

Significance
An atomic layer deposition (ALD) procedure is described for stabilizing surface binding of a water oxidation catalyst to the surfaces of nanostructured films of indium tin oxide. The catalyst is stabilized on the surface of electrodes by ALD of an overlayer of TiO2. Stabilization of surface binding allows use of basic solutions where a rate enhancement for water oxidation of ∼106 is observed compared with acidic conditions. There are important implications for stabilizing surface-bound molecular assemblies for applications in dye sensitized solar cells, electrocatalysis, and photoelectrocatalysis.
Keywords: electrocatalysis, surface stabilization
Abstract
Enhancing the surface binding stability of chromophores, catalysts, and chromophore–catalyst assemblies attached to metal oxide surfaces is an important element in furthering the development of dye sensitized solar cells, photoelectrosynthesis cells, and interfacial molecular catalysis. Phosphonate-derivatized catalysts and molecular assemblies provide a basis for sustained water oxidation on these surfaces in acidic solution but are unstable toward hydrolysis and loss from surfaces as the pH is increased. Here, we report enhanced surface binding stability of a phosphonate-derivatized water oxidation catalyst over a wide pH range (1–12) by atomic layer deposition of an overlayer of TiO2. Increased stability of surface binding, and the reactivity of the bound catalyst, provides a hybrid approach to heterogeneous catalysis combining the advantages of systematic modifications possible by chemical synthesis with heterogeneous reactivity. For the surface-stabilized catalyst, greatly enhanced rates of water oxidation are observed upon addition of buffer bases  and with a pathway identified in which O-atom transfer to OH− occurs with a rate constant increase of 106 compared to water oxidation in acid.
and with a pathway identified in which O-atom transfer to OH− occurs with a rate constant increase of 106 compared to water oxidation in acid.
Heterogeneous catalysis plays an important role in industrial chemical processing, fuel reforming, and energy-producing reactions. Examples include the Haber–Bosch process, steam reforming, Ziegler–Natta polymerization, and hydrocarbon cracking (1–8). Research in heterogeneous catalysis continues to flourish (9–15) but iterative design and modification are restricted by limitations in materials preparation and experimental access to surface mechanisms. By contrast, synthetic modification of molecular catalysts is possible by readily available routes; a variety of experimental techniques is available for monitoring rates and mechanism in solution for the investigation of homogeneous catalysis (16–23). Transferring this knowledge and the reactivity of homogeneous molecular catalysts to a surface could open the door to heterogeneous applications in fuel cells, dye sensitized photoelectrochemical cells, and multiphase industrial reactions.
Procedures are available for immobilization of organometallic and coordination complexes on the surfaces of solid supports. Common strategies include surface derivatization of metal oxides by carboxylate, phosphonate, and siloxane bindings (24–27), carbon-grafted electrodes (28–30), and electropolymerization (31–33). These approaches provide a useful bridge to the interface and a way to translate mechanistic understanding and ease of synthetic modification of solution catalysts to heterogeneous applications with a promise of higher reactivity under milder conditions.
A significant barrier to this approach arises from the limited stability of surface binding. Surface-bound carboxylates are typically unstable to hydrolysis in water, whereas phosphonates are unstable in neutral or basic solutions (27, 34). For water oxidation catalysis this is particularly detrimental given the accelerated rates that are accessible for catalytic water oxidation as the pH is increased due to the intervention of base-catalyzed pathways with concerted atom–proton transfer accompanying O—O bond formation (35).
