

Significance
Cellular interactions that regulate the immune response of T cells to viral infection are poorly understood. Here we report that in the absence of activity of CD8 regulatory T-cells (CD8 Treg cells), antiviral immunity is enhanced and the deleterious effects of viral infection are constrained. Using a genetically modified mouse model that displays defective regulatory activity of CD8 Treg cells, the immune response against viruses was substantially enhanced during the acute and chronic phase of viral infection; this enhanced antiviral response prevented tissue damage associated with viral infection. This suggests that protective immunity to viral infection can be achieved by blocking the inhibitory effects of CD8 Treg cells and opens the possibility of unique approaches to treat viral infection.
Keywords: T-cell exhaustion, killer cell Ig-like receptor, immune regulation
Abstract
The immunological interactions that regulate the T-cell response to chronic viral infection are insufficiently understood. Here we study a cellular interaction that may enhance the antiviral immune response and constrain immunopathology. We analyze the contribution of Qa-1-restricted CD8+ regulatory T cells (Treg cells) to antiviral immunity after infection by lymphocytic choriomeningitis virus. These CD8+ Treg cells recognize and eliminate target cells through an interaction with the murine class Ib MHC molecule Qa-1 (HLA-E in humans). Using Qa-1 mutant mice (B6.Qa-1-D227K [B6-DK]) that harbor a single mutation that abrogates binding of Qa-1 peptide to the CD8–TCR (T-cell receptor) complex, we show that disruption of immune suppression mediated by CD8+ Treg cells results in robust antiviral immune responses in both acute and chronic viral infection. Enhanced antiviral responses of B6-DK mice were accompanied by increased control of virus, reduced tissue inflammation in the acute phase, and dramatic alleviation of disease in the chronic phase. In addition, CD8+ effector T cells in B6-DK mice displayed a less exhausted phenotype characterized by decreased expression of programmed cell death 1 (PD-1), LAG3 (CD223), and 2B4 (CD244) and increased expression of NKG2D (CD314) and killer cell lectin-like receptor subfamily G member 1 (KLRG1). Enhanced antiviral immunity in B6-DK mice reflected, in part, reduced inhibition of CD8+ effector cells by CD8+ Treg cells. These findings indicate that direct inhibition of effector CD8+ T cells by Qa-1-restricted CD8+ Treg cells results in increased disease severity and delayed recovery. These data suggest that depletion or inactivation of CD8+ Treg cells represents a potentially effective strategy to enhance protective immunity to chronic viral infection.
Viral infection continues to be a major worldwide health burden, as indicated by recent estimates that more than 500 million people suffer from chronic viral hepatitis B and C (1). Although vaccines have led to the eradication of smallpox (2) and the prevention of many other viral infections, major obstacles to progress include an incomplete understanding of the interactions between the immune system and viral pathogens that underlie viral evasion, T-cell exhaustion, and immunopathology (3). Further definition of immunological mechanisms that promote robust antiviral immune responses with minimal host tissue damage requires a better understanding of the host immune response during the progression, spread, and clearance of virus (4, 5).
Lymphocytic choriomeningitis virus (LCMV) is a widely used disease model for studies of the immune response to viral infection (6). LCMV-Armstrong (Arm), first described in the 1930s after an encephalitis epidemic in St. Louis, Missouri (7), results in an acute viral infection, the rapid resolution of which depends largely on an efficient, virus-specific CD8+ cytotoxic T-cell response. Although LCMV-Arm is noncytopathic, an excessive host immune response to the virus can lead to severe tissue damage, including inflammatory hepatitis and pneumonitis (8, 9). The LCMV strain clone 13 (LCMV-Cl13) is a genetic variant of LCMV-Arm that can persist in the host as a chronic infection (10), which is marked by CD8+ T-cell exhaustion, continued tissue inflammation, and destruction (11, 12).
The role of regulatory T cells during viral infection has been the subject of studies aimed at understanding mechanisms that control this process. The majority of these studies in both mice and humans have focused on CD4+CD25+FoxP3+ (forkhead box P3) regulatory T cells (Treg cells) (13). Coevolution of microorganisms and CD4+ Treg cells have led to a range of outcomes, including protection of the host through inhibition of collateral inflammatory damage as well as exacerbation of disease through promotion of pathogen persistence (13). The contribution of CD4+ Treg cells to the outcome of LCMV infection has not been clearly defined (14–16).
In addition to FoxP3+ CD4+ cells, a regulatory lineage within the CD8+ T-cell subset has recently been identified (17). CD8+ Treg cells display a memory phenotype and express CD44 and CD122 receptors along with the natural killer (NK) cell receptor Ly49. This small subset of CD8+ T cells is IL-15-dependent and is essential for both maintenance of self-tolerance and prevention of autoimmune disease in mice (18). CD8+ Treg cells target mainly CD4+ follicular T-helper cells through recognition of the murine class Ib MHC molecule Qa-1, resulting in perforin-dependent elimination and diminished autoimmune responses (18). This analysis is based in part on the generation of Qa-1 knock-in mice that harbor a single D→K amino acid exchange point mutation at position 227 [B6.Qa-1-D227K (B6-DK)] that abrogates binding of Qa-1 peptide to the CD8–TCR complex (19). B6-DK mutant mice develop severe autoimmune disease marked by the generation of autoantibodies to multiple tissues, lymphocyte infiltration into nonlymphoid tissues, and lethal glomerulonephritis (19). The contribution of CD8+ Treg cells to the antiviral immune response has not been directly examined.
