

Abstract
A functional high throughput screen and subsequent multi-dimensional, iterative parallel synthesis effort identified the first muscarinic acetylcholine receptor (mAChR) negative allosteric modulator (NAM) selective for the M5 subtype. ML375 is a highly selective M5 NAM with sub-micromolar potency (human M5 IC50 = 300 nM, rat M5 IC50 = 790 nM, M1–4 IC50 >30 μM), excellent multi-species PK, high CNS penetration, and enantiospecific inhibition.
Keywords: Muscarinic acetylcholine receptor, M5, negative allosteric modulator (NAM), ML375, MLPCN probe
INTRODUCTION
The five G protein-coupled muscarinic acetylcholine receptors (mAChRs or M1-M5) utilize acetylcholine as their endogenous agonist and are broadly distributed throughout the periphery and central nervous system (CNS) where they regulate a diverse array of physiological processes. 1–4 M1 and M4 are predominantly expressed within the CNS and have the highest expression levels, whereas M2 and M3 are expressed in both the periphery and moderately within the CNS, while M5 expression is low (<2% of the total CNS mAChR population).2,5 Localization studies have found low levels of M5 expression in multiple brain regions, but M5 mRNA is the only mAChR transcript identified in dopaminergic neurons of the substantia nigra pars compacta (SNc) and ventral tegmental area (VTA).4–7 Here, M5 is co-expressed with D2 dopamine mRNA, which has led to the hypothesis that M5 might modulate dopaminergic neurotransmission and function in addiction/reward mechanisms.6 Subsequent studies in M5 −/− mice confirmed this hypothesis, with M5 −/− mice showing reduced morphine and cocaine-conditioned place preference and self-administration, with no effect on food intake, suggesting preferential abuse-related effects. 8–10 Thus, much of our current understanding of the function of M5 has come from M5 receptor localization, M5 −/− mice and experiments conducted with non-selective, orthosteric muscarinic ligands, as no M5-selective antagonists or negative allosteric modulators (NAMs) have been reported.4 Recently, targeting allosteric sites on mAChRs has led to the discovery of highly selective positive allosteric modulators (PAMs) of M1, M4 and M5;11–16 however, highly selective NAMs for individual subtypes have not yet been identified for any of the five mAChRs - and only one M1-selective orthosteric antagonist chemotype has been reported.17 Therefore, to address this limitation in small molecule tools to study M5 function, we elected to pursue the discovery and development of selective M5 NAMs to enable the dissection of the physiological role and therapeutic potential of M5 inhibition.
RESULTS AND DISCUSSION
High-Throughput Screen
We performed a triple-add, functional high-throughput screen to identify M5 modulator leads.16,18 For this effort, we screened the MLPCN19 collection (360,000 compounds) in Chinese hamster ovary (CHO) cells stably expressing human M5 (hM5) and measuring intracellular calcium mobilization. This effort identified 3,920 M5 primary hits (1.07% hit rate). Counter-screening against the parental CHO cell line as well as CHO cells expressing human M1 and human M4 and reconfirmation of powders in 10-point concentration-response curves (CRCs) resulted in 9 confirmed, selective antagonists of hM5.20 At this point, it was not clear if these confirmed hits were selective M5 orthosteric antagonists or negative allosteric modulators (NAMs).
Chemistry
Of the confirmed hits (Figure 1), our attention focused on 1, a unique 2,3-dihydro-1H-imidazo[2,1-a] isoindol-5(9bH)-one-based scaffold, which was inactive on M1 and M4, but displayed weak inhibition of M5 (IC50 >10 μM, 41% ACh Max). Upon a simple two-step resynthesis involving condensation of ethylene diamine and 2-benzoylbenzoic acid 2 to provide 3 and subsequent acylation (Scheme 1), we were pleased to observe enhanced activity of fresh powder of 1 at M5 (hM5 IC50 = 3.5 μM, rM5 IC50 = 5.7 μM) and no activity at the other four mAChRs (hM1-M4 IC50 > 30 μM).20 Figure 1 also highlights the chemical optimization strategy for 1, evaluating multiple dimensions simultaneously through iterative parallel synthesis, necessitated by the often shallow nature of allosteric SAR.11
Figure 1.