We report here the results of a designed strategy for the systematic surface stabilization of molecular catalysts on solid oxide surfaces. In the strategy we use indium tin oxide (ITO) electrodes and first bind a phosphonate-derivatized molecular water oxidation catalyst to the surface of the electrode. The derivatized electrode is then coated with a conformal nanoscale TiO2 overlayer applied by layer-by-layer atomic layer deposition (ALD). The overlayer of TiO2 acts to block hydrolysis of the phosphonate groups from the surface, Fig. 1. ALD stabilization has been used previously to demonstrate significantly enhanced photostability of surface-bound chromophores in acidic and neutral solutions (36–38). In this article we apply the ALD stabilization procedure to surface stabilization of a known water oxidation catalyst and show remarkably enhanced surface binding stability even in basic solutions. Retention of electrocatalytic reactivity on the surface is demonstrated and water oxidation catalysis investigated over a wide pH range. Clear evidence is found in these studies that added proton acceptor bases enhance the kinetic pathways in the key, rate-limiting step (O—O bond formation) via an atom–proton transfer (APT) mechanism (22, 35). In addition, a facile pathway has been identified with direct attack by OH− on an activated oxo form of the catalyst with rate enhancements of up to 106 for water oxidation.
Fig. 1.

Schematic representation of the ALD overlayer protection strategy for a catalyst surface-attached to nanoITO protected by TiO2. (A) Illustrating the electrochemical device architecture showing the surface derivatized electrode and water oxidation. (B) Underivatized electrodes exposed to basic aqueous conditions showing detachment of the catalysts from the electrode surface. (C) ALD protection of surface attachment even basic aqueous conditions. This figure was adapted from ref. 38.
Results
Single-site, phosphonic acid-derivatized (-PO3H2) polypyridyl complexes of ruthenium have been shown to act as water oxidation electrocatalysts on planar ITO and FTO (fluorine-doped SnO2) electrodes (24, 25, 39) and in mesocopic, nanostructured films of ITO (nanoITO) (40). The structure of the surface-bound catalyst [Ru(Mebimpy)(4,4′-((HO)2OPCH2)2bpy)(OH2)]2+ (Mebimpy is 2,6-bis(1-methylbenzimidazol-2yl); 4,4′-((HO)2OPCH2)2bpy is 4,4′-bis-methlylenephosphonato-2,2′-bipyridine), nanoITO-Ru-OH22+, and the mechanism of interfacial water oxidation are shown in Scheme 1 (25).
The catalytic cycle in Fig. 2 is initiated by accumulating multiple oxidative equivalents at the catalyst through stepwise proton coupled electron transfer (PCET) reactions,  , followed by 1e− oxidation of RuIV = O2+ to RuV(O)3+ which occurs at E1/2 ∼ 1.6 V vs. the normal hydrogen electrode (NHE). The RuV(O)3+ intermediate is reactive toward O—O bond formation with the initial product being a hydroperoxide intermediate, RuIII-OOH2+. This O—O bond forming step is rate determining in acid solution with k = 0.009 s−1 (22). Oxidation of RuIII-OOH2+ to the peroxide RuIV(OO)2+ is followed by slow O2 evolution. Further oxidation of the RuIV peroxide to RuV(OO)3+, which is accessible under oxidative conditions where RuV = O3+ can be generated, increases the lability of O2, leading to reentry into the catalytic cycle through O2 loss and regeneration of RuIII-OH2+ (22, 25, 41).
, followed by 1e− oxidation of RuIV = O2+ to RuV(O)3+ which occurs at E1/2 ∼ 1.6 V vs. the normal hydrogen electrode (NHE). The RuV(O)3+ intermediate is reactive toward O—O bond formation with the initial product being a hydroperoxide intermediate, RuIII-OOH2+. This O—O bond forming step is rate determining in acid solution with k = 0.009 s−1 (22). Oxidation of RuIII-OOH2+ to the peroxide RuIV(OO)2+ is followed by slow O2 evolution. Further oxidation of the RuIV peroxide to RuV(OO)3+, which is accessible under oxidative conditions where RuV = O3+ can be generated, increases the lability of O2, leading to reentry into the catalytic cycle through O2 loss and regeneration of RuIII-OH2+ (22, 25, 41).
Fig. 2.

Water oxidation mechanism for nanoITO-Ru-OH22+.
