

Abstract
A simple protocol, involving the green synthesis for the construction of novel bis-pyrimidine derivatives, 3a–i and 4a–e are accomplished by the aqueous diethylamine media promoted tandem Aldol-Michael reaction between two molecules of barbituric acid derivatives 1a,b with various aldehydes. This efficient synthetic protocol using an economic and environmentally friendly reaction media with versatility and shorter reaction time provides bis-pyrimidine derivatives with high yields (88%–99%).
Keywords: tandem Aldol-Michael reactions, MCRs, barbituric acid, aqueous media, green chemistry
1. Introduction
Derivatives of the 1,3-dimethylbarbituric acid (1) framework represent an important structural motif that is embodied in a number of bioactive natural products [1–5]. In particular, 5-Alkylated barbituric acids are one class of such compounds that possess diverse biological activity, with anticancer, HIV-1 and HIV-2 protease inhibitors [6], sedative-hypnotic [7,8], and anticonvulsant [9] properties. Many of their representatives have clinical use as antiinflammatory and hypnotic drugs, such as bucolome (Figure 1), veronal, phenobarbital, seconal, and sodium pentothal [10–13]. A number of compounds having these systems are synthesized with diverse pharmacological activity [14,15]. New routes for the synthesis of these molecules have therefore attracted considerable attention in the search for a rapid entry to these heterocycles with diverse biological properties. In accordance with the principles of green chemistry, the design of easily separable, non-toxic and low cost synthesis obviating the isolation and purification of intermediates and thus leading to a reduction in pollution has become an important area of research in chemistry [16]. A potential approach toward this goal is to combine two or more distinct reactions into a single transformation, thereby affecting tandem reactions [17–27]. Tandem reactions are therefore of increasing importance in modern organic chemistry. One-pot tandem reactions by virtue of their convergence, elegance, and super-economy are particularly appealing in the context of rapid target-oriented synthesis [28–30].
Figure 1.

Bioactive compounds containing the barbituric acid framework.
We have investigated a new, simple and efficient synthesis of novel bis-pyrimidines derivatives based on tandem Aldol-Michael reactions using aqueous diethylamine medium.
2. Results and Discussion
Upon treatment of 2 molecules from 1,3-dimethylbarbituric acid 1a, with benzaldehyde 2a, water (1.5 mL) and in presence of one equivalent diethylamine, a one-pot three-component reaction proceeded spontaneously at room temperature. “Trimolecular adduct salts” 3a was obtained in 99% yield by simple filtration. The structure of the compound 3a was elucidated using spectroscopic data and elemental analyses. The NMR spectra confirmed the absence of the methylene proton of the 1,3-dimethylbarbituric acid moiety and the presence of a proton at δ 5.86 detected ppm revealing it to be the benzylic proton. The 3D structure was confirmed by single crystal structure determination (Figure 2a).
Figure 2.

ORTEP representation of the structure of 3a–c.
Encouraged by this result, we proceeded to study the effect of different amines and reaction conditions on the tandem Aldol-Michael reactions for the synthesis of bis-pyrimidine derivatives.
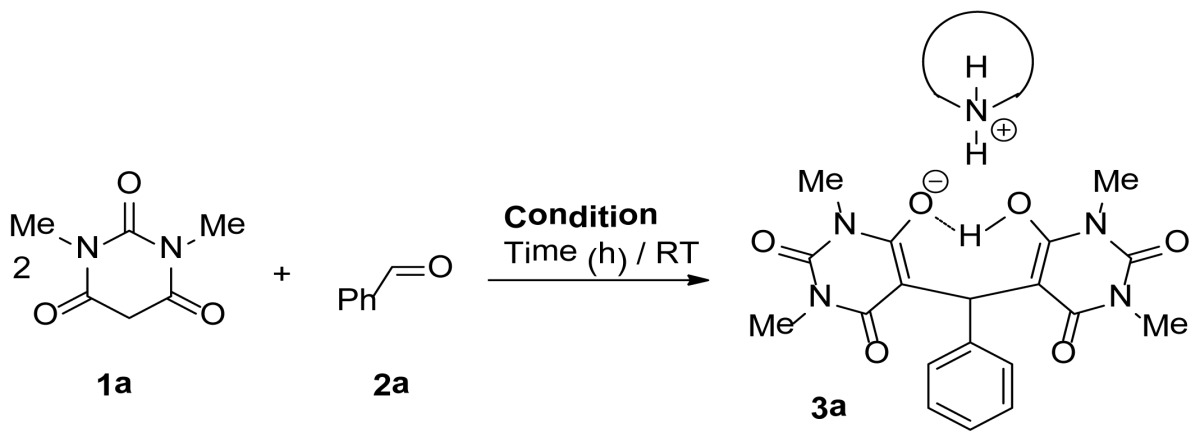
Due to the fact that the aqueous diethylamine medium gave the desired product 3a with quantitative yield, several secondary amines were therefore tested. Compared to aqueous diethylamine, aqueous diisopropylamine, aqueous dicyclohexylamine, and aqueous morpholine, which contain a bulkier amine, also gave the product but with less yield and lower reaction rates (Table 1, entries 2–4 respectively). Furthermore, NaOH was also examined and was found to be less efficient in the reaction. Only moderate yield was obtained (Table 1, entry 5). We also found that in the absence of water (entry 6) or with only water as a reactant (entry 7), the reaction either could not be processed or preceded very slowly.
Table 1.