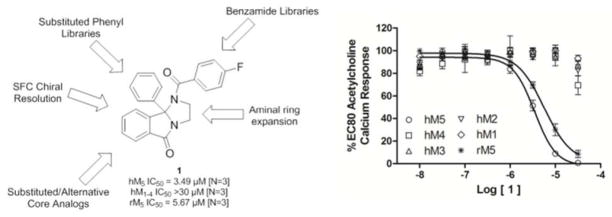
Structure, pharmacology (hM5 IC50 = 3.49 μM, rM5 IC50 = 5.67 μM, M1-M4 IC50>30 μM) and chemical optimization plan for 1.
Scheme 1.
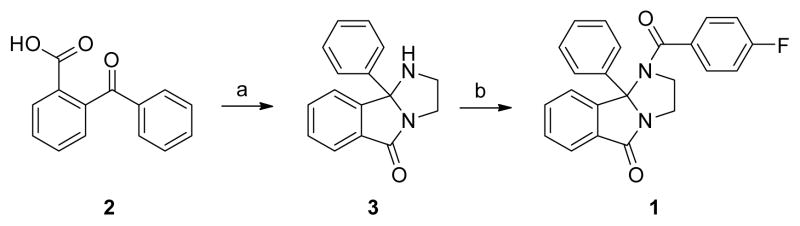
Synthesis of 1 and route for analog synthesisa
aReagents and conditions: (a) ethylene diamine, p-TSA, toluene, reflux, Dean-Stark trap, 80%; (b) 4-fluorobenzoyl chloride, DCM, DIPEA, 93%.
The first round of library synthesis evaluated alternate amides; thus, 3 was subsequently acylated with an array of 40 acid chlorides (aryl, heteroaryl and aliphatic) according to scheme 1 to provide analogs 4. The library was efficiently triaged in a 10 μM single point assay against an EC80 of ACh on human M5.15 SAR was shallow, with only seven analogs significantly reducing the EC80, and only benzamide congeners were active. As shown in Table 1, a 3,4-difluro analog, 4g, proved the most active (hM5 IC50 = 1.0 μM, rM5 IC50 = 2.1 μM), and it also maintained excellent mAChR selectivity (M1-M4 IC50s >30 μM).
Table 1.
Structures and activities of analogs 4.
| ||||
|---|---|---|---|---|
| Entry | R | hM5 pIC50* | hM5 IC50 (μM) | ACh Min* (%) |
| 4a | 4-OCF3 | 5.35±0.03 | 4.47 | 0.2±2.4 |
| 4b | 4-SCF3 | 5.86±0.02 | 1.38 | 0.5±1.5 |
| 4c | 4-CF3 | 5.71±0.03 | 1.95 | −2.4±1.4 |
| 4d | 3-CF3 | 5.52±0.06 | 3.02 | −0.7±5.2 |
| 4e | 3,5-diCl | 5.24±0.17 | 5.75 | 0.0±18.9 |
| 4f | 3,5-diF | 5.42±0.06 | 3.80 | −2.2±5.6 |
| 4g | 3,4-diF | 5.98±0.02 | 1.05 | 0.0±1.3 |
hM5 pIC50 and ACh Min data reported as averages±SEM from our calcium mobilization assay; n = 3
Other library efforts led exclusively to inactive analogs. 20 Expansion of the aminal ring to a six-membered congener was inactive, conversion of the isoindolinone core to an azaindolinone proved inactive, as did urea, sulfonamide and tertiary amine derivatives as replacements for the amide linkage. These data suggested that both the 2,3-dihydro-1H-imidazo[2,1-a] isoindol-5(9bH)-one core and the benzamide moiety were critical for M5 activity.
Our attention then focused on introducing substituents into the 9b phenyl ring of 1, and assessing the impact on M5 activity. Once again, analogs 5–7 were prepared following Scheme 1, but employing functionalized congeners of 2 in a matrix library (3 × 10). As Shown in Table 2, SAR was shallow and unpredictable, with the data suggesting a cooperative relationship between benzamide and 9b phenyl substituents. However, one analog, 5g, possessing the 3,4-difluorobenzamide and a 9b 4-chlorophenyl moiety, afforded sub-micromolar potency at M5 (hM5 IC50 = 0.48 μM, rM5 IC50 = 1.1 μM) and no activity at the other four mAChRs (hM1-M4 IC50 > 30 μM). This was an exciting result, as this was the racemic form of the analog with potential for enantioselective inhibition of M5.20
Table 2.