In acidic conditions, the rate-limiting step for this catalytic process is O—O bond formation, 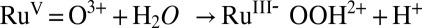 . Based on kinetic isotope effects (35) and quantum mechanics/molecular mechanics (QM/MM)-minimal free-energy path method calculations (42), O—O bond formation occurs in concert with solvation of the released proton by a neighboring water molecule or water cluster (35). However, water is a poor proton acceptor base (pKa = −1.74 for
. Based on kinetic isotope effects (35) and quantum mechanics/molecular mechanics (QM/MM)-minimal free-energy path method calculations (42), O—O bond formation occurs in concert with solvation of the released proton by a neighboring water molecule or water cluster (35). However, water is a poor proton acceptor base (pKa = −1.74 for  ) and catalysis is enhanced with the added proton acceptor bases
) and catalysis is enhanced with the added proton acceptor bases 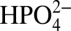 and
and  . These proton acceptor bases activate the concerted APT pathway shown in Eq. 1 in which O—O bond formation occurs in concert with proton loss to the added base (35). Attempts to exploit APT pathways for phosphonate-derivatized catalysts on oxide and semiconductor electrodes have been greatly limited by hydrolytic loss of the catalyst from the surface of the electrode as the pH is increased above 5 with added coordinating buffer base anions such as acetate and
. These proton acceptor bases activate the concerted APT pathway shown in Eq. 1 in which O—O bond formation occurs in concert with proton loss to the added base (35). Attempts to exploit APT pathways for phosphonate-derivatized catalysts on oxide and semiconductor electrodes have been greatly limited by hydrolytic loss of the catalyst from the surface of the electrode as the pH is increased above 5 with added coordinating buffer base anions such as acetate and 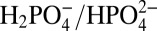 (27, 34, 43).
(27, 34, 43).
Stabilization by ALD.
We reported earlier on the utilization of Al2O3 ALD layers to stabilize surface binding of the chromophore [Ru(bpy)2(4,4′-(PO3H2)bpy)]2+on TiO2 surfaces (36). ALD is a self-limiting thin-film deposition technique in which reactive vapor phase precursors are sequentially exposed to a substrate surface. Through controlled surface reactions and sequential precursor delivery, ALD permits exquisite control over layer thickness and conformality even over complex 3D nanoarchitectures (44), and is useful in a variety of electrochemical system applications (45). Here, we use TiCl4/H2O precursor chemistry to deposit TiO2 films with a thickness control of 0.5 Å per ALD cycle.
The TiO2 overlayer has no effect on E1/2 values or on the pH dependences for the surface-bound Ru(III/II) couple. As shown by cyclic voltammetry (CV) measurements, E1/2 values are consistent with previously reported values and the pH dependence of the surface-bound RuIII/II varied as expected with the surface pKa values of 1.8 for  and 11.5 for
and 11.5 for 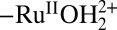 (24, 25, 41). Distortions observed in the waveforms for the
(24, 25, 41). Distortions observed in the waveforms for the 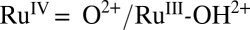 couple are consistent with prior observations and are caused by kinetic inhibitions arising from PCET effects (46).
couple are consistent with prior observations and are caused by kinetic inhibitions arising from PCET effects (46).
 |
Stabilization Toward Redox Cycling.
Repetitive CV scans through the RuIII/II redox couple were used to evaluate the surface stabilization effect of the ALD overlayers (Fig. S1) (47). Surface coverage values (Γ in mol/cm2) were determined by integration of the current-potential waveform for the RuIII/II redox couple and by use of Eq. 2 with QCV the integrated charge from the current-potential waveform, n (=1) the number of electrons transferred for the redox couple, F the Faraday constant, and A the surface area of the electrode (39).