Screening of conditions for the Aldol-Michael addition reaction of model substrate a.
| |||
|---|---|---|---|
|
| |||
| Entry | Condition | Time (h) | Yield (%) b |
| 1 | Et2NH/H2O | 1 | 99 |
| 2 | iPr2NH/H2O | 5 | 85 |
| 3 | (Cyclohexyl)2NH/H2O | 4 | 82 |
| 4 | Morpholine/H2O | 3 | 78 |
| 5 | NaOH/H2O | 8 | 65 |
| 6 | Et2NH | 12 | 10 |
| 7 | H2O | 12 | 0 |
All reactions were carried out with 1,3-dimethylbarbituric acid 1a (3 mmol), benzaldehyde 2a (1.5 mmol) and amine (1.5 mmol) in water (1.5 mL) for the specified time;
Yield of isolated product.
The X-ray structure of diethylaminium salt 3b (Figure 2b) was obtained from a single crystal grown from DCM/Et2O as a solvent, and the structure shows interesting characteristics. We were unable to determine the location of the C4 and C8 hydrogens by 1H-NMR analysis. The hydrogen from C4, rather than from C8 of the barbituric acid moiety, is removed by the basicity of diethylamine. We determined that this was due to the fact that one hydrogen is on the diethylamine and the other is involved in hydrogen bonding interactions between both barbituric acid rings. It is noteworthy to mention that 1H-NMR also have shown a singlet signal at δ 17.64 pmm, which was revealed to belong to the OH group that is making a hydrogen bond. This hypothesis was further unambiguously confirmed by the elucidation the 3D chemical structure of product 3c by single crystal X-ray crystallography (Figure 2c).
A possible mechanism for the tandem Aldol-Michael reaction is shown in Figure 3. First, the hydrogen bonding activation of the C=O group by water eases the deprotonation of 1a by diethylamine. The aldehyde is then attacked by the enolate to form the Aldol condensation product after dehydration of the intermediate. The second Michael addition under these conditions produces the final product 3 (Figure 3) [31–37].
Figure 3.

A possible mechanistic pathway.
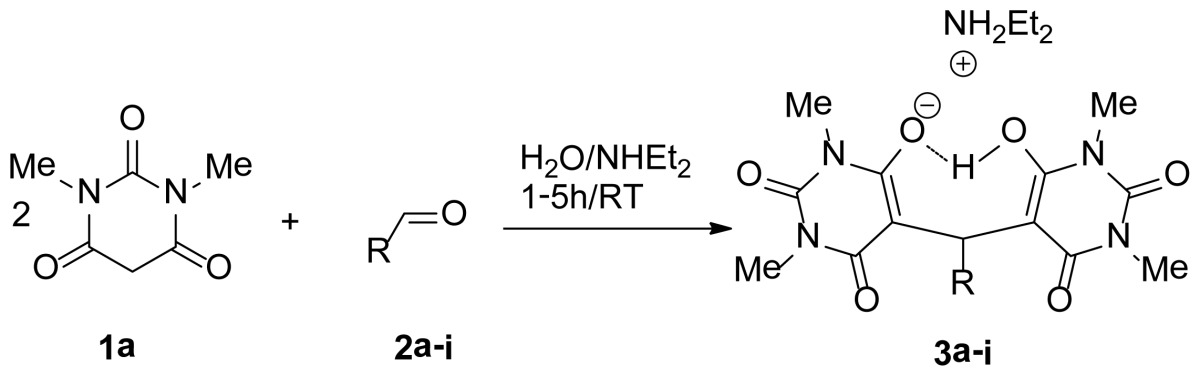
With the optimal reaction conditions established, the generality of the Aldol-Michael reactions was next investigated by using a series of aldehydes (Table 2). Various aldehydes derivatives with either electron-withdrawing or electron-donating groups at the para-, meta-, or even sterically hindered ortho-position on the aromatic ring were tolerated and gave the corresponding bis-pyrimidine derivatives 3a–i in excellent chemical yield up to 99% (Table 2). In addition, reactions with substrates 2, which contain sterically hindered 1-naphthyl proceeded smoothly to give products with very good results (Table 2, entry 9).
Table 2.
Tandem Aldol-Michael reactions of 1,3-dimethylbarbituric acid 1a with aldehydes in aqueous diethylamine medium a.
| |||
|---|---|---|---|
|
| |||
| Entry | 3 | R | Yield (%) b |
| 1 | 3a | Ph | 99 |
| 2 | 3b | p-CH3Ph | 97 |
| 3 | 3c | p-ClPh | 95 |
| 4 | 3d | p-BrPh | 92 |
| 5 | 3e | m-BrPh | 92 |
| 6 | 3f | p-CH3OPh | 90 |
| 7 | 3g | p-NO2Ph | 88 |
| 8 | 3h | m-CH3Ph | 92 |
| 9 | 3i | 2-Naphthaldehyde | 94 |
All reactions were carried out with 1,3-dimethyl barbituric acid 1a (3 mmol), aldehydes 2a–i (1.5 mmol) and amine (1.5 mmol) in water (1.5 mL) for the specified time;
Yield of isolated product.
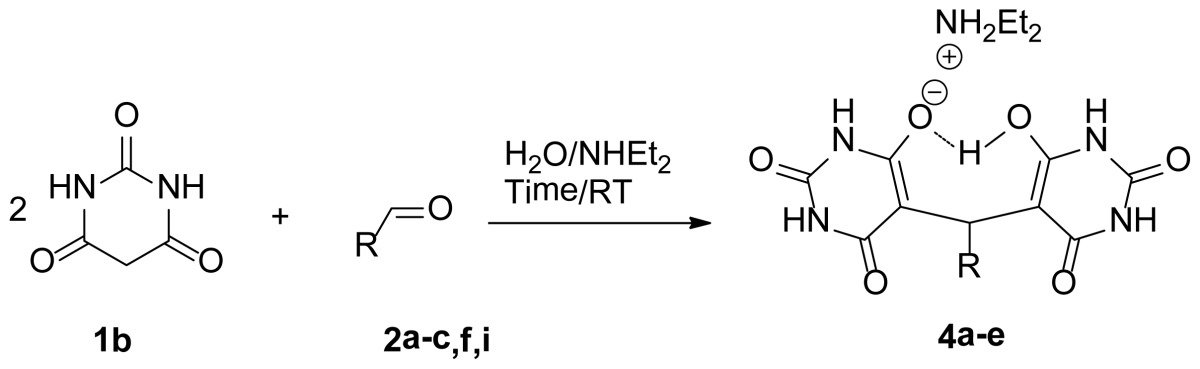
Finally, to confirm the general applicability of this novel reaction method in the preparation of a wide variety of bis-pyrimidines derivatives, we used barbituric acid itself. In every case, the corresponding bis-pyrimidines derivatives 4a–e were isolated in excellent quantitative yields (Table 3).