Structures and activities of analogs 5–7.
| ||||||
|---|---|---|---|---|---|---|
| Entry | R1 | R2 | hM5 pIC50* | hM5 IC50 (μM) | ACh Min* (%) | |
| 5a | Cl |
|
4-OCF3 | 5.63±0.06 | 2.3 | −5.3±4.4 |
| 5b | 4-SCF3 | 5.52±0.13 | 3.0 | −9.2±8.7 | ||
| 5c | 4-CF3 | 5.69±0.04 | 2.0 | 0.7±2.3 | ||
| 5d | 3-CF3 | 5.31±0.11 | 4.9 | −7.2±8.0 | ||
| 5e | 4-Me | 5.58±0.04 | 2.6 | 0.1±2.8 | ||
| 5f | 3,5-diF | 6.00±0.04 | 1.0 | 2.8±2.0 | ||
| 5g | 3,4-diF | 6.32±0.02 | 4.8 | 0.1±1.0 | ||
| 6a | F |
|
4-OCF3 | --- | >10 | --- |
| 6b | 4-SCF3 | 5.89±0.03 | 1.3 | −0.2±1.7 | ||
| 6c | 4-CF3 | --- | >10 | --- | ||
| 6d | 3-CF3 | 5.68±0.03 | 2.1 | −2.4±1.8 | ||
| 6e | 4-Me | --- | >10 | --- | ||
| 6f | 3,5-diF | --- | >10 | --- | ||
| 6g | 3,4-diF | --- | >10 | --- | ||
| 7a | Me |
|
4-OCF3 | 5.46±0.06 | 3.5 | −0.2±4.3 |
| 7b | 4-SCF3 | 5.39±0.08 | 4.1 | 4.1±5.5 | ||
| 7c | 4-CF3 | --- | >10 | --- | ||
| 7d | 3-CF3 | --- | >10 | --- | ||
| 7e | 4-Me | --- | >10 | --- | ||
| 7f | 3,5-diF | 5.63±0.05 | 2.3 | −1.1±3.3 | ||
| 7g | 3,4-diF | 5.58±0.06 | 2.6 | −6.8±4.3 | ||
hM5 pIC50 and ACh mMin data reported as averages±SEM from our calcium mobilization assay; n = 3; ---, not determined
We were able to quickly develop SFC conditions to separate the pure enantiomers of 5g to provide 8 and 9, both in >99% ee.20 Optical rotations were recorded, and the (−)-enantiomer 8 proved to be active (hM5 IC50 = 300 nM), where the (+)-enantiomer 9 was devoid of M5 activity (hM5 IC50 >30 μM) as shown in Figure 2. However, the absolute stereochemistry was unknown. Ultimately, single X-ray crystallography indicated that the active (−)-enantiomer 8 possessed the (S)-stereochemistry. Thus, this core showed enantiospecific activity for the inhibition of M5.
Figure 2.

Structures and activities of the (S)-and (R)-enantiomers, 8 (hM5 IC50 = 300 nM) and 9 (hM5 IC50 >30 μM) respectively of 5g (hM5 IC50 = 480 nM). A) M5 CRC for racemic 5g, 8 and 9. B) X-ray crystal structure of 8 (CCDC 953105); C) structures of 5g, 8 and 9.