Here we find that CD8+ Treg cells inhibit the immune response to LCMV infection. Effector T cells are suppressed by Qa-1-restricted CD8+ Treg cells during both acute and chronic LCMV infection. Genetic disruption of CD8+ Treg cell activity in B6-DK mice results in alleviated clinical disease, enhanced viral clearance, and reduced tissue pathology. Moreover, disruption of the suppressive activity of CD8+ Treg cells prevents development of an exhausted phenotype by effector CD8+ T cells and allows enhanced expression of antiviral CD8 memory T-cell activity. Characterization of the inhibitory interaction between regulatory and effector CD8+ T cells may allow the development of unique therapeutic approaches to chronic viral infection.
Results
Genetic Disruption of CD8 Treg Cell Activity Results in Enhanced Antiviral CD8 Responses During Acute LCMV Infection.
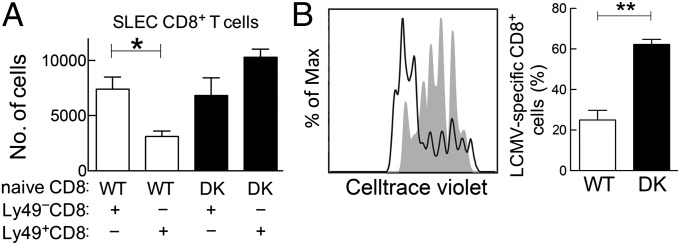
To assess the effect of Qa-1-restricted CD8+ Treg cells during acute viral infection, we compared the immune response to LCMV-Arm of B6.Qa-1 wild-type (B6-WT) and B6.Qa-1-D227K knock-in (B6-DK) mice. Analysis of Qa-1 expression by LCMV-specific CD8+ effector T cells showed increased levels by B6-WT and B6-DK effector CD8+ T cells after infection. The former WT Qa-1+ CD8 cells are potential targets of Qa-1-restricted CD8+ Treg cells (Fig. 1A), whereas CD8+ cells from B6-DK mice contain a Qa-1 point mutation that disrupts the inhibitory interaction between CD8 Treg cells and Qa-1+ target cells (20). We observed a significantly enhanced polyclonal and antiviral CD8+ T-cell response in B6-DK mice. The numbers of CD62L–CD44+ effector CD8+ T cells were approximately threefold higher in B6-DK mice compared with B6-WT mice by day 3 after infection; this increase persisted through the immune response at day 8 (Fig. 1B). We also analyzed killer cell lectin-like receptor subfamily G member 1 (KLRG1)+CD127– CD8+ T cells, which are “short-lived effector cells” (SLECs) that can contribute to the early adaptive antiviral immune response. By day 5–8 after infection, B6-DK mice displayed an approximate fivefold increase in LCMV-specific SLECs (gp33-tetramer+CD62L–CD44+CD127–KLRG1+CD8+) compared with wild-type mice (Fig. 1C).
Fig. 1.

Genetic disruption of CD8+ Treg cell activity results in enhanced antiviral CD8+ response during acute LCMV infection. (A) B6-WT and B6-DK mice were infected i.p. with LCMV-Arm (2 × 105 pfu). On day 5 postinfection, LCMV-specific H-2/Db-gp33+ CD44+CD62L−CD3+CD8+ T cells and naive gp33–CD44–CD62L+CD3+CD8+ T cells from spleen were evaluated by flow cytometry for expression of Qa-1. Expression of Qa-1 on naive CD8+ T cells (black line), B6-WT LCMV-specific CD8+ T cells (gray line), and B6-DK LCMV-specific CD8+ T cells (dotted line), as well as Qa-1-isotype staining (gray shading), are shown (n > 3 per group). (B) B6-WT and B6-DK mice were infected i.p. with LCMV-Arm (2 × 105 pfu). On days 3, 5, and 8 postinfection, CD3+CD8+ T cells from the spleens were evaluated by flow cytometry. Total CD62L–CD44+CD3+CD8+ effector T-cell numbers in B6-WT and B6-DK mice on day 3 (Left) and fold increase in B6-DK mice of CD62L–CD44+CD3+CD8+ effector T cells during the further course of infection (Right) are shown (n > 3 per group per time). (C) B6-WT and B6-DK mice were infected i.p. with LCMV-Arm (2 × 105 pfu). On days 5 and 8 postinfection, virus-specific H-2/Db-gp33+ CD3+CD8+ splenocytes were evaluated by flow cytometry for expression of CD44, CD62L, KLRG1, and CD127. Data shown represent total cell numbers of H-2/Db-gp33+ CD62L–CD44+CD127–KLRG1+CD8+ T cells at day 5 (Left) and day 8 (Right) postinfection. (D) B6-WT and B6-DK mice were infected i.p. with LCMV-Arm (2 × 105 pfu). On day 8 postinfection, virus-specific H-2/Db-gp33+ CD3+CD8+ splenocytes were evaluated by flow cytometry for expression of granzyme B in virus-specific CD8+ T cells. Data shown represent mean fluorescence intensity day 8 postinfection. (E) (Upper) Representative dot plots of B6-WT and B6-DK mice 8 d after infection with LCMV-Arm . Cells were gated on CD3+ cells, and IFN-γ+ CD8+ splenocytes were compared between the two groups. IFN-γ+CD8+ cell percentage (Left) and number (Right) are shown (n = 3 per group). (Right) IFN-γ production in SLECs from B6-WT versus B6-DK mice was compared. For analysis, splenocytes were gated on CD3+CD8+CD127low cells, and KLRG1 expression was plotted against IFN-γ. Representative dot plots are shown, and the KLRG1highIFN-γ+ cell percentage (Left) and number (Right) are shown (n = 3 per group).