Table 3.
Tandem Aldol-Michael reactions of barbituric acid 1b with aldehydes in aqueous diethylamine medium a.
| |||
|---|---|---|---|
|
| |||
| Entry | 4 | R | Yield (%) b |
| 1 | 4a | Ph | 98 |
| 2 | 4b | p-CH3Ph | 95 |
| 3 | 4c | p-ClPh | 95 |
| 4 | 4d | p-CH3OPh | 91 |
| 5 | 4e | 2-Naphthaldehyde | 93 |
All reactions were carried out with barbituric acid 1b (3 mmol), aldehydes 2 (1.5mmol) and amine (1.5mmol) in water (1.5 mL) for the specified time;
Yield of isolated product.
3. Experimental Section
General: Chemicals were purchased from Sigma-Aldrich, Fluka (Seelze, Germany), and were used without further purification, unless otherwise stated. Melting points were measured on a Gallenkamp melting point apparatus (Weiss Gallenkamp Ltd., Leicestershire, UK) in open glass capillaries and are uncorrected. IR Spectra were measured as KBr pellets on a Nicolet 6700 FT-IR spectrophotometer (Madison, WI, USA). The NMR spectra were recorded on a Varian Mercury Jeol-400 NMR spectrometer (Tokyo, Japan).1H-NMR (400 MHz), and 13C-NMR (100 MHz) were run in either deuterated dimethylsulphoxide (DMSO-d6) or deuterated chloroform (CDCl3). Chemical shifts (δ) are referred in terms of ppm and J-coupling constants are given in Hz. Mass spectra were recorded on a Jeol of JMS-600 H (Tokyo, Japan). Elemental analysis was carried out on an Elmer 2400 elemental analyzer, CHN mode (Vernon Hills, IL, USA).
3.1. General Procedure for Aldol Condensation Michael Addition for the Synthesis of 3a–i and 4a–e (GP1)
A mixture of aldehyde 2 (1.5mmol), barbituric acid derivatives 1a,b (3 mmol) and Et2NH (1.5 mmol, 155 μL) in 3 mL of degassed H2O (bubbling nitrogen through the water) was stirred at room temperature for 1–5 h until TLC showed complete disappearance of the reactants. The precipitate was removed by filtration and washed with ether (3 × 20 mL). The solid was dried to afford pure product 3a–i and 4a–e.
3.1.1. 5,5′-(Phenylmethylene)bis(1,3-dimethylpyrimidine-2,4,6(1H,3H,5H)-trione) Diethylaminium Salt 3a
3a was prepared from 1,3-dimethylbarbituric acid 1a, and benzaldehyde 2a according to the general procedure (GP1) yielding colorless crystalline materials (1.4 g, 2.97 mmol, 99%). m.p.: 192 °C; IR (KBr, cm−1): 3450, 3222, 3019, 2814, 1707, 1655, 1524, 1442, 1345, 1274; 1H-NMR (400 MHz, DMSO-d6): δ 17.64 (s, 1H, OH), 7.58 (d, 2H, J = 7.3 Hz, NH2), 7.19 (m, 5H, Ph), 5.86 (s, 1H, benzyl-H), 3.05 (s, 12H, 4CH3), 2.93 (q, 4H, J = 7.3 Hz, CH2CH3), 1.16 (t, 6H, J = 7.3 Hz, CH2CH3); 13C-NMR (100 MHz, DMSO-d6): δ = 161.6, 153.2, 145.5, 141.6, 129.1, 128.2, 127.8, 125.8, 88.5, 49.1, 41.9, 27.5, 11.5; LC/MS (ESI): 473 [M]+; Anal. for C23H31N5O6; Calcd: C, 58.34; H, 6.60; N, 14.79; Found: C, 58.35; H, 6.62; N, 14.80.
The structure of 3a was confirmed by X-ray crystal structure analysis (Bruker AXS GmbH, Karlsruhe, Germany). CCDC-933457 contains the supplementary crystallographic data for this compound. These data can be obtained free of charge from the Cambridge Crystallographic Data Centre via http://www.ccdc.cam.ac.uk/data_request/cif. A colorless crystal suitable for X-ray analysis was obtained from recrystallization of the compound from DCM/Et2O at room temperature after 5 days.
3.1.2. 5,5′-(p-Tolylmethylene)bis(1,3-dimethylpyrimidine-2,4,6(1H,3H,5H)-trione) Diethylaminium Salt 3b
3b was prepared from 1,3-dimethylbarbituric acid 1a, and p-tolualdehyde 2b according to the general procedure (GP1) yielding colorless needle materials (1.41 g, 2.91 mmol, 97%). m.p.: 152 °C; IR (KBr, cm−1): 3455, 3210, 2984, 2820, 1560, 1449, 1359; 1H-NMR (400 MHz, CDCl3): δ 17.64 (s, 1H, OH), 6.99–6.96 (m, 4H, Ph), 5.80 (s, 1H, benzyl-H), 3.32 (s, 12H, 4CH3), 3.03 (q, 4H, J = 7.3 Hz, CH2CH3), 2.25 (s, 3H, CH3), 1.28 (t, 6H, J = 7.3 Hz, CH2CH3); 13C-NMR (100 MHz, CDCl3): δ = 165.3, 164.3, 151.8, 138.6, 134.8, 128.9, 126.3, 92.1, 42.0, 34.2, 28.9, 28.6, 21.0, 11.4; LC/MS (ESI): 487[M]+; Anal. for C24H35N5O6; Calcd: C, 59.12; H, 6.82; N, 14.36; Found: C, 59.13; H, 6.81; N, 14.35.