Fig. 3A shows a plot of Γ versus number of CV scans for 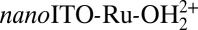 at pH 7. With no overlayer protection, >60% of the catalyst is desorbed from the electrode surface after 50 CV scans from 0 to 1.2 V vs. NHE at a scan rate of 20 mVs−1. With 10 ALD cycles of TiO2 (∼5.0 Å), only 20% of the surface-bound catalyst is lost. Stability is further increased at 20 ALD cycles (10 Å) where only ∼10% of the catalyst is lost after 50 CVs. Above 30 ALD cycles (≥15 Å), the RuIII/II wave is difficult to detect, which is attributed to the 12–14-Å diameter catalyst being “buried” beneath the ALD overlayer. Thus, all subsequent experiments are carried out at an ALD thickness of ∼10 Å (20 ALD cycles).
at pH 7. With no overlayer protection, >60% of the catalyst is desorbed from the electrode surface after 50 CV scans from 0 to 1.2 V vs. NHE at a scan rate of 20 mVs−1. With 10 ALD cycles of TiO2 (∼5.0 Å), only 20% of the surface-bound catalyst is lost. Stability is further increased at 20 ALD cycles (10 Å) where only ∼10% of the catalyst is lost after 50 CVs. Above 30 ALD cycles (≥15 Å), the RuIII/II wave is difficult to detect, which is attributed to the 12–14-Å diameter catalyst being “buried” beneath the ALD overlayer. Thus, all subsequent experiments are carried out at an ALD thickness of ∼10 Å (20 ALD cycles).
Fig. 3.

(A) Variation in fractional surface coverage Γ/Γo with number of CV scans for ALD(TiO2)-protected 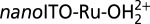 as a function of ALD TiO2 cycles.
as a function of ALD TiO2 cycles. 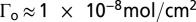 , ALD cycles as labeled on the figure at pH = 7.1, I = 0.1 M,
, ALD cycles as labeled on the figure at pH = 7.1, I = 0.1 M,  , total I adjusted to 0.25 M with added LiClO4, T = 23 °C, 20 mV/s. (B) Variation of surface coverage with number of CV scans for ALD(TiO2)-protected
, total I adjusted to 0.25 M with added LiClO4, T = 23 °C, 20 mV/s. (B) Variation of surface coverage with number of CV scans for ALD(TiO2)-protected 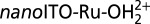 as a function of pH. Γo ∼ 1 × 10−8 mol/cm2; number of ALD cycles and pH as labeled on the figure. pH = 7.1, I = 0.1 M,
as a function of pH. Γo ∼ 1 × 10−8 mol/cm2; number of ALD cycles and pH as labeled on the figure. pH = 7.1, I = 0.1 M,  ; pH = 9.0, I = 0.1 M, B(OH)3/B(OH)2O−; pH = 11.1, I = 0.1 M
; pH = 9.0, I = 0.1 M, B(OH)3/B(OH)2O−; pH = 11.1, I = 0.1 M 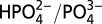 . Total I adjusted to 0.25 M with added LiClO4, T = 23 °C, 20 mV/s.
. Total I adjusted to 0.25 M with added LiClO4, T = 23 °C, 20 mV/s.
Fig. 3B shows that ALD stabilization is effective over a wide pH range even in basic solutions. 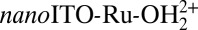 coated with 10 Å of TiO2 (20 cycles) exhibited similar stability in solutions from pH 7 to pH 11. Under these conditions, the catalyst is nearly completely lost from the electrode surface at pH 11 without ALD stabilization. Surface binding stability and catalyst complex stability are separate issues. Both surface-binding and catalyst complex stability are observed during CV redox cycling through the wave for the RuIII/RuII couple at all pH values. However, at pH ≥11, a more rapid loss of current response for the catalyst is observed with application of the high positive potentials required for water oxidation. The loss of catalytic activity appears to be due to ligand decomposition, consistent with known reactivity in solution (48, 49), and not to surface detachment due to hydrolysis.
coated with 10 Å of TiO2 (20 cycles) exhibited similar stability in solutions from pH 7 to pH 11. Under these conditions, the catalyst is nearly completely lost from the electrode surface at pH 11 without ALD stabilization. Surface binding stability and catalyst complex stability are separate issues. Both surface-binding and catalyst complex stability are observed during CV redox cycling through the wave for the RuIII/RuII couple at all pH values. However, at pH ≥11, a more rapid loss of current response for the catalyst is observed with application of the high positive potentials required for water oxidation. The loss of catalytic activity appears to be due to ligand decomposition, consistent with known reactivity in solution (48, 49), and not to surface detachment due to hydrolysis.
