

Abstract
Phacomatosis Pigmentovascularis is a rare syndrome characterized by capillary malformation and pigmentary nevus. A case of a 2-year-old patient is reported, who presented extensive nevus flammeus and an aberrant Mongolian spot, without systemic disease, manifestations that allow us to classify this case as type IIa Phacomatosis Pigmentovascularis, according to Hasegawa's classification.
Keywords: Hemangioma; Nevus of Ota; Nevus, pigmented
Abstract
A Facomatose Pigmentovascular, síndrome rara, é caracterizada pela presença concomitante de malformação capilar e nevos pigmentares. Relata-se o caso de um paciente de dois anos de idade com malformação capilar extensa e mancha mongólica aberrante sem comprometimento sistêmico, manifestações que o incluem no tipo IIa na classificação da Facomatose Pigmentovascular, segundo Hasegawa.
INTRODUCTION
Phacomatosis Pigmentovascularis (PPV) represents a rare cutaneous congenital malformation syndrome, characterized mainly by the presence of capillary malformation and pigmentary nevi.1 Around 245 cases have been published since it was described in 1947. The present case describes a patient with type IIa Phacomatosis Pigmentovascularis or cesioflammea.
CASE REPORT
We evaluated a male patient, 2 years and 5 months old, white, born full-term through vaginal delivery, weighing 3330g. Apgar score was 9 at first and fifth minutes. He presented since birth erythema-to-violaceous spots on the face, anterior trunk and upper and lower limbs that did not stop at midline (Figures 1 and 2). Moreover, aberrant Mongolian spots on the back, abdomen, buttocks and lower limbs (Figures 3 and 4) were noted. The lesions were asymptomatic and did not change over time.
FIGURE 1.
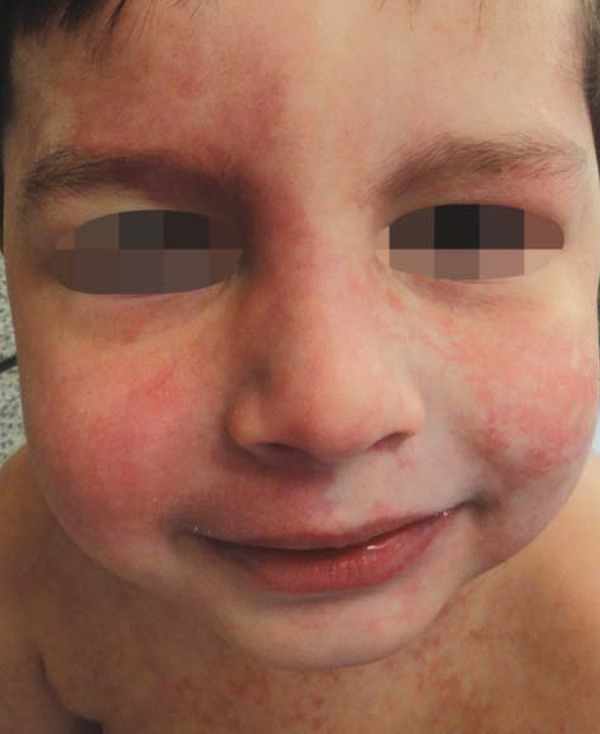
Bilateral erythemato-violaceous spots on the face
FIGURE 2.