The structure of 3b was confirmed by X-ray crystal structure analysis (Bruker AXS GmbH). CCDC-957025 contains the supplementary crystallographic data for this compound. These data can be obtained free of charge from the Cambridge Crystallographic Data Centre via www.ccdc.cam.ac.uk/data_request/cif. A colorless crystal suitable for X-ray analysis was obtained from recrystallization the compound from DCM/Et2O at room temperature after 2 days.
3.1.3. 5,5′-((4-Chlorophenyl)methylene)bis(1,3-dimethylpyrimidine-2,4,6(1H,3H,5H)-trione) Diethylaminium Salt 3c
3c was prepared from 1,3-dimethylbarbituric acid 1a, and p-chlorobenzaldehyde 2c according to the general procedure (GP1) yielding colorless crystals (1.44 g, 2.85 mmol, 95%). m.p.: 103 °C; IR (KBr, cm−1): 3450, 3198, 2988, 1698, 1603, 1479, 1385; 1H-NMR (400 MHz, CDCl3): δ 17.66 (s, 1H, OH), 7.18 (d, 2H, J = 8.8 Hz, Ph), 7.05 (d, 2H, J = 8.8 Hz, Ph), 5.80 (s, 1H, benzyl-H), 3.34 (s, 12H, 4CH3), 3.06 (q, 4H, J = 7.3 Hz, CH2CH3), 1.30 (t, 6H, J = 7.3 Hz, CH2CH3); 13C-NMR (100 MHz, CDCl3): δ = 165.3, 164.3, 151.7, 138.6, 134.8, 128.2, 126.3, 91.7, 42.1, 34.2, 28.9, 28.7, 11.5; LC/MS (ESI): 507[M]+; Anal. for C23H30ClN5O6; Calcd: C, 54.38; H, 5.95; Cl, 6.98; N, 13.79; Found: C, 54.38; H, 5.94; Cl, 7.01; N, 13.81.
The structure of 3c was confirmed by X-ray crystal structure analysis (Bruker AXS GmbH). CCDC-957026 contains the supplementary crystallographic data for this compound. These data can be obtained free of charge from the Cambridge Crystallographic Data Centre via www.ccdc.cam.ac.uk/data_request/cif. A colorless crystal suitable for X-ray analysis was obtained from recrystallization the compound from DCM/Et2O at room temperature after 2 days.
3.1.4. 5,5′-((4-Bromophenyl)methylene)bis(1,3-dimethylpyrimidine-2,4,6(1H,3H,5H)-trione) Diethylaminium Salt 3d
3d was prepared from 1,3-dimethylbarbituric acid 1a, and p-bromobenzaldehyde 2d according to the general procedure (GP1) yielding beige powder (1.5 g, 2.76 mmol, 92%). m.p.: 125 °C; IR (KBr, cm−1): 3454, 3200, 3019, 2814, 1707, 1650, 1518, 1442, 1377, 1274; 1H-NMR (400 MHz, CDCl3): δ 17.62 (s, 1H, OH), 7.31 (d, 2H, J = 8.8 Hz, Ph), 6.99 (d, 2H, J = 8.8 Hz, Ph), 5.79 (s, 1H, benzyl-H), 3.33 (s, 12H, 4CH3), 3.03 (q, 4H, J = 7.3 Hz, CH2CH3), 1.27 (t, 6H, J = 7.3 Hz, CH2CH3) ; 13C-NMR (100 MHz, CDCl3): δ = 165.3, 164.3, 151.7, 141.1, 131.1, 128.4, 119.3, 91.7, 42.1, 34.2, 28.9, 28.7, 11.4; LC/MS (ESI): 552[M]+; Anal. for C23H30BrN5O6; Calcd: C, 50.01; H, 5.47; Br, 14.46; N, 12.68; Found: C, 50.00; H, 5.47; Br, 14.49; N, 12.70.
3.1.5. 5,5′-((3-Bromophenyl)methylene)bis(1,3-dimethylpyrimidine-2,4,6(1H,3H,5H)-trione) Diethylaminium Salt 3e
3e was prepared from 1,3-dimethylbarbutric acid 1a, and m-bromobenzaldehyde 2e according to the general procedure (GP1) yielding colorless crystalline materials (1.5g, 2.76 mmol, 92%). m.p.: 169 °C; IR (KBr, cm−1): 3450, 3120, 2982, 1694, 1667, 1615, 1577, 1445, 1250; 1H-NMR (400 MHz, CDCl3): δ 17.63 (s, 1H, OH), 7.22 (d, 1H, J = 7.3 Hz, Ph), 7.19 (s, 1H, Ph), 7.07 (d, 1H, J = 7.3 Hz, Ph), 7.05 (d, 1H, J = 7.3 Hz, Ph), 5.84 (s, 1H, benzyl-H), 3.34 (s, 6H, 2CH3), 3.32 (s, 6H, 2CH3), 3.02 (q, 4H, J = 7.3 Hz, CH2CH3), 1.27 (t, 6H, J = 7.3 Hz, CH2CH3); 13C-NMR (100 MHz, CDCl3): δ = 165.2, 164.4, 151.7, 144.7, 129.7,129.6, 128.7, 125.3, 91.5, 42.1, 34.4, 28.9, 28.7, 11.5; LC/MS (ESI): 552[M]+; Anal. for C23H30BrN5O6; Calcd: C, 50.01; H, 5.47; Br, 14.46; N, 12.68; Found: C, 50.03; H, 5.48; Br, 14.47; N, 12.71.