Water Oxidation Catalysis.
With a stabilization protocol in place, we investigated water oxidation catalysis by surface-stabilized 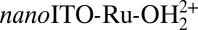 (TiO2) (1-ALD). Following Scheme 1, water oxidation in acidic solution is triggered by oxidation of
(TiO2) (1-ALD). Following Scheme 1, water oxidation in acidic solution is triggered by oxidation of  to
to 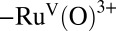 at E ∼ 1.6 V in 0.1 M HNO3 (22). The results of a controlled potential electrolysis experiment for 1-ALD at 1.40 V vs. NHE at pH = 7.1, I = 0.25 M,
at E ∼ 1.6 V in 0.1 M HNO3 (22). The results of a controlled potential electrolysis experiment for 1-ALD at 1.40 V vs. NHE at pH = 7.1, I = 0.25 M, 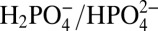 ,
, 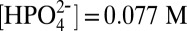 are shown in Fig. S2. Gas chromatography analysis of the head space in the electrolysis cell, Fig. S3, revealed that O2 was produced with a 74% Faradaic efficiency over a period of 1,000 s with a turnover number of 260 moles of O2 per moles of catalyst, which corresponds to a turnover frequency of 0.26 turnovers per catalyst/s. This value is 2 orders of magnitude greater than rates reported at pH 1 at a constant overpotential of 0.58 V (22). The rate enhancement is consistent with the appearance of the concerted APT mechanism in Eq. 1 with
are shown in Fig. S2. Gas chromatography analysis of the head space in the electrolysis cell, Fig. S3, revealed that O2 was produced with a 74% Faradaic efficiency over a period of 1,000 s with a turnover number of 260 moles of O2 per moles of catalyst, which corresponds to a turnover frequency of 0.26 turnovers per catalyst/s. This value is 2 orders of magnitude greater than rates reported at pH 1 at a constant overpotential of 0.58 V (22). The rate enhancement is consistent with the appearance of the concerted APT mechanism in Eq. 1 with 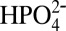 as the proton acceptor base.
as the proton acceptor base.
Contributions from APT depend both on the concentration of buffer base and the pKa of the conjugate acid. At a fixed pH of 7.1 and fixed 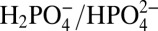 ratio (pKa = 7.2), the catalytic current icat increases linearly with
ratio (pKa = 7.2), the catalytic current icat increases linearly with  (Fig. S4). This observation is consistent with the relationship between icat and kobs in Eq. 3 where kobs = kH2O + kB . In this expression, kH2O is the catalytic rate constant in the absence of buffer base, kB is the first-order rate constant for the APT pathway, and V is the volume of the nanoITO electrode. A similar buffer base effect, but with accelerated kinetics compared with pH 7, was observed at pH 9 with B(OH)2O− as the buffer base [pKa = 9.2 for B(OH)3] (Fig. S5).
(Fig. S4). This observation is consistent with the relationship between icat and kobs in Eq. 3 where kobs = kH2O + kB . In this expression, kH2O is the catalytic rate constant in the absence of buffer base, kB is the first-order rate constant for the APT pathway, and V is the volume of the nanoITO electrode. A similar buffer base effect, but with accelerated kinetics compared with pH 7, was observed at pH 9 with B(OH)2O− as the buffer base [pKa = 9.2 for B(OH)3] (Fig. S5).