We then asked whether enhanced CD8+ T-cell responses during acute viral infection secondary to genetic disruption of Qa-1-restricted CD8+ Treg cells activity was associated with enhanced CD8+ functional activity. Cytotoxic activity of effector CD8+ T cells toward target cells was mediated in part by up-regulated perforin and granzymes. Assessment of granzyme B in LCMV-specific gp33-tet+CD8+ T cells ex vivo 8 d after infection with LCMV-Arm revealed elevated granzyme B expression in B6-DK CD8+ T-cells consistent with more efficient effector T-cell responses compared with B6-WT mice (Fig. 1D). Expression of IFN-γ, an additional hallmark of protective CD8+ T-cell responses to viral infection, was increased in B6-DK CD8+ T cells (Fig. 1E).
Increased Virus-Specific CD8 T Cells on Chronic LCMV Infection of B6-DK Mice.
Next we asked whether enhanced CD8+ T-cell immunity could influence chronic infection. B6-WT and B6-DK mice were inoculated with LCMV-Cl13 to produce a chronic viral infection, and the antiviral immune response was evaluated at the early (day 10) and late (day 45–55) stages of disease. We noted that B6-DK mice developed enhanced immune responses during chronic viral infection, as noted earlier for acute LCMV infection. The LCMV-specific CD8+ T-cell response was increased in B6-DK mice by day 10 and became more pronounced at later stages of chronic infection (Fig. 2A). Effector CD8+ T-cell activity was also increased in B6-DK mice, as judged from enhanced intracellular expression of IFN-γ, granzyme B, and CD107a by CD8+ cells (Fig. 2B). Moreover, B6-DK mice harbored increased proportions of CD8+ T cells that coexpressed either two or all three markers of effector function (Fig. 2B).
Fig. 2.

Increased virus-specific CD8+ T cells on chronic LCMV infection. (A) B6-WT and B6-DK mice were infected i.v. with LCMV clone 13 (1 × 106 pfu). On days 10 and between 45–55 d postinfection, virus-specific H-2/Db-gp33+ CD3+CD8+ splenocytes were evaluated by flow cytometry. Data shown represent total cell numbers during early (Left) and late (Right) stages of chronic infection. (B) B6-WT and B6-DK mice were infected i.v. with LCMV clone 13 (1 × 106 pfu). On day 30 postinfection, total CD8+ T cells were evaluated for degranulation (CD107a) and coexpression of IFN-γ and granzyme B by flow cytometry. (Left) Dot plots for coexpression of CD107a and IFN-γ in B6-WT and B6-DK mice are shown. (Center) Pie charts represent relation of coexpression of IFN-γ, CD107a, and both in B6-WT and B6-DK mice. (Right) Percentages of single-, double-, and triple-coexpression of CD107a, granzyme B, and IFN-γ in CD8+ T cells are compared between WT (white) and B6-DK (black) mice (n > 3 per group). (C) Virus-specific H-2/Db-gp33+ CD3+CD8+ splenocytes were further assessed on day 45 postinfection with LCMV clone 13 for expression of surface markers for CD8+ T-cell memory/exhaustion. Representative histograms gated on WT H-2/Db-gp33+ CD3+CD8+ T cells (dotted line), B6-DK H-2/Db-gp33+ CD3+CD8+ T cells (solid line), and gp33–CD44–CD62L+CD8+ T (naive) T cells (gray), as well as mean fluorescence intensity (MFI), are shown. (D) Virus-specific H-2/Db-gp33+ CD3+CD8+ splenocytes were assessed on d45 postinfection with LCMV clone 13 for expression of transcription factor Eomes. Representative histograms gated on WT H-2/Db-gp33+ CD3+CD8+ T cells (dotted line), B6-DK H-2/Db-gp33+ CD3+CD8+ T cells (solid line), and gp33–CD44−CD62L+CD8+ T (naive) T cells (gray), as well as MFI, are shown. (E) IFN-γ expression in CD3+CD8+ splenocytes from WT and B6-DK mice 45 d postinfection with LCMV clone 13 was measured by flow cytometry. Representative dot plots gated on CD3+CD8+ T cells (Upper), as well as comparison between WT and B6-DK CD3+CD8+IFN-γ+ percentage (Lower, Left) and cell number (Lower, Right).
Advanced chronic LCMV-Cl13 viral infection is accompanied by effector CD8+ T cells that display an exhausted phenotype and poor effector function and express higher levels of inhibitory receptors. We asked whether the elevated cellular immune response of B6-DK mice was associated with reduced levels of exhausted CD8+ T cells. We compared LCMV-specific gp33-tet+CD62L–CD44+CD8+ T cells from B6-WT and B6-DK mice 45 d after infection with LCMV clone 13. LCMV-specific CD8+ T cells from B6-DK mice expressed a phenotype skewed toward fully functioning memory cells, whereas CD8+ T-cells from B6-WT mice displayed a phenotype more characteristic of exhausted CD8+ T cells (Fig. 2C) (21). LCMV-specific CD8+ T cells from B6-WT mice expressed elevated levels of the inhibitory receptors LAG-3 (CD223), PD-1 (programmed cell death 1), and 2B4 (CD244) and reduced levels of activating receptors NKG2D (CD314) and KLRG1 compared with LCMV-specific CD8+ cells from B6-DK mice (Fig. 2C). B6-DK effector CD8+ T cells also expressed reduced levels of Eomesodermin, a transcription factor expressed by exhausted CD8+ T cells (Fig. 2D). Further analysis revealed that B6-DK KLRG1+ effector CD8+ T cells expressed reduced levels of inhibitory receptors (SI Appendix, Fig. S1). Increased expression of IFN-γ by B6-DK mice at the late stage of infection (day 45) was consistent with the observation that virus-specific CD8+ T cells in B6-DK mice display a less exhausted phenotype (Fig. 2E).
Effective Clearance During Acute Infection by B6-DK Mice Is Associated with Diminished Lymphocyte Infiltration into Peripheral Tissues.