3.1.6. 5,5′-((4-Methoxyphenyl)methylene)bis(1,3-dimethylpyrimidine-2,4,6(1H,3H,5H)-trione) Diethylaminium Salt 3f
3f was prepared from 1,3-dimethylbarbutric acid 1a, and p-methoxybenzaldehyde 2f according to the general procedure (GP1) yielding rose-colored crystalline materials (1.35 g, 2.7 mmol, 90%). m.p.: 160 °C; IR (KBr, cm−1): 3445, 3195, 2977, 2836, 1689, 1664, 1613, 1504, 1447, 1378, 1242; 1H-NMR (400 MHz, CDCl3): δ 17.67 (s, 1H, OH), 7.01 (d, 2H, J = 8.8 Hz, Ph), 6.75 (d, 2H, J = 8.8 Hz, Ph), 5.79 (s, 1H, benzyl-H), 3.33 (s, 12H, 4CH3), 2.99 (q, 4H, J = 7.3 Hz, CH2CH3), 1.26 (t, 6H, J = 7.3 Hz, CH2CH3); 13C-NMR (100 MHz, CDCl3): δ = 165.3, 164.3, 157.4, 151.7, 133.6, 132.0, 127.4, 114.3, 92.1, 55.6, 42.1, 33.8, 28.9, 11.5; LC/MS (ESI): 503[M]+; Anal. for C24H33N5O7; Calcd: C, 57.25; H, 6.61; N, 13.91; Found: C, 57.26; H, 6.61; N, 13.90.
3.1.7. 5,5′-((4-Nitrophenyl)methylene)bis(1,3-dimethylpyrimidine-2,4,6(1H,3H,5H)-trione) Diethylaminium Salt 3g
3g was prepared from 1,3-dimethylbarbutric acid 1a, and p-nitrobenzaldehyde 2g according to the general procedure (GP1) yielding a yellow powder (1.35 g, 2.61 mmol, 87%); m.p.: 195 °C; IR (KBr, cm−1): 3453, 3205, 2987, 2904, 1675, 1608, 1576, 1511, 1438, 1343, 1254; 1H-NMR (400 MHz, CDCl3): δ 17.58 (s, 1H, OH), 8.08 (d, 2H, J = 8.8 Hz, Ph), 7.29 (d, 2H, J = 8.8 Hz, Ph), 5.95 (s, 1H, benzyl-H), 3.34 (s, 12H, 4CH3), 3.07 (q, 4H, J = 7.3 Hz, CH2CH3), 1.29 (t, 6H, J = 7.3 Hz, CH2CH3); 13C-NMR (100 MHz, CDCl3): δ = 165.2, 164.4, 151.6, 150.8, 146.1, 127.5, 123.5, 91.4, 42.2, 34.9, 28.9, 28.7, 11.5; LC/MS (ESI): 518[M]+; Anal. for C23H30N6O8; Calcd: C, 53.28; H, 5.83; N, 16.21; Found: C, 53.29; H, 5.85; N, 16.23.
3.1.8. 5,5′-(3-Tolylmethylene)bis(1,3-dimethylpyrimidine-2,4,6(1H,3H,5H)-trione) Diethylaminium Salt 3h
3h was prepared from 1,3-dimethylbarbituric acid 1a, and m-tolualdehyde 2h according to the general procedure (GP1) yielding rose-colored crystalline materials. (1.41 g, 2.91 mmol, 97%). m.p.: 135 °C; IR (KBr, cm−1): 3455, 3201, 2988, 1693, 1667, 1611, 1573, 1443; 1H-NMR (400 MHz, CDCl3): δ 17.62 (s, 1H, OH), 7.10 (t, 1H, J = 7.3 Hz, Ph), 6.92 (d, 1H, J = 7.3 Hz, Ph), 6.88 (d, 1H, J = 7.3 Hz, Ph), 5.82 (s, 1H, benzyl-H), 3.32 (s, 12H, 4CH3), 3.01 (q, 4H, J = 7.3 Hz, CH2CH3), 2.25 (s, 3H, CH3), 1.26 (t, 6H, J = 7.3 Hz, CH2CH3); 13C-NMR (100 MHz, CDCl3): δ = 165.3, 164.4, 151.8, 141.7, 137.4, 127.9, 127.1, 126.4, 123.6, 92.1, 42.0, 34.4, 28.9, 28.6, 21.8, 11.4; LC/MS (ESI): 487[M]+; Anal. for C24H35N5O6; Calcd: C, 59.12; H, 6.82; N, 14.36; Found: C, 59.13; H, 6.81; N, 14.35.
3.1.9. 5,5′-(Naphthalen-2-ylmethylene)bis(1,3-dimethylpyrimidine-2,4,6(1H,3H,5H)-trione) Diethylaminium Salt 3i
3i was prepared from 1,3-dimethylbarbutric acid 1a, and 2-naphthaldehyde 2i according to the general procedure (GP1) yielding beige powder (1.47 g, 2.82 mmol, 94%). m.p.: 146 °C; IR (KBr, cm−1): 3454, 3200, 2967, 1668, 1585, 1438, 1250; 1H-NMR (400 MHz, CDCl3): δ 17.33 (s, 1H, OH), 8.10 (d, 2H, J = 8.8 Hz, naphthyl-H), 7.99 (d, 2H, J = 8.8 Hz, naphthyl-H), 7.92 (d, 2H, J = 8.8 Hz, naphthyl-H), 7.90 (d, 2H, J = 8.8 Hz, naphthyl-H), 7.84 (d, 2H, J = 8.8 Hz, naphthyl-H), 7.68–7.38 (m, 3H, naphthyl-H), 6.37 (s, 1H, benzyl-H), 3.39 (s, 12H, 4CH3), 3.01 (q, 4H, J = 7.3 Hz, CH2CH3), 1.30 (t, 6H, J = 7.3 Hz, CH2CH3); 13C-NMR (100 MHz, CDCl3): δ = 164.9, 151.7, 136.8, 135.3, 134.3, 131.5, 129.1, 128.5, 127.0, 125.2 124.9, 123.8, 93.2, 41.8, 33.2, 28.8, 11.4; LC/MS (ESI): 523 [M]+; Anal. for C27H33N5O6; Calcd: C, 61.94; H, 6.35; N, 13.38; Found: C, 61.95; H, 6.34; N, 13.40.