Molecular Pharmacology
The SAR was driven using a human M5 functional assay, and, since we desired an in vivo tool compound, we also evaluated 8 against rat M5. There was a slight species difference, with 8 displaying a >2-fold loss in activity at the rat M5 receptor (rM5 IC50 = 790 nM). However, 8 was inactive at human M1-M4 (Figure 3A) as well as rat M1-M4 (Figure 3B), representing the first M5 selective small molecule inhibitor. To determine the mechanism of action of 8, whether orthosteric or allosteric, we first performed competition binding experiments with the orthosteric mAChR antagonist [3H]-NMS and compared this to atropine. Compound 8 displayed no competition [3H]-NMS for binding to hM5, suggesting an allosteric mode of receptor inhibition (Figure 3C).13–15,20 To further investigate an allosteric mechanism, we also performed [3H]-NMS dissociation kinetic experiments (Figure 3D), with hM5 cell membranes, which revealed that 8 decreased the dissociation rate of [3H-NMS], further confirming an allosteric effect of 8 on the orthosteric site.13–15,19 Thus, 8 is the first M5-selective negative allosteric modulator (NAM).
Figure 3.

Molecular pharmacology profile of 8. A) Human mAChR selectivity. 8 is selective for hM5 (hM5 IC50 = 300 nM, hM1-M4 IC5os >30 μM); B) rat mAChR selectivity. 8 is selective for rM5 (rM5 IC50 = 790 nM rM1-M4 IC5os >30 μM); C) [3H]-NMS competition binding [N=3] in membranes prepared from hM5 cells. This stongly suggests that 8 does not bind at the M5 orthosteric site; D) [3H]-NMS dissociative kinetics [N=3}. Atropine (alone) t1/2 = 49.9 min, atropine (+8) t1/2 = 68.8 min. 8 exerts an allosteric effect on the orthosteric site.
Metabolism and Disposition
Evaluation of the in vitro and in vivo DMPK profile20,21 of 8 (Table 3) revealed the compound to possess high metabolic stability with low hepatic microsomal intrinsic clearance (CLint;; human 2.6 mL/min/kg, cynomolgus monkey (cyno), 20 mL/min/kg, rat, 24 mL/min/kg) and a corresponding low predicted hepatic clearance in multiple species (CLhep; human, 2.3 mL/min/kg, cyno, 14 mL/min/kg rat, 18 mL/min/kg).
Table 3.
DMPK profile of 8.
| Parameter/Species | Rat (male, Sprague-Dawley) | NHP (male, cynomolgus) | Human |
|---|---|---|---|
| Hepatic microsome CLint(mL/min/kg) | 24 | 20 | 2.6 |
| Predicted CLhep* | 18 | 14 | 2.3 |
| fu plasma, fu brain | 0.029, 0.003 | 0.001, - | 0.013, - |
| CYP inhibition (P450, IC50) | - | - | 3A4, 2D6, 1A2: >15; 2C9: 7.4 |
| CLp (mL/min/kg), Elimination t1/2 (hr) | 2.5, 80 | 3.0,10 | - |
| Vdss(IV) | 16 L/kg | 1.9 L/kg | - |
| %F (PO) | 80 | - | - |
| Brain-plasma Kp, Kp,uu (1.0hr, PO) | 1.8, 0.2 | - | - |
determined using CLint values in well-stirred model of organ clearance uncorrected for fraction unbound in plasma
Correspondingly, 8 exhibited low clearance (CLp, 2.5 mL/min/kg) and a long elimination half-life (t1/2, 80hr) in rodents (male, Sprague-Dawley rat, 1 mg/kg IV, n = 2) and nonhuman primates (male, cynomolgus monkey, 1 mg/kg, CLp, 3.0 mL/min/kg, t1/2, 10 hr, n = 3). Consistent with a low clearance compound, 8 also demonstrated high oral bioavailability (%F, 80) following administration of a suspension-dose to male SD rats (n=2) with a maximal plasma concentration (Cmax) of 1.4 μM and a corresponding time to reach Cmax (Tmax) of 7 hours. The distribution of compound 8 was characterized by a low fraction unbound in plasma (fu,p; human: 0.013, cyno: 0.001, rat: 0.029) and a high nonspecific binding in brain homogenate (fu,br; rat: 0.003). Following an oral CNS distribution study in rat (male, Sprague-Dawley, n = 2; 10 mg/kg) we observed total and unbound brain-plasma partition coefficients of 1.8 and 0.2 (Kp, Kp,uu, respectively) one hour post-administration.