Effector CD8+ T cells play a central role in the clearance of LCMV-Arm and in the context of tissue damage resulting from aberrant or excessive immune responses to viral infection. We asked whether enhanced CD8+ T-cell antiviral responses of B6-DK mice were associated with altered tissue pathology. Histopathological assessment of lung and liver did not show signs of pathology in B6-WT and B6-DK mice early in infection (day 3 and 5). By day 8 after LCMV-Arm infection, B6-DK mice developed elevated CD8+ T-cell responses that were not accompanied by significant tissue pathology. In contrast, the weaker B6-WT CD8 T-cell response was accompanied by severe pneumonitis and hepatitis characterized by strong leukocyte infiltration into these organs (Fig. 3A). Measurement of leukocyte infiltrates revealed an approximate fivefold increase in lungs and a >10-fold increase in the liver of B6-WT mice compared with B6-DK mice. The inflammatory changes observed in WT mice may be a result of reduced clearance of virus, as their relatively inefficient CD8+ T-cell response was associated with increased serum virus titers on day 5 after LCMV-Arm infection (Fig. 3B). These data suggest that enhanced antiviral responses in mice secondary to genetic disruption of CD8+ Treg cell activity allowed more efficient viral clearance and decreased tissue pathology.
Fig. 3.

Effective clearance during acute infection by B6-DK mice is associated with diminished leukocyte infiltration into peripheral tissue. (A) Histopathology of the liver and lung of B6-WT and B6-DK mice is shown. (Right) Leukocyte infiltration of liver and lung per field was enumerated and compared between B6-WT and B6-DK mice. (B) On day 5 after infection with LCMV-Arm (2 × 105 pfu), virus titer in organs of B6-WT and B6-DK mice was determined via standard plaque assay. Viral titer of kidney, lung, and serum are shown as plaque-forming units.
B6-DK Mice Develop Enhanced Antiviral Response During Chronic Infection.
Infection with LCMV-Cl13 is associated with severe clinical symptoms, persistent viral infection, and tissue pathology. To assess the effect of CD8+ Treg cell-dependent suppression on chronic LCMV infection, we compared the clinical course of disease, tissue pathology, and viral titers of B6-WT and B6-DK mice after LCMV-Cl13 infection. Body weight measurements showed no difference between B6-WT and B6-DK mice during the early phase of infection (Fig. 4A). Around day 10, however, B6-DK mice began to gain weight and show signs of recovery, whereas weight loss in WT mice continued until day 20 (Fig. 4A). A more comprehensive approach to evaluate the clinical disease status depended on illness scores. B6-DK mice showed reduced levels of disease intensity and an accelerated recovery and were free of clinical symptoms by day 23, whereas WT mice remained moderately ill and did not fully recover (Fig. 4B).
Fig. 4.

B6-DK mice develop enhanced antiviral response during chronic infection. (A) B6-WT (○) and B6-DK (●) mice were infected i.v. with 1 × 106 pfu LCMV clone 13 on day 0 or not infected (WT control [□] and DK control [■]). Body weight was measured daily before (day 0) and after infection. Percentage of original body weight is shown (n > 4 per group). (B) Illness scores of B6-WT (○) and B6-DK (●) mice after infection with LCMV-Cl13 are shown. Scores were evaluated as described in Methods (n > 4/group). (C) Histopathology of liver and lung from B6-WT and B6-DK mice on day 55 after LCMV clone 13 infection is shown. (Right) Leukocyte infiltration of liver and lung per field was enumerated and compared between B6-WT and B6-DK mice. (D) During chronic LCMV infection, virus titers in B6-WT (○) and B6-DK (●) mice at the indicated times after infection with LCMV-Cl13 were assessed by standard plaque assay. Course of viral titer is shown in pfu in kidney, lung, and serum.
A similar pattern was observed for viral titers and tissue pathology. Both groups showed severe tissue pathology in lung and liver in the early stage of chronic disease with highly active pneumonitis and hepatitis. However, by day 55 postinfection, lung and liver showed no sign of pathological changes in B6-DK mice, whereas WT mice displayed immune cell infiltration and histopathology (Fig. 4C). Consistent with these histological findings at late stages of chronic infection, significant reduction of viral titers was seen in organs of B6-DK mice compared with B6-WT mice (Fig. 4D).
Qa-1-Restricted Inhibition of CD8+ Effector Cells by CD8+ Treg Cells Underlies the Attenuated Antiviral Response.
Next we tested whether the enhanced CD8+ T-cell response reflected direct suppression of CD8+ effector cells by CD8+ Treg cells. CD45.1+Ly49+CD8+ Treg cells or CD45.1+Ly49−CD8+ T cells were adoptively transferred along with naive CD45.2+CD62L+CD44– B6-WT or B6-DK CD8+ T cells into Rag2−/− hosts. One day later, Rag2−/− hosts were infected with LCMV-Arm, and 5 d later, the CD45.1–CD45.2+CD8+ T-cell response was measured. Reduced CD8+ T-cell responses were observed after cotransfer of Ly49+CD8+ Treg cells and WT CD8+ target cells, but not B6-DK CD8 effector cells (Fig. 5A).
Fig. 5.