3.1.10. 5,5′-(Phenylmethylene)bis(6-hydroxypyrimidine-2,4(1H,3H)-dione) Diethylaminium Salt 4a
4a was prepared from barbituric acid 1b, and benzaldehyde 2a according to the general procedure (GP1) yielding an oil product (1.22g, 2.94mmol, 98%); IR (KBr, cm−1): 3450, 3200, 2976, 2839, 1668, 1615, 1505, 1383, 1247; 1H-NMR (400 MHz, DMSO-d6): δ 17.22 (s, 1H, OH), 10.10 (bs, 4H, NH), 7.14 (t, 3H, J = 7.3 Hz, Ph), (d, 2H, J = 7.3 Hz, Ph), 7.29 (d, 2H, J = 8.8 Hz, Ph), 5.95 (s, 1H, benzyl-H), 2.93 (q, 4H, J = 7.3 Hz, CH2CH3), 1.15 (t, 6H, J = 7.3 Hz, CH2CH3); 13C-NMR (100 MHz, DMSO-d6): δ = 165.3, 164.4, 151.7, 142.1.3, 134.3, 129.9, 127.3, 91.6, 42.6, 30.1, 11.6; LC/MS (ESI): 417[M]+; Anal. for C19H23N5O6; Calcd: C, 54.67; H, 5.55; N, 16.78; Found: C, 54.68; H, 5.54; N, 16.79.
3.1.11. 5,5′-(p-Tolylmethylene)bis(6-hydroxypyrimidine-2,4(1H,3H)-dione) Diethylaminium Salt 4b
4b was prepared from barbituric acid 1b, and p-tolualdehyde 2b according to the general procedure (GP1) yielding white powder (1.22 g, 2.85 mmol, 95%); m.p.: 205 C; IR (KBr, cm−1): 3459, 3120, 2978, 2811, 1689, 1612, 1325, 1252; 1H-NMR (400 MHz, DMSO-d6): δ 17.18 (s, 1H, OH), 10.09 (bs, 4H, NH), 6.93 (m, 4H, Ph), 5.90 (s, 1H, benzyl-H), 2.79 (q, 4H, J = 7.3 Hz, CH2CH3), 2.20 (s, 3H, CH3), 1.07 (t, 6H, J = 7.3 Hz, CH2CH3); 13C-NMR (100 MHz, DMSO-d6): δ = 164.8, 164.1, 151.3, 142.1, 133.5, 128.5, 127.1, 91.6, 42.6, 30.6, 21.1, 13.0; LC/MS (ESI): 431[M]+; Anal. for C20H25N5O6; Calcd: C, 55.68; H, 5.84; N, 16.23; Found: C, 55.67; H, 5.83; N, 16.22.
3.1.12. 5,5′-((4-Chlorophenyl)methylene)bis(6-hydroxypyrimidine-2,4(1H,3H)-dione) Diethylaminium Salt 4c
4c was prepared from barbituric acid 1b, and p-chlorobenzaldehyde 2c according to the general procedure (GP1) yielding a white powder (1.28 g, 2.85 mmol, 95%); m.p.: 221 °C; IR (KBr, cm−1): 3435, 3185, 2978, 2830, 1677, 1548, 1448, 1345, 1250; 1H-NMR (400 MHz, DMSO-d6): δ 17.17 (s, 1H, OH), 10.00 (bs, 4H, NH), 7.18 (m, 4H, Ph), 5.93 (s, 1H, benzyl-H), 2.88 (q, 4H, J = 7.3 Hz, CH2CH3), 1.12 (t, 6H, J = 7.3 Hz, CH2CH3); 13C-NMR (100 MHz, DMSO-d6): δ = 164.7, 164.0, 151.2, 144.6, 133.5, 129.9, 129.1, 127.8, 91.3, 42.1, 30.7, 11.8; LC/MS (ESI): 451[M]+; Anal. for C19H22ClN5O6; Calcd C, 50.50; H, 4.91; Cl, 7.85; N, 15.50; Found: C, 50.51; H, 4.90; Cl, 7.83; N, 15.51.
3.1.13. 5,5′-((4-Methoxyphenyl)methylene)bis(6-hydroxypyrimidine-2,4(1H,3H)-dione) Diethylaminium Salt 4d
4d was prepared from barbituric acid 1b, and p-methoxybenzaldehyde 2f according to the general procedure (GP1) yielding a beige powder (1.22 g, 2.73 mmol, 91%); m.p.: 195 °C; IR (KBr, cm−1): 3449, 3190, 2991, 2835, 1688, 1592, 1505, 1383, 1247; 1H-NMR (400 MHz, DMSO-d6): δ 17.26 (s, 1H, OH), 9.99 (bs, 4H, NH), 6.92 (d, 2H, J = 8.0 Hz, Ph), 6.72 (d, 2H, J = 8.0 Hz, Ph), 5.88 (s, 1H, benzyl-H), 2.90 (q, 4H, J = 7.3 Hz, CH2CH3), 1.14 (t, 6H, J = 7.3 Hz, CH2CH3); 13C-NMR (100 MHz, DMSO-d6): δ = 164.6, 164.0, 157.0, 151.2, 137.2, 132.4, 115.1, 91.7, 55.4, 42.1, 30.7, 11.6; LC/MS (ESI): 447[M]+; Anal. for C20H25N5O7; Calcd C, 53.69; H, 5.63; N, 15.65; Found: C, 53.69; H, 5.63; N, 15.66.