Compound 8 displayed an acceptable human cytochrome P450 inhibition profile producing acceptable IC50 values for 3A4 (16 μM), 1A2 (25 μM, 2C9 (7.4 μM) and 2D6 (26 μM). Moreover, in a Eurofins radioligand binding panel of 68 GPCRs, ion channels and transporter,20 compound 8 displayed significant binding (>50% inhibition @10 μM) at only 1 target (CB1, 66%), but no functional activity at this target in a subsequent assay.
Conclusion
In summary, we have developed 8 (also referred to as ML375 or VU0483253), the first mAChR NAM that selectively targets M5 (hM5 IC50 = 300 nM, hM1-M4 IC50 >30 μM), with a favorable DMPK profile and CNS penetration. Enantiospecific M5 activity was noted, with all activity residing in the (S)-enantiomer, 8. Due to the unexpected human-rodent species difference in regards to M5 potency and brain homogenate binding, 8 is not suitable for in vivo work in rodents, but may achieve sufficient exposure in non-human primate. Current efforts are focused on a new optimization program, driving the SAR on rat M5 to deliver an in vivo tool for rodent addiction studies, and progress will be reported in due course.
EXPERIMENTAL SECTION
Chemistry
The general chemistry, experimental information, and syntheses of all other compounds are supplied in the Supporting Information.
(S)-9b-(4-chlorophenyl)-1-(3,4-difluorobenzoyl)-2,3-dihydro-1H-imidazo[2,1-a]isoindol-5(9bH)-one (8)
To a mixture of 2-(4-chlorobenzoyl)benzoic acid (5.21 g, 20.0 mmol, 1 eq.) and ethylenediamine (2.67 mL, 40.0 mmol, 2 eq.) in toluene (30 mL, 0.67 M) was added p-toluenesulfonic acid monohydrate (~0.1 g, 3 mol%). A Dean-Stark trap was used to remove water while the mixture was allowed to stir at reflux for 4 hours. After cooling to ambient temperature, the reaction mixture was dissolved in dichloromethane. The organic layer was washed with a saturated aqueous solution of sodium bicarbonate and then with brine. Solvent was removed under reduced pressure and the crude product was recrystallized from ethanol to give 2.86 g of pure 9b-(4-chlorophenyl)-2,3-dihydro-1H-imidazo[2,1-a]isoindol-5(9bH)-one (50.2% yield). To a solution of 9b-(4-chlorophenyl)-2,3-dihydro-1H-imidazo[2,1-a]isoindol-5(9bH)-one (15 mg, 0.053 mmol, 1.0 eq.) and DIPEA (18 μL, 0.105 mmol, 2.0 eq.) in DCM (0.53 mL, 0.1 M) was added 3,4-difluorobenzoyl chloride (9.9 μL, 0.079 mmol, 1.5 eq.). The reaction was allowed to stir at ambient temperature for 2 hours. The reaction was quenched with methanol and the organics were concentrated on a heated air-drying block. Crude product was purified via Gilson preparative LC to obtain 12.0 mg of 5g (53.3 % yield). The second eluting pure enantiomer of 5g was separated via CO2 supercritical fluid chromatography (Lux cellulose-3 10 × 250 mm column at 40 °C, backpressure regulated at 100 bar, MeOH co-solvent, 10% isocratic prep over 7 minutes at 15 mL/min) and was determined to have an ee of >98% by chiral HPLC analysis (Lux cellulose-3 4.6 × 250 mm column at 40 °C, backpressure regulated at 100 bar, MeOH co-solvent, 5–50% over 7 minutes at 3.5 mL/min). 1H NMR (400.1 MHz, CDCl3) δ (ppm): 8.04-7.99 (m, 1H); 7.90-7.85 (m, 1H); 7.65-7.56 (m, 2H); 7.38-7.30 (m, 3H); 7.25-7.19 (m, 2H); 7.18-7.14 (m, 2H); 4.38-4.30 (m, 1H); 4.01-3.93 (m, 1H); 3.82-3.75 (m, 1H); 3.34-3.25 (m, 1H). 13C NMR (100.6 MHz, CDCl3) δ (ppm): 172.07, 166.84, 151.81 (dd, JC=F = 254 Hz, 12.7 Hz), 150.33 (dd, JC–F = 252 Hz, 13 Hz) 145.77, 136.65, 134.94, 133.55, 132.91 (t, J = 4.8 Hz), 131.88, 130.61, 129.06, 128.97, 127.53, 124.03, 123.62 (dd, J = 6.8 Hz, 4 Hz), 117.94 (d, J = 17 Hz), 116.83 (d, J = 18 Hz), 87.37, 52.24, 39.70. SFC (214 nM) RT = 3.591 min (>98%). HRMS (TOF, ES+) C23H16N2O2F2Cl [M+H]+ calc. mass 425.0868, found 425.0872. Specific rotation (c = 0.75, CHCl3).