Fig. 4 shows CVs for 1-ALD illustrating a decrease in the onset potential for water oxidation of 0.25 V from pH = 7.1 with 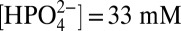 to pH = 11.1 with
to pH = 11.1 with  . The pH-dependent waves in the CVs from 0.3 to 0.6 V are due to the -
. The pH-dependent waves in the CVs from 0.3 to 0.6 V are due to the -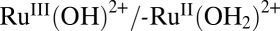 coupled with E1/2 = 0.58 V at pH = 7.1, 0.46 V at pH = 9, and 0.33 at pH = 11.1. The shifts in the E1/2 values with pH follow the expected Nernstian response for this PCET couple and agree with previously reported potentials for the complex in solution (41).
coupled with E1/2 = 0.58 V at pH = 7.1, 0.46 V at pH = 9, and 0.33 at pH = 11.1. The shifts in the E1/2 values with pH follow the expected Nernstian response for this PCET couple and agree with previously reported potentials for the complex in solution (41).
Fig. 4.

CVs for 1-ALD at 23 °C and 20 mV/s in I = 0.1 M of the added buffers at pH values indicated in figure. The dashed line is the background current at pH 11.1 from a nanoITO electrode with 20 ALD cycles of TiO2 and no catalyst loading.
By pH 11, the concentration of OH− is no longer negligible and a dependence on [OH−] appears in the rate of water oxidation. The dependence on [OH−] was investigated between pH 11 and 12.5 by varying the buffer ratio  at constant
at constant  at fixed ionic strength. A Tafel analysis, similar to the recently developed foot of the wave analysis by Savéant and coworkers (50), was used to analyze the data. In this procedure, catalytic currents at the potential for the –RuV(O)3+/–RuIV = O2+ couple in Scheme 1 (1.6 V) are determined by extrapolation to minimize complications from the electrochemical background.
at fixed ionic strength. A Tafel analysis, similar to the recently developed foot of the wave analysis by Savéant and coworkers (50), was used to analyze the data. In this procedure, catalytic currents at the potential for the –RuV(O)3+/–RuIV = O2+ couple in Scheme 1 (1.6 V) are determined by extrapolation to minimize complications from the electrochemical background.
A plot of icat vs. [OH−] is shown in Fig. 5. The first-order dependence on [OH−] is consistent with direct attack on the reactive intermediate –RuV(O)3+by OH− to give the hydroperoxide intermediate as shown in Eq. 4. At the highest concentration of OH− investigated, 0.013 M (pH = 12.1), the catalytic current enhancement compared with acidic solution (pH = 1) is ∼106 with an increase in kobs from ∼10−2 ⋅s−1 at pH 1 to ∼104 s−1 at pH 12 at an overpotential of 1.02 V.
Fig. 5.

Variation of icat normalized for surface coverage with [OH−] in 0.25 M LiClO4 with 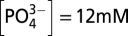 . The icat values at 1.6 V were obtained by Tafel extrapolation; see text and Fig. S6 for details.
. The icat values at 1.6 V were obtained by Tafel extrapolation; see text and Fig. S6 for details.
Discussion
We demonstrate here surface stabilization and enhanced performance toward water oxidation catalysis by a surface-attached molecular catalyst. There are important findings in these results: 1) the retention of properties, including reactivity, on the surface; 2) a high degree of stability both toward surface binding and water oxidation reactivity at higher pH values; 3) a general basis for synthesizing tailored surfaces combining features of molecular and heterogeneous catalysis.
Our results have clear implications for utilization of the ALD overlayer strategy for stabilization of molecular catalysts and assemblies on oxide electrode and semiconductor surfaces for possible applications in catalysis, electrocatalysis, and photoelectrochemistry.
Methods
All aqueous solutions were prepared with Milli-Q ultrapure water (>18 MΩ ITO glass, Rs = 4–8 Ω/G) was purchased from Delta Technologies. nanoITO powder (40 nm diameter) was obtained from Lihochem. Optically transparent, electrically conductive, high surface area nanoITO films were prepared as described previously (40). The light blue nanoITO films were prepared with an average thickness of 5 μm with a resistance of ∼200 Ω across a 1-cm section of the film. Synthesis of 1 has been previously reported (41). Stable phosphonate surface binding of 1 on nanoITO electrodes occurred following immersion of the films in solutions containing 0.1 mM catalyst in methanol. Electrodes were immersed for >12 h in the catalyst solution, and immediately upon removal were rinsed with Milli-Q water and loaded into the ALD reactor chamber.
