat Cell Biol 9: 347–353 [DOI] [PubMed] [Google Scholar]
- Yamamoto S, Tateishi K, Kudo Y, Yamamoto K, Isagawa T, Nagae G, Nakatsuka T, Asaoka Y, Ijichi H, Hirata Y, et al. 2013. Histone demethylase KDM4C regulates sphere formation by mediating the cross talk between Wnt and Notch pathways in colonic cancer cells. Carcinogenesis 34: 2380–2388 [DOI] [PubMed] [Google Scholar]
- Zhang J, Baran J, Cros A, Guberman JM, Haider S, Hsu J, Liang Y, Rivkin E, Wang J, Whitty B, et al. 2011. International Cancer Genome Consortium Data Portal: A one-stop shop for cancer genomics data. Database 2011: bar026. [DOI] [PMC free article] [PubMed] [Google Scholar]