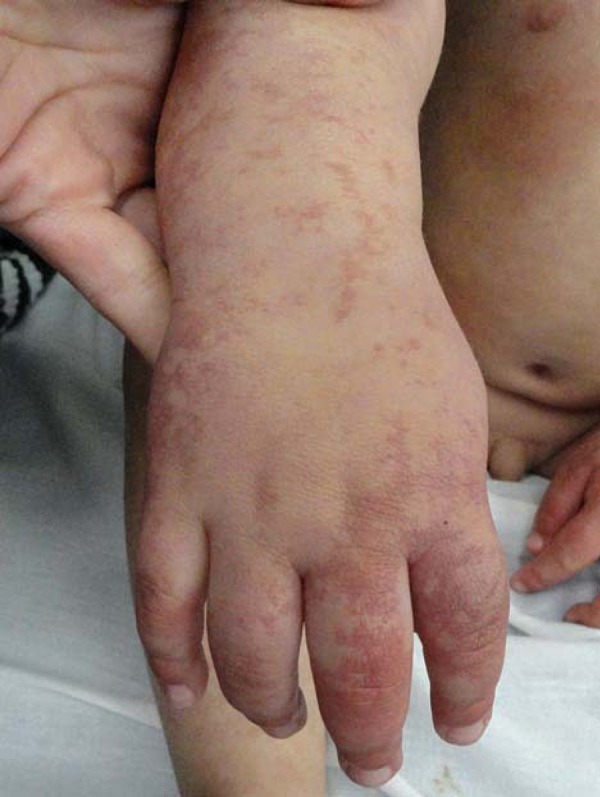
Erythemato-violaceous spots on hand and right forearm
FIGURE 3.
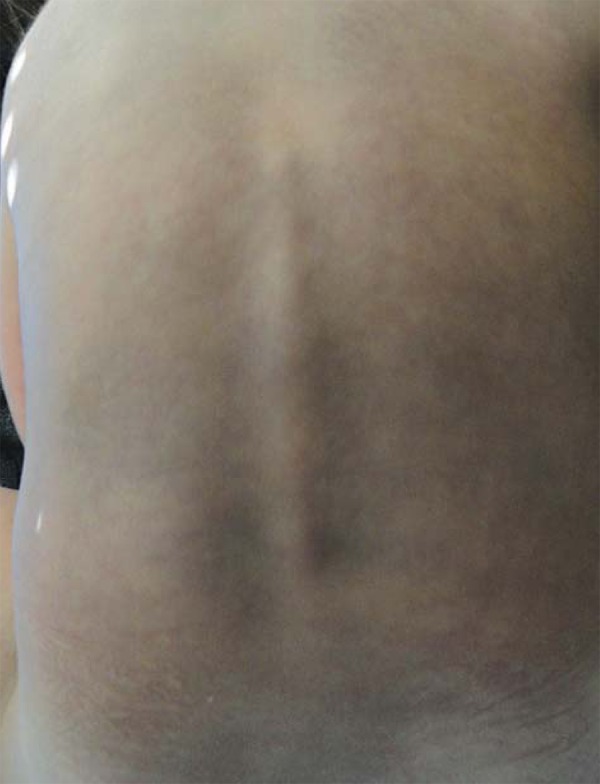
Aberrant Mongolian spot on the dorsum
FIGURE 4.

Mongolian spot on buttocks
The child was not suffering from chronic diseases and there was no history of parental consanguinity or similar cases in the family. He was not taking any medication. During the exam no alterations in the neuropsychomotor development were observed and the extremities were symmetrical, without atrophy or hypertrophy signs in the soft tissues.
The opthalmic exam revealed Nevus of Ota on the left, without criteria for diagnosis of glaucoma or other anomalies (Figure 5). Laboratory exams were normal. Furthermore, bilateral head and orbital magnetic resonance imaging, total abdominal ultrasonography, electroencephalogram during sleep and vigil and echocardiogram did not show alterations.
FIGURE 5.