Qa-1–restricted inhibition of CD8+ effector cells by CD8+ Treg cells underlies the attenuated antiviral response. (A) CD45.1+Ly49+CD8+ Treg cells and CD45.1+Ly49–CD8+ T cells (as controls), as well as B6-WT and B6-DK CD8+CD44–CD62L+-naive T cells, were highly purified via FACS sorting and 4 groups of Rag2−/− hosts were transferred with CD45.1+Ly49+CD8+ Treg cells and WT CD8+CD44–CD62L+-naive T cells, CD45.1+Ly49+CD8+ Treg cells and B6-DK CD8+CD44–CD62L+-naive T cells, CD45.1+Ly49−CD8+ T cells and B6-WT CD8+CD44–CD62L+-naive T cells, or CD45.1+Ly49–CD8+ T cells and B6-DK CD8+CD44–CD62L+-naive T cells. After cell transfer mice were infected with LCMV-Arm i.p. (2 × 105 pfu). On day 5, CD45.1– splenic mononuclear cells were assessed for antiviral immune response. Shown are total cell numbers of CD45.1–CD3+CD8+CD62L–CD44+CD127–KLRG1+ SLECs (n = 3 per group). (B) In vivo LCMV-Arm activated Thy1.1+Ly49+CD122+CD8+ Treg cells were cotransferred with in vivo activated and Celltrace Violet-labeled B6-WT CD45.1+P14+CD8+ T cells and Celltrace Violet-labeled B6-DK CD45.2+P14+CD8+ T cells into the same Rag2−/− host at a 2:1:1 ratio. Twelve hours after transfer, host mice were immunized i.v. with 0.5 μg gp33-peptide/mouse. (Left) Seventy-two hours after immunization, proliferation of LCMV-specific B6-WT (gray shading) and B6-DK (solid line) CD8+ T cells were evaluated by Celltrace Violet dilution. (Right) Percentage of B6-WT and B6-DK LCMV-specific CD8+ T cells that proliferated for >6 generations is shown (n = 3 per group).
We then examined the effect of CD8+ Treg cells on virus-specific CD8+ T cells, using transgenic mice that express a TCR specific for LCMV viral epitope gp33 associated with H-2Db (P14 mice) (22). CD45.1+CD8+ T cells from wild-type P14 mice were cotransferred with CD45.2+CD8+ T cells from B6-DK P14 mice, along with gp33 peptide, into Rag2−/− hosts along with Thy1.1+Ly49+CD8+ Treg cells. Comparison of B6-WT and B6-DK P14 CD8+ T-cell numbers and dye dilution revealed that B6-DK LCMV-specific CD8+ T cells underwent strong proliferation detected by Celltrace Violet dilution, whereas the level of proliferation by B6-WT P14 CD8+ cells was significantly reduced (Fig. 5B). These data indicate that Qa-1-restricted inhibition of LCMV-specific responses by CD8+ cells reflects, at least in part, direct targeting of LCMV-specific CD8+ cells by CD8+ Treg cells.
Discussion
We report here that the antiviral immune response after acute and chronic LCMV infection is enhanced by genetic disruption of Qa-1-restricted CD8+ Treg cell activity. Enhanced immune responses were apparent from increased effector CD8+ T-cell responses, reduced clinical disease, more effective virus control, and reduced tissue inflammation. These findings expand the regulatory reach of CD8+ Treg cells to the control of host antiviral immune responses.
B6-DK mice, which harbor a Qa-1 point mutation that disrupts recognition of Qa-1+ target cells by Qa-1-restricted CD8+ Treg cells, displayed a substantial increase in the effector CD8+ T-cell response to LCMV infection associated with reduced virus load and diminished tissue inflammation. Over the course of chronic infection, enhanced CD8+ T-cell responses of B6-DK mice were accompanied by increased viral control, reduced tissue pathology, and diminished disease symptoms. Although persistent exposure to viral antigens during chronic infection can lead to exhaustion of effector CD8+ T cells, B6-DK mice displayed a CD8+ T-cell phenotype consistent with a memory response rather than T-cell exhaustion. Adoptive transfer experiments indicated that one target of Qa-1-restricted CD8+ Treg cells was virus-specific effector CD8+ T cells.
Experimental definition of regulatory mechanisms that may affect the outcome of inflammatory and infectious disease has gained substantial traction during the last several years. A major focus has been the potential contribution of CD4+C25+FoxP3+ regulatory T cells, which may play a role in controlling immune-mediated pathology and consequent tissue damage in the context of infection (23). In contrast, regulation of immune responses by CD4+CD25+ Treg cells can also lead to enhanced pathogen survival and persistence in some cases (24, 25) and can allow immune evasion (26). Understanding the mechanisms that underlie the protective and pathogenic effects of CD4+CD25+ Treg cells to different pathogens is incomplete. Although there are several studies of the effect of CD4+CD25+ Treg cells (14, 15), NK cells, and B cells in the context of LCMV infection (27–29), the contribution of CD8 Treg cells to host protection is not clear (17).
Studies of the contribution of Qa-1-restricted CD8+ Treg cells to this antiviral immune response indicate that recognition of Qa-1–peptide complexes on activated effector CD8+ T cells by CD8+ Treg cells allows targeting and suppression of virus-specific effector T-cells. Genetic disruption of this inhibitory interaction resulted in decreased tissue pathology and diminished levels of CD8+ T-cell exhaustion. The extent of clinical improvement after chronic infection secondary to genetic disruption of CD8+ Treg cell function was striking. It is likely that diminished clinical symptoms reflect an enhanced immune response that more effectively clears the virus. An adequate virus-specific CD8+ T-cell response is crucial for the control and clearance of acute infection, and ineffective antiviral CD8+ T-cell responses can lead to chronic infection (30). Genetic disruption of CD8+ Treg cell suppression resulted in an increased CD8+ T-cell response and reduced numbers of exhausted CD8+ T cells. Although the factors that govern the development of exhausted CD8+ T cells during chronic infection are not fully understood (31), it is likely that enhanced viral control by B6-DK mice promoted increased differentiation into memory, rather than exhausted CD8+ T cells, and enhanced protective immunity in the context of chronic viral infection. Further definition of the interaction between CD8+ Treg cells and Qa-1-expressing target cells may allow development of new approaches to enhancing antiviral immunity in the context of chronic infection.