Supplementary Material
Acknowledgments
Funding Sources
This work was generously supported by the NIH/MLPCN U54 MH084659 (C.W.L.) and U54 MH084512 (Scripps).
ABBREVIATIONS USED
- M5
muscarinic acetylcholine receptor subtype 5
- CRC
concentration-response-curve
- NAM
negative allosteric modulator
- MLPCN
Molecular Libraries Probe Production Centers Network
Footnotes
Supporting Information. Experimental procedures and spectroscopic data for selected compounds, detailed pharmacology and DMPK methods. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Smythies J. Section I. The cholinergic system. Int Rev Neurobiol. 2005;64:1–122. doi: 10.1016/S0074-7742(05)64001-9. [DOI] [PubMed] [Google Scholar]
- 2.Wess J, Eglen RM, Gautam D. Muscarinic acetylcholine receptors: mutant mice provide new insights for drug development. Nat Rev Drug Discov. 2007;6:721–733. doi: 10.1038/nrd2379. [DOI] [PubMed] [Google Scholar]
- 3.Langmead CJ, Watson J, Reavill C. Muscarinic acetylcholine receptors as drug targets. Pharmacol Ther. 2008;117:232–243. doi: 10.1016/j.pharmthera.2007.09.009. [DOI] [PubMed] [Google Scholar]
- 4.Denker D, Thomsen M, Wortwein G, Weikop G, Cui Y, Jeon J, Wess J, Fink-Jensen A. Muscarinic acetylcholine receptor subtypes as potential drug targets for the treatment of schizophrenia, drug abuse and Parkinson’s disease. ACS Chem Neurosci. 2012;3:80–89. doi: 10.1021/cn200110q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Caulfield MP, Birdsall NJM. Classification of muscarinic acetylcholine receptors. Pharmacol Rev. 1998;50:279–290. [PubMed] [Google Scholar]
- 6.Weiner DM, Levey AI, Brann MR. Expression of muscarinic acetylcholine and dopamine receptor messenger-RNAs in rat basal ganglia. Proc Natl Acad Sci US A. 1990;87:7050–7054. doi: 10.1073/pnas.87.18.7050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Yasuda RP, Ciesla W, Flores LR, Wall SJ, Li M, Satkus SA, Weisstein JS, Spagnola BV, Wolfe BB. Development of antisera selective for M4 and M5 muscarinic cholinergic receptors -distribution of M4 and M5 receptors in ratbrain. Mol Pharmacol. 1993;43:149–157. [PubMed] [Google Scholar]
- 8.Basile AS, Fedorova I, Zapata A, Liu X, Shippenberg T, Duttaroy A, Yamada M, Wess J. Deletion of the M5 muscarinic acetylcholine receptor attenuates morphine reinforcement and withdrawal but not morphine analgesia. Proc Natl Acad Sci US A. 2002;99:11452–11457. doi: 10.1073/pnas.162371899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Fink-Jensen A, Fedorova I, Wörtwein; G, Woldbye DP, Rasmussen T, Thomsen M, Bolwig TG, Knitowski KM, McKinzie DL, Yamada M, Wess J, Basile A. Role for M5 muscarinic acetylcholine receptors in cocaine addiction. J Neurosci Res. 2003;74:91–96. doi: 10.1002/jnr.10728. [DOI] [PubMed] [Google Scholar]
- 10.Thomsen M, Woldbye DP, Wortwein G, Fink-Jensen A, Wess J, Caine SB. Reduced cocaine self-administration in muscarinic M5 acetylcholine receptor-deficient mice. J Neurosci. 2005;25:8141–8149. doi: 10.1523/JNEUROSCI.2077-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Melancon BJ, Hopkins CR, Wood MR, Emmitte KA, Niswender CM, Christopoulos A, Conn PJ, Lindsley CW. Allosteric Modulation of 7 Transmembrane Spanning Receptors: Theory, Practice and Opportunities for CNS Drug Discovery. J Med Chem. 2012;55:1445–1464. doi: 10.1021/jm201139r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Conn PJ, Jones C, Lindsley CW. Subtype selective allosteric modulators of muscarinic receptors for the treatment of CNS disorders. Trends in Pharm Sci. 2009;30:148–156. doi: 10.1016/j.tips.2008.