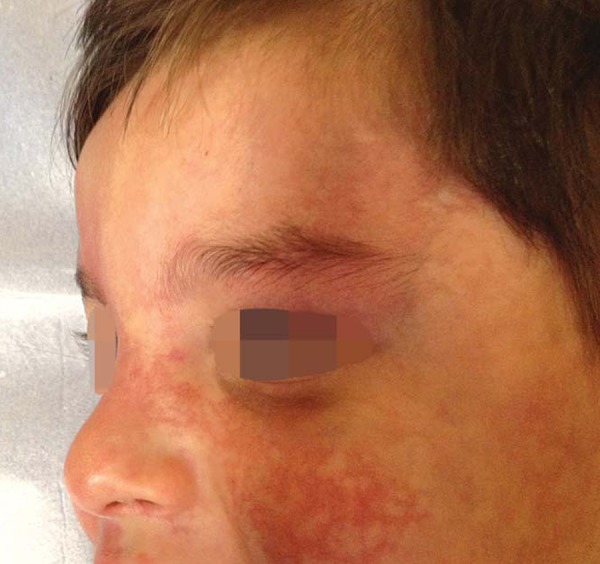
Nevus of Ota
DISCUSSION
Phacomatosis Pigmentovascularis was first described by Ota, in 1947, as an association of dermal melanocytosis with congenital vascular nevi, mainly capillary malformation.2
It was classified by Hasegawa and Yasuhara into four types according to the pigmentary lesion associated with nevus flammeus and in 2003 a fifth type was described, characterized by cutis marmorata telangiectatica and dermal melanocytosis.3,4 Each type was further subdivided according to the presence of cutaneous-only manifestations (subtype a) or if associated to systemic manifestations (subtype b), the majority of which are ocular, vascular, skeletal and neurological.5
In 2005, Happle proposed a new simplified classification into four groups, utilizing descriptive terms: Phacomatosis Cesioflamea (association of the nevus flameus and aberrant Mongolian spots or blue nevus); Phacomatosis Spilorosea (coexistence of Nevus Spilus and telangiectatic nevus, which for Happle is different than nevus flameus); Phacomatosis Cesiomarmorata (association of aberrant Mongolian spots or blue nevus with Cutis Marmorata Telangiectatica Congenita) and Phacomatosis Pigmentovascularis of non-classifiable type.6 On this last classification, there is no subdivision between presentation with or without systemic symptoms. Chart 1 synthesizes the two classifications.
CHART 1.
Information about PPV classifications
| Type | Type | Vascular lesion | Pigmentary lesion |
| (according to Happle) | (according to Hasegawa*) | ||
| (inexistent**) | I | Nevus flammeus | Verrucous and pigmented nevus |
| Cesioflammea | II | Nevus flammeus ± anemic nevus | Mongolian spot |
| Spilorosea | III | Nevus flammeus ± anemic nevus | Nevus Spilus |
| Non-classifiable | IV | Nevus flammeus ± anemic nevus | Mongolian spot, Nevus Spilus |
| Cesiomarmorata | V | Cutis Marmorata Telangiectatica Congenita | Mongolian spot |
Until now, about 245 cases have been reported, the majority on Oriental patients. Type II is the most frequently found, related or not to systemic symptoms. The most common association with extra cutaneous presentations is with the Sturge-Weber syndrome and with the Klippel-Trénauney syndrome, individually or combined.7
In 2008, the Fernández-Guarino revision showed that 77% of the 222 cases of PPV described until then were of type II or cesioflammea, of which around 60% presented systemic involvement. Series of 15 PPV cases presented similar percentages to previous series regarding the distribution of presentations.5
The pathogenesis of PPV is not fully known yet, but it is believed to be an abnormality in the development of melanocytic nevus cells and vasomotor neural cells derived from the neural crest.3 The best accepted genetic model to explain PPV is didymosis (twin spots), a well known phenomenon in plants and animals. Didymosis is characterized by two areas of adjacent cutaneous lesions, formed by mutant tissues which also differ from the normal surrounding tissue.