3.1.14. 5,5′-(Naphthalen-2-ylmethylene)bis(6-hydroxypyrimidine-2,4(1H,3H)-dione) Diethylaminium Salt 4e
4e was prepared from barbituric acid 1b, and 2-naphthaldehyde 2i according to the general procedure (GP1) yielding a beige powder (1.3 g, 2.79 mmol, 93%); m.p.: 192 °C; IR (KBr, cm−1): 3459, 3208, 2994, 1677, 1579, 1448, 1386, 1354; 1H-NMR (400 MHz, DMSO-d6): δ 16.92 (s, 1H, OH), 10.41 (bs, 4H, NH), 8.13 (d, 1H, J = 8.8 Hz, naphthyl), 7.81(d, 1H, J = 8.8Hz, naphthyl), 7.63 (d, 1H, J = 8.8 Hz, naphthyl), 7.38–7.32 (m, 4H, naphthyl), 6.46 (s, 1H, benzyl-H), 2.79 (q, 4H, J = 7.3 Hz, CH2CH3), 1.08 (t, 6H, J = 7.3 Hz, CH2CH3); 13C-NMR (100 MHz, DMSO-d6): δ = 164.9, 151.1,141.5, 135.8, 134.0,132.4, 129.3, 128.7, 126.0,125.8, 125.5, 125.2, 124.9, 123.8, 92.3, 42.5, 29.7, 12.7; LC/MS (ESI): 467[M]+; Anal. for C23H25N5O6; Calcd C, 59.09; H, 5.39; N, 14.98; Found: C, 59.12; H, 5.40; N, 15.01.
4. Conclusions
In summary, we have shown that the multicomponent tandem Aldol condensation/Michael addition reaction of aromatic aldehydes with barbituric acid 1a,b by aqueous diethylamine medium is a powerful and efficient method for the synthesis of novel bis-pyrimidine derivatives 3a–i and 4a–e, which are of biological significance. In addition to its efficiency, simplicity and milder reaction conditions, this method provides excellent yields of the products with selectivity. Further studies on expanding the application of this method and the biological evaluation of these pyrimidine derivatives are in progress.
Acknowledgments
The authors extend their appreciation to the Deanship of Scientific Research at King Saud University for funding the work through the research group project Number RGP-VPP-257.
Conflicts of Interest
The authors declare no conflict of interest.
References
- 1.Bojarski J.T., Mokrocz J.L., Barton H.J., Paluchowska M.H. Recent progress in barbituric acid chemistry. Adv. Heterocycl. Chem. 1985;38:229–297. [Google Scholar]
- 2.Undheim K., Bennecke T., Katritzky A.R., Rees C.W., Scriven E.F.V., Boulton A.J. Comprehensive Heterocyclic Chemistry. Suppl 93 Vol. 6. Elsevier Pergamon; Oxford, UK: 1996. (Hrsg.) [Google Scholar]
- 3.Von Angerer S. Product class 12: Pyrimidines. Sci. Synth. 2004;16:379–572. [Google Scholar]
- 4.Sans S.R.G., Chosaz M.G. Historical aspects and applications of barbituric acid derivatives. Pharmazie. 1988;43:827–829. [PubMed] [Google Scholar]
- 5.Taylor J.B. Modern Medical Chemistry. Prentice Hall; New York, NY, USA: 1994. [Google Scholar]
- 6.Guerin D.J., Mazeas D., Musale M.S., Naguib F.N.M., Safarjalani O.N.A., Kouni M.H., Panzica R.P. Uridine phosphorylase inhibitors: Chemical modification of benzyloxybenzyl barbituric acid and its effects on UrdPase inhibition. Bioorg. Med. Chem. Lett. 1999;9:1477–1480. doi: 10.1016/s0960-894x(99)00238-3. [DOI] [PubMed] [Google Scholar]
- 7.Andrews G. Medical Pharmacology. The CV Mosby Co; St. Louis, MO, USA: 1976. pp. 243–250. [Google Scholar]
- 8.Foye W.O. Principles of Medicinal Chemistry. Lea & Febiger; Pennsylvania, PA, USA: 1989. pp. 143–237. [Google Scholar]
- 9.Goodman L.S., Gilman A. The Pharmacological Basis of Therapeutics. Mc Graw-Hill; New Delhi, India: 1991. pp. 358–360. [Google Scholar]
- 10.Fisher E., von Mering J. Ueber eine neue Klasse von Schlafmitteln. Ther. Ggw. 1903;44:97–105. [Google Scholar]
- 11.Doran W.J. Barbituric acid hypnotics. Med. Chem. 1959;4:164–167. [Google Scholar]
- 12.Bobranski B. Progress in chemistry of barbituric acid. Wiad. Chem. 1977;31:231–278. [Google Scholar]
- 13.Senda S., Izumi H., Fujimura H. Uracil derivatives and related compounds. VI. Derivatives of 5-alkyl-2,4,6-trioxoperhydropyrimidine as anti-inflammatory agents. Arzneim. Forsch. 1967;17:1519–1523. [PubMed] [Google Scholar]
- 14.Anderson G.L., Shim J.L., Brown A.D. Pyrido[2,3-d]pyrimidines. IV. Synthetic studies leading to various oxopyrido[2,3-d]pyrimidines. J. Org. Chem. 1976;41:1095–1099. doi: 10.1021/jo00869a003. [DOI] [PubMed] [Google Scholar]
- 15.Grivaky E.M., Lee S., Siyal C.W., Duch D.S., Nichol C.A. Synthesis and antitumor activity of 2,4-diamino-6-(2,5-dimethoxybenzyl)-5-methylpyrido[2,3-d]pyrimidine. J. Med. Chem. 1980;23:327–329. doi: 10.1021/jm00177a025. [DOI] [PubMed] [Google Scholar]