Recent studies of simian immunodeficiency virus (SIV) infection of nonhuman primates report that HLA-E-restricted CD8+ T cells directly target SIV-infected CD4 cells and strongly inhibit SIV infection (32). Definition of the human homolog of murine Qa-1-restricted CD8+ Treg cells represents a critical step toward a mechanistic understanding of clinical viral infections. Identification of the Ly49 receptor as a key marker of murine CD8+ Treg cells suggests that killer Ig receptors (KIRs), the functional homologs of Ly49, may help identify human HLA-E restricted CD8+ Treg cells (17, 33) and lead to more direct approaches to defining the contribution of CD8+ Treg cells to the human antiviral immune response.
Definition of specific KIR subtypes expressed by human CD8 cells that display HLA-E-dependent inhibitory activity may allow development of KIR-specific Abs that interrupt CD8+ Treg cell-mediated suppression in the setting of established disease. This approach may allow therapeutic intervention during the course of disease, rather than simply a potential preventative measure. Because HLA-E (Qa-1) is up-regulated by activated effector cells, antibody blockade of HLA-E (Qa-1)-restricted suppression by CD8 Treg cells may allow rescue of effector cells and enhanced antiviral activity during the course of a chronic infection. In contrast to other therapeutic approaches, such as immune checkpoint blockade and CD4+ Treg cell depletion, selective enhancement of antiviral effector cells after interruption of CD8+ Treg cell-mediated suppression may avoid nonspecific activation that can lead to an autoimmune response.
In sum, these findings extend previous analyses suggesting that Qa-1-restricted CD8+ Treg cells regulate a wide range of immune responses to include chronic viral infections. Additional analysis of this inhibitory interaction may yield new approaches to regulate antiviral immunity.
Methods
Animals.
C57BL/6 and Thy1.1+ mice (B6.PL-Thy1a/CyJ) were purchased from Jackson Laboratory, and Rag2−/− (B6.129S6-Rag2tm1Fwa N12) mice were purchased from Taconic Farms. B6.Qa-1-D227K (B6-DK) mice were generated as described previously (19). P14 mice [B6.Cg-Tcratm1Mom Tg(TcrLCMV)327Sdz; Taconic Farms] were crossed with B6.Qa-1-D227K mice and with CD45.1+ mice (B6.SJL-Ptprca-BoyAiTac; Taconic Farms). Mice were housed in a specific pathogen-free animal facility at the Dana-Farber Cancer Institute. All experiments were done in compliance with federal laws and institutional guidelines and were approved by the Dana-Farber Cancer Institute Animal Care and Use Committee.
Viral Infection.
Age- and sex-matched animals were infected intraperitoneally with 2 × 105 pfu LCMV-Arm (acute) or i.v. with 1 × 106 pfu LCMV clone 13 (chronic). LCMV clone 13 and Arm were propagated on BHK cells, as previously described (34). After infection, mice were weighed daily, and body weight was calculated as a percentage of initial weight on day 0. Illness scores were measured by giving 1 point for each of the following conditions: ruffled fur, weight loss exceeding 15%, visually lethargic, lethargic to the touch, and a hunched back (35). Virus titers were assessed by plaque assay on Vero cells (36) after lysing whole organs, using a Qiagen TissueLyser II according to the manufacturer’s instructions.
Flow Cytometry.
For surface phenotype analyses by flow cytometry (Fortessa), the following antibodies were used: anti-CD3 (145-2C11), anti-CD8 (53-6.7), anti-CD44 (IM7), anti-CD62L (MEL-14), anti-KLRG1 (2F1), anti-CD127 (A7R34), anti-CD122 (TM-b1), anti-Ly49 (14B11), anti-Qa-1 (6A8.6F10.1A6), gp33 H-2Db Tetramer (KAVYNEFATC), anti-LAG-3 (eBioC9B7W), anti-NKG2D (CX5), anti-PD-1 (RMP1-30), anti-2B4 (2B4), anti-CD45.1 (A20), anti-CD45.2 (104), anti-Thy1.1 (HIS51), and anti-Thy1.2 (53-2.1).
For intracellular staining, splenocytes were incubated in RPMI medium (supplemented with FCS, penicillin-streptomycin, sodium pyruvate, Hepes buffer, and l-glutamine) for 3 h with either 1 μg/mL glycoprotein 33–41 (GP33–41) peptide or 50 ng/mL phorbol 12-myristate 13-acetate (PMA) + 500 ng/mL ionomycin in the presence of 3 μg/mL brefeldin and 2 μM monensin (BD Biosciences). For degranulation assays, anti-CD107a antibody was also present during stimulation. After incubation, viability, surface, and intracellular staining were performed using a BD cytofix/cytoperm kit, according to manufacturer’s instructions, using the following antibodies: anti-granzyme-B (GB11), anti-IFN-γ (XMG1.2), anti-CD107a (1D4B), anti-Eomes (Dan11mag), Celltrace Violet, and fixable viability dye.
Histology.
At individual points, mice were killed and organs were immediately fixed in Bouin’s solution or snap frozen in O.C.T. Compound (Tissue-Tek) before sectioning and staining. More than 5 sections of each sample were analyzed by microscopy, and cell numbers were counted per field.
Adoptive Transfer.