Dermal melanocytoses occur when fetal melanocytes present a migration failure, from the neural crest to the dermal basal layer. There are five types, the Mongolian spot being the most frequent, followed by blue nevus and Nevus of Ota. In Brazil, the Mongolian spot has an estimated prevalence of 65% in blacks and 52% in mixed ethnicities. The pattern of inheritance is autosomal dominant and it is located on the lumbar and gluteal areas, disappearing with age. It is considered aberrant when it is on an ectopic site (dorsum, shoulders, extremities and face). In this case, it is generally extensive, may involve over 50% of body surface and does not disappear with age. The Nevus of Ota and the Nevus of Ito can be considered aberrant Mongolian spots8
The vascular component is usually extensive in PPV patients and, when compared with other capillary malformations, there are histopathological differences, like the presence of vasomotor neural cells and prominent endothelial cells.8
The case reported presents manifestations that classify it as type IIa, that is, extensive bilateral lesions of capillary malformation and aberrant Mongolian spots. To this day, the patient has not presented systemic involvement. However, the importance of follow-up should be emphasized, since systemic alterations can become evident with time, changing the classification and prognosis.
PPV without systemic involvement has a benign course e does not require treatment. Nonetheless, due to the aesthetic impact, some procedures have been considered in order to enhance the quality of life of PPV patients, using pulsed laser light for treatment of nevus flammeus and Q-switched laser for treatment of pigmented nevi. The exact moment for the intervention has also been discussed. Today it is suggested that the combined laser treatment should be done in childhood, preferably before school age, to diminish the extension of lesions to be treated and their effect on the self-esteem of patients.9
Footnotes
* Work performed at Sanitary Dermatology Outpatient Clinic (ADS)- Secretariat of Health - Porto Alegre (RS), Brazil.
Conflict of interest: None
Financial funding: None
REFERENCES
- 1.Chang BP, Hsu CH, Chen HC, Hsieh JW. An infant with extensive Mongolian spot, naevus flammeus and cutis marmorata telangiectatica congenita: a unique case of phakomatosis pigmentovascularis. Br J Dermatol. 2007;156:1068–1071. doi: 10.1111/j.1365-2133.2007.07798.x. [DOI] [PubMed] [Google Scholar]
- 2.Ota N, Kawarnura T, Ito N. Phakomatosis pigmentovascularis. Jpn J Dermatol B. 1947;57:1–3. [Google Scholar]
- 3.Hasegawa Y, Yasuhara M. Phakomatosis pigmentovascularis type VIa. Arch Dermatol. 1985;121:651–655. [PubMed] [Google Scholar]
- 4.Torrelo A, Zambrano A, Happle R. Cutis marmorata telangiectasica congenita and extensive Mongolian spots: type V phakomatosis pigmentovascularis. Br J Dermatol. 2003;148:342–345. doi: 10.1046/j.1365-2133.2003.05118.x. [DOI] [PubMed] [Google Scholar]
- 5.Fernández-Guarino M, Boixeda P, de Las Heras E, Aboin S, García-Millán C, Olasolo PJ. Phakomatosis pigmentovascularis: Clinical findings in 15 patients and review of the literature. J Am Acad Dermatol. 2008;58:88–93. doi: 10.1016/j.jaad.2007.08.012. [DOI] [PubMed] [Google Scholar]
- 6.Happle R. Phakomatosis pigmentovascularis revisited and reclassified. Arch Dermatol. 2005;141:385–388. doi: 10.1001/archderm.141.3.385. [DOI] [PubMed] [Google Scholar]
- 7.Finklea LB, Mohr MR, Warthan MM, Darrow DH, Williams JV. Two reports of phacomatosis pigmentovascularis type IIb, one in association with Sturge-Weber Syndrome and Klippel-Trenaunay Syndrome. Pediatr Dermatol. 2010;27:303–305. doi: 10.1111/j.1525-1470.2010.01144.x. [DOI] [PubMed] [Google Scholar]
- 8.Ribas J, Ribas CBR, Schettini APM. Facomatose Pigmentovascular: relato de caso. An Bras Dermatol. 1997;72:163–166. [Google Scholar]
- 9.Kono T, Erçöçen AR, Chan HH, Kikuchi Y, Hori K, Uezono S, et al. Treatment of Phacomatosis Pigmentovascularis: A Combined Multiple Laser Approach. Dermatol Surg. 2003;29:642–646. doi: 10.1046/j.1524-4725.2003.29154.x. [DOI] [PubMed] [Google Scholar]