- 16.Gawande M.B., Bonifacio V.D.B., Luque R., Branco P.S., Varma R.S. Benign by design: Catalyst-free in-water, on-water green chemical methodologies in organic synthesis. Chem. Soc. Rev. 2013;42:5522–5551. doi: 10.1039/c3cs60025d. [DOI] [PubMed] [Google Scholar]
- 17.Ho T.L. Tandem Organic Reactions. Wiley; New York, NY, USA: 1992. [Google Scholar]
- 18.Nicolaou K.C., Yue E.W., Oshima T. New Roads to Molecular Complexity. In: Hall N., editor. The New Chemistry. Cambridge University Press; Cambridge, UK: 2001. pp. 168–198. [Google Scholar]
- 19.Tietze L.F., Hautner F. Domino Reaction in Organic Synthesis. An Approach to Efficiency, Elegance, Ecological Benefit, Economic Advantage, and Preservation of Our Resources in Chemical Transformation. In: Vögtle F., Stoddart J., Shibasaki F.M., editors. Stimulating Concepts in Chemistry. Wiley-VCH; Weinheim, Germany: 2000. pp. 38–64. [Google Scholar]
- 20.Tietze L.F., Beifuss U. Sequential transformations in organic chemistry: A synthetic strategy with a future. Angew. Chem. Int. Ed. Engl. 1993;32:131–163. [Google Scholar]
- 21.Tietze L.F. Domino reactions in organic synthesis. Chem. Rev. 1996;96:115–136. doi: 10.1021/cr950027e. [DOI] [PubMed] [Google Scholar]
- 22.Thompson L.A. Recent applications of polymer-supported reagents and scavengers in combinatorial, parallel, or multistep synthesis. Curr. Opin. Chem. Biol. 2000;4:324–337. doi: 10.1016/s1367-5931(00)00096-x. [DOI] [PubMed] [Google Scholar]
- 23.Nefzi A., Ostresh J.M., Houghten R.A. The current status of heterocyclic combinatorial libraries. Chem. Rev. 1997;97:449–472. doi: 10.1021/cr960010b. [DOI] [PubMed] [Google Scholar]
- 24.Yanovskaya L.A., Dombrovsky V.A., Khusid A. Cyclopropanes with Functional Groups. Synthesis and Application. Nauka; Moscow, Russia: 1980. [Google Scholar]
- 25.Tsuji T., Nishida S. The Chemistry of the Cyclopropyl Group. Wiley and Sons; New York, NY, USA: 1987. [Google Scholar]
- 26.Boche G., Walbirsky H.M. Cyclopropane Derived Intermediates. John Wiley and Sons; New York, NY, USA: 1990. [Google Scholar]
- 27.Rappoport Z. The Chemistry of the Cyclopropyl Group. Wiley and Sons; New York, NY, USA: 1996. [Google Scholar]
- 28.Posner G.H. Multicomponent one-pot annulations forming 3 to 6 bonds. Chem. Rev. 1986;86:831–834. [Google Scholar]
- 29.Bunce R.A. Recent advances in the use of tandem reactions for organic synthesis. Tetrahedron. 1995;48:13103–13159. [Google Scholar]
- 30.Anastas P.T., Warner J.C. Green Chemistry: Theory and Practice. Oxford University Press; New York, NY, USA: 2000. [Google Scholar]
- 31.Gruttadauria M., Giacalone F., Marculesco A.M., Meo P.L., Riela S., Noto R. Hydrophobically directed Aldol reactions: Polystyrene-supported l-proline asa recyclable catalyst for direct asymmetric Aldol reactions in the presence of water. Eur. J. Org. Chem. 2007:4688–4698. [Google Scholar]
- 32.Breslow R. Determining the geometries of transition states by use of antihydrophobic additives in water. Acc. Chem. Res. 2004;37:471–478. doi: 10.1021/ar040001m. [DOI] [PubMed] [Google Scholar]
- 33.Blackmond D.G., Armstrong A., Coombe V., Wells A. Water in organocatalytic processes: Debunking the myths. Angew. Chem. Int. Ed. Engl. 2007;46:3798–3800. doi: 10.1002/anie.200604952. [DOI] [PubMed] [Google Scholar]
- 34.Abaee M.S., Cheraghi S., Navidipoor S., Mojtahedi M.M., Forghani S. An efficient tandem aldol condensation-thia-Michael addition process. Tetrahedron Lett. 2012;53:4405–4408. [Google Scholar]
- 35.Barakat A., Al-Majid A.A., Shahidul Islam M., Al-Othman Z.A. Highly enantioselective Friedel–Crafts alkylations of indoles with α,β-unsaturated ketones under Cu(II)-simple oxazoline-imidazoline catalysts. Tetrahedron. 2013;69:5185–5192. [Google Scholar]
- 36.Jursic B.S. Preparation of unsubstituted dipyridine-dibarbituric acid ylide through dimerization of pyridin-2-ylmethylenepyrimidinetrione as a reactive intermediate. J. Heterocyclic Chem. 2003;40:167–170. [Google Scholar]
- 37.Jursic B.S., Neumann D.M. Preparation of 5,5′-pyrilidene and 5,5′-quinolidene bis-barbituric acid derivatives. J. Heterocycl. Chem. 2003;40:465–474. [Google Scholar]