Ten days after in vivo stimulation with Keyhole limpet hemocyanin (KLH)-Complete Freund's Adjuvant (CFA) i.p., splenocytes from CD45.1+ WT mice were isolated, CD8+ T cells were enriched using magnetic beads, and subsequently, Ly49+CD122+CD3+CD8+ Treg cells and Ly49–CD122+CD3+CD8+ T cells were stained as described and sorted using a BD Aria I. CD8+ T cells from naive B6-WT and B6-DK mice were enriched using BDiMAG, stained for CD3+, CD8+, CD44+, and CD62L+, and naive (CD3+CD8+CD44–CD62L+) T cells from B6-WT and B6-DK mice were sorted as described earlier. Next, 5 × 105 Ly49+CD8+ Treg cells or Ly49–CD8+ control effector cells were cotransferred with either B6-WT or B6-DK naive target CD8+ T cells into Rag2−/− hosts. Twelve hours later, mice were infected with 2 × 105 pfu LCMV-Arm, and on day 5, cell numbers of CD45.1– CD8+ T cells were enumerated.
For analysis of suppression of LCMV-specific CD8+ T cells, Thy1.1 mice were infected with 5 × 105 pfu LCMV-Arm. On day 8, CD8+ Treg cells were isolated as described earlier and cotransferred into Rag2−/− hosts with Celltrace Violet-labeled activated B6-WT CD45.1+CD8+ P14 cells and Celltrace Violet-labeled activated B6-DK CD45.2+CD8+ P14 cells. Twelve hours later, mice were immunized with 0.5 μg/mouse gp33–peptide; proliferation in Thy1.1– cells was assessed on day 3.
Statistical Analyses.
Statistical analyses were performed using Student’s t test or Mann–Whitney test for comparison of two conditions. Error bars denote mean ± SD. A P value of <0.05 was considered to be statistically significant (*P < 0.05; **P < 0.005; ***P < 0.0005).
Supplementary Material
Acknowledgments
We thank U. von Andrian for LCMV Armstrong and clone 13; R. Bronson (Dana-Farber/Harvard Cancer Center Rodent Histopathology Core) for histology analysis; J. Daley, S. Lazo-Kallanian, and R. Smith for cytometry support; and A. Angel for manuscript and figure preparation. This work was supported in part by National Institutes of Health Research Grant (AI 037562) and a gift from the LeRoy Schecter Research Foundation (to H.C.), the Alexander von Humboldt Foundation (SKA2010; to P.A.L.), and National Research Service Award Fellowship (T32CA070083; to H.-J.K.).
Footnotes
The authors declare no conflict of interest.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1320999110/-/DCSupplemental.
References
- 1.Rehermann B. Pathogenesis of chronic viral hepatitis: Differential roles of T cells and NK cells. Nat Med. 2013;19(7):859–868. doi: 10.1038/nm.3251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Jenner E. On the Origin of the Vaccine Inoculation. London, UK: Shury; 1801. [PMC free article] [PubMed] [Google Scholar]
- 3.Rouse BT, Lukacher AE. Some unmet challenges in the immunology of viral infections. Discov Med. 2010;10(53):363–370. [PMC free article] [PubMed] [Google Scholar]
- 4.Jost S, Altfeld M. Control of human viral infections by natural killer cells. Annu Rev Immunol. 2013;31:163–194. doi: 10.1146/annurev-immunol-032712-100001. [DOI] [PubMed] [Google Scholar]
- 5.Terilli RR, Cox AL. Immunity and hepatitis C: A review. Curr HIV/AIDS Rep. 2013;10(1):51–58. doi: 10.1007/s11904-012-0146-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zhou X, Ramachandran S, Mann M, Popkin DL. Role of lymphocytic choriomeningitis virus (LCMV) in understanding viral immunology: Past, present and future. Viruses. 2012;4(11):2650–2669. doi: 10.3390/v4112650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Armstrong C, Lillie RD. Experimental Lymphocytic Choriomeningitis of Monkeys and Mice Produced by a Virus Encountered in Studies of the 1933 St. Louis Encephalitis Epidemic. Public Health Reports (1896-1970) 1934;49(35):1019–1027. [Google Scholar]
- 8.Buchmeier MJ, Welsh RM, Dutko FJ, Oldstone MB. The virology and immunobiology of lymphocytic choriomeningitis virus infection. Adv Immunol. 1980;30:275–331. doi: 10.1016/s0065-2776(08)60197-2. [DOI] [PubMed] [Google Scholar]
- 9.Lang PA, et al. Aggravation of viral hepatitis by platelet-derived serotonin. Nat Med. 2008;14(7):756–761. doi: 10.1038/nm1780. [DOI] [PubMed] [Google Scholar]
- 10.Matloubian M, Somasundaram T, Kolhekar SR, Selvakumar R, Ahmed R. Genetic basis of viral persistence: Single amino acid change in the viral glycoprotein affects ability of lymphocytic choriomeningitis virus to persist in adult mice. J Exp Med. 1990;172(4):1043–1048. doi: 10.1084/jem.172.4.1043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wherry EJ. T cell exhaustion. Nat Immunol. 2011;12(6):492–499. doi: 10.1038/ni.2035. [DOI] [PubMed] [Google Scholar]
- 12.Moskophidis D, Lechner F, Pircher H, Zinkernagel RM. Virus persistence in acutely infected immunocompetent mice by exhaustion of antiviral cytotoxic effector T cells. Nature. 1993;362(6422):758–761. doi: 10.1038/362758a0. [DOI] [PubMed] [Google Scholar]
- 13.Belkaid Y, Tarbell K. Regulatory T cells in the control of host-microorganism interactions (*) Annu Rev Immunol. 2009;27:551–589. doi: 10.1146/annurev.immunol.021908.132723. [DOI] [PubMed] [Google Scholar]