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Tarr JC, Turlington ML, Reid PR, Utely TJ, Sheffler DJ, Cho HP, Klar R, Pancani T, Klein MT, Bridges TM, Morrison RD, Xiang Z, Daniels SJ, Niswender CM, Conn PJ, Wood MR, Lindsley CW. Targeting selective activation of M1 for the treatment of Alzheimer’s Disease: further chemical optimization and pharmacological characterization of the M1 positive allosteric modulators ML169. ACS Chem Neurosci. 2012;3:884–895. doi: 10.1021/cn300068s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Brady A, Jones CK, Bridges TM, Kennedy PJ, Thompson AD, Breininger ML, Gentry PR, Yin H, Jadhav SB, Shirey J, Conn PJ, Lindsley CW. Centrally active allosteric potentiators of the M4 muscarinic acetylcholine receptor reverse amphetamine-induced hyperlocomotion behavior in rats. J Pharm & Exp Ther. 2008;327:941–953. doi: 10.1124/jpet.108.140350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bridges TM, Marlo JE, Niswender CM, Jones JK, Jadhav SB, Gentry PR, Weaver CD, Conn PJ, Lindsley CW. Discovery of the first highly M5-preferring muscarinic acetylcholine receptor ligand, an M5 positive allosteric modulator derived from a series of 5-trifluoromethoxy N-benzyl isatins. J Med Chem. 2009;52:3445–3448. doi: 10.1021/jm900286j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gentry PR, Bridges TM, Lamsal A, Vinson PN, Smith E, Chase P, Hodder PS, Engers JL, Niswender CM, Daniels JS, Conn PJ, Wood MR, Lindsley CW. Discovery of ML326: The first sub-micromolar, selective M5 PAM. Bioorg Med Chem Lett. 2013;23:2996–3000. doi: 10.1016/j.bmcl.2013.03.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sheffler DJ, Williams R, Bridges TM, Lewis LM, Xiang Z, Zheng F, Kane AS, Byum NE, Jadhav S, Mock MM, Zheng F, Lewis LM, Jones CK, Niswender CM, Weaver CD, Conn PJ, Lindsley CW, Conn PJ. Novel Selective Muscarinic Acetylcholine Receptor Subtype 1 (M1 mAChR) Antagonist Reduces Seizures without Impairing Hippocampal-Dependent Learning. Mol Pharmacol. 2009;76:356–368. doi: 10.1124/mol.109.056531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Marlo JE, Niswender CM, Luo Q, Brady AE, Shirey JK, Rodriguez AL, Bridges TM, Williams R, Days E, Nalywajko NT, Austin C, Williams M, Xiang Y, Orton D, Brown HA, Kim K, Lindsley CW, Weaver CD, Conn PJ. Discovery and characterization of novel allosteric potentiators of M1 muscarinic receptors reveals multiple modes of activity. Mol Pharm. 2009;75:577–588. doi: 10.1124/mol.108.052886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.For the MLPCN see: http://mli.nih.gov/mli/mlpcn/; ML375 is an MLPCN probe and freely available upon request.
- 20.See Supporting Information for full details.
- 21.Wenthur CJ, Niswender CM, Morrison R, Daniels JS, Conn PJ, Lindsley CW. Discovery of (R)-(2-fluoro-4-((-4-methoxyphenyl)ethynyl)phenyl)(3-hydroxypiperidin-1-yl)methanone (ML337), an mGlu3 selective and CNS penetrant negative allosteric modulator (NAM) J Med Chem. 2013;56:3713–3718. doi: 10.1021/jm400439t. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.