- 14.Punkosdy GA, et al. Regulatory T-cell expansion during chronic viral infection is dependent on endogenous retroviral superantigens. Proc Natl Acad Sci USA. 2011;108(9):3677–3682. doi: 10.1073/pnas.1100213108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lund JM, Hsing L, Pham TT, Rudensky AY. Coordination of early protective immunity to viral infection by regulatory T cells. Science. 2008;320(5880):1220–1224. doi: 10.1126/science.1155209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Schmitz I, et al. IL-21 restricts virus-driven Treg cell expansion in chronic LCMV infection. PLoS Pathog. 2013;9(5):e1003362. doi: 10.1371/journal.ppat.1003362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kim HJ, et al. CD8+ T regulatory cells express the Ly49 Class I MHC receptor and are defective in autoimmune prone B6-Yaa mice. Proc Natl Acad Sci USA. 2011;108(5):2010–2015. doi: 10.1073/pnas.1018974108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kim HJ, Verbinnen B, Tang X, Lu L, Cantor H. Inhibition of follicular T-helper cells by CD8(+) regulatory T cells is essential for self tolerance. Nature. 2010;467(7313):328–332. doi: 10.1038/nature09370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lu L, Kim HJ, Werneck MB, Cantor H. Regulation of CD8+ regulatory T cells: Interruption of the NKG2A-Qa-1 interaction allows robust suppressive activity and resolution of autoimmune disease. Proc Natl Acad Sci USA. 2008;105(49):19420–19425. doi: 10.1073/pnas.0810383105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lu L, et al. Regulation of activated CD4+ T cells by NK cells via the Qa-1-NKG2A inhibitory pathway. Immunity. 2007;26(5):593–604. doi: 10.1016/j.immuni.2007.03.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Richter K, Agnellini P, Oxenius A. On the role of the inhibitory receptor LAG-3 in acute and chronic LCMV infection. Int Immunol. 2010;22(1):13–23. doi: 10.1093/intimm/dxp107. [DOI] [PubMed] [Google Scholar]
- 22.Pircher H, Bürki K, Lang R, Hengartner H, Zinkernagel RM. Tolerance induction in double specific T-cell receptor transgenic mice varies with antigen. Nature. 1989;342(6249):559–561. doi: 10.1038/342559a0. [DOI] [PubMed] [Google Scholar]
- 23.Suvas S, Azkur AK, Kim BS, Kumaraguru U, Rouse BT. CD4+CD25+ regulatory T cells control the severity of viral immunoinflammatory lesions. J Immunol. 2004;172(7):4123–4132. doi: 10.4049/jimmunol.172.7.4123. [DOI] [PubMed] [Google Scholar]
- 24.Taylor MD, et al. Removal of regulatory T cell activity reverses hyporesponsiveness and leads to filarial parasite clearance in vivo. J Immunol. 2005;174(8):4924–4933. doi: 10.4049/jimmunol.174.8.4924. [DOI] [PubMed] [Google Scholar]
- 25.Belkaid Y, Piccirillo CA, Mendez S, Shevach EM, Sacks DL. CD4+CD25+ regulatory T cells control Leishmania major persistence and immunity. Nature. 2002;420(6915):502–507. doi: 10.1038/nature01152. [DOI] [PubMed] [Google Scholar]
- 26.Suffia I, Reckling SK, Salay G, Belkaid Y. A role for CD103 in the retention of CD4+CD25+ Treg and control of Leishmania major infection. J Immunol. 2005;174(9):5444–5455. doi: 10.4049/jimmunol.174.9.5444. [DOI] [PubMed] [Google Scholar]
- 27.Majlessi L, Lo-Man R, Leclerc C. Regulatory B and T cells in infections. Microbes Infect. 2008;10(9):1030–1035. doi: 10.1016/j.micinf.2008.07.017. [DOI] [PubMed] [Google Scholar]
- 28.Waggoner SN, Cornberg M, Selin LK, Welsh RM. Natural killer cells act as rheostats modulating antiviral T cells. Nature. 2012;481(7381):394–398. doi: 10.1038/nature10624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lang PA, et al. Natural killer cell activation enhances immune pathology and promotes chronic infection by limiting CD8+ T-cell immunity. Proc Natl Acad Sci USA. 2012;109(4):1210–1215. doi: 10.1073/pnas.1118834109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Missale G, et al. Different clinical behaviors of acute hepatitis C virus infection are associated with different vigor of the anti-viral cell-mediated immune response. J Clin Invest. 1996;98(3):706–714. doi: 10.1172/JCI118842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Utzschneider DT, et al. T cells maintain an exhausted phenotype after antigen withdrawal and population reexpansion. Nat Immunol. 2013;14(6):603–610. doi: 10.1038/ni.2606. [DOI] [PubMed] [Google Scholar]
- 32.Lu W, et al. Induction of CD8+ regulatory T cells protects macaques against SIV challenge. Cell Rep. 2012;2(6):1736–1746. doi: 10.1016/j.celrep.2012.11.016. [DOI] [PubMed] [Google Scholar]
- 33.Schenkel AR, Kingry LC, Slayden RA. The ly49 gene family. A brief guide to the nomenclature, genetics, and role in intracellular infection. Front Immunol. 2013;4:90. doi: 10.3389/fimmu.2013.00090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. von Herrath M, Whitton JL (2001) Animal models using lymphocytic choriomeningitis virus. Curr Protocols Immunol 36:19.10.1–19.10.19. [DOI] [PubMed]
- 35.Stamm A, Valentine L, Potts R, Premenko-Lanier M. An intermediate dose of LCMV clone 13 causes prolonged morbidity that is maintained by CD4+ T cells. Virology. 2012;425(2):122–132. doi: 10.1016/j.virol.2012.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ahmed R, Salmi A, Butler LD, Chiller JM, Oldstone MB. Selection of genetic variants of lymphocytic choriomeningitis virus in spleens of persistently infected mice. Role in suppression of cytotoxic T lymphocyte response and viral persistence. J Exp Med. 1984;160(2):521–540. doi: 10.1084/jem.160.2.521. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.