ALD was conducted in a home-built, hot-walled, flow tube reactor. The main reaction chamber is a 24-inch-long, 4-inch inner diameter stainless steel tube. Precursors were delivered into the reaction zone through a manifold constructed from 1/4-inch stainless steel tubing. Nitrogen carrier gas (99.999% purity, National Welders) was metered through a mass flow controller at 300 standard cubic centimeters per minute and exhausted through a rotary vane pump. Precursor gases were pulsed into the reactor using three-way pneumatically actuated diaphragm valves controlled electronically by a LabVIEW sequencer. Gate valves were positioned on either side of the reaction zone to conduct hold steps that permit precursor infiltration into nanoITO structure. For TiO2 deposition, 99% pure TiCl4 (Strem Chemicals) is used as the metal precursor and reagent-grade water (Ricca Chemicals) is used as the oxidant. Standard ALD coating conditions were 120 °C and 2 Torr of N2 carrier gas with a sequence of 0.3-s metal precursor dose, 60-s hold, 180-s N2 purge, 0.3-s H2O dose, 60-s hold, 180-s N2 purge. A 120 °C deposition temperature was selected to improve water desorption during purge steps. Silicon monitor wafers included during deposition on nanoITO electrodes confirmed the expected 0.5 Å per cycle deposition rate for each run.
Electrochemical measurements were performed with a model 601D electrochemical workstation from CH Instruments. The three-electrode system consisted of a nanoITO film on a glass slide working electrode (roughly 0.5 cm2 area), a Pt wire auxiliary electrode, and a saturated calomel electrode reference. The potential of the reference electrode was adjusted by 0.24 V for the reported potentials versus NHE. GC analyses were performed on a custom-made Varian 450-GC 220-MS spectrometer with capillary column.
Supplementary Material
Acknowledgments
Funding by the Center for Catalytic Hydrocarbon Functionalization, an Energy Frontier Research Center (EFRC) funded by the US Department of Energy (DOE), Office of Science, Office of Basic Energy Sciences, under Award DE-SC0001298 supporting A.K.V., and the UNC EFRC: Center for Solar Fuels, an EFRC funded by the US DOE, Office of Science, Office of Basic Energy Sciences, under Award DE-SC0001011 supporting L.A. and J.J.C., is gratefully acknowledged. M.D.L. was supported by the Research Triangle Solar Fuels Institute. B.K. was supported by the National Science Foundation under Award CBET-1034374.
Footnotes
The authors declare no conflict of interest.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1319832110/-/DCSupplemental.
References
- 1.van Santen RA, Leurock M. Molecular Heterogeneous Catalysis: A Conceptual and Computational Approach. New York: Wiley; 2006. [Google Scholar]
- 2.Ertl G. Molecules at surfaces: 100 years of physical chemistry in Berlin-Dahlem. Angew Chem Int Ed Engl. 2013;52(1):52–60. doi: 10.1002/anie.201205401. [DOI] [PubMed] [Google Scholar]
- 3. Thomas JM (2012) The societal significance of catalysis and the growing practical importance of single-site heterogeneous catalysts. Proc R Soc A 468(2143):1884–1903.
- 4.Somorjai GA. Molecular concepts of heterogeneous catalysis. J Mol Struct THEOCHEM. 1998;424(1-2):101–117. [Google Scholar]
- 5.Somorjai GA, Kliewer CJ. Reaction selectivity in heterogeneous catalysis: An invited review. React Kinet Catal Lett. 2009;96(2):191–208. [Google Scholar]
- 6.Somorjai GA, Li Y-M. Major successes of theory-and-experiment-combined studies in surface chemistry and heterogeneous catalysis. Top Catal. 2010;53(5-6):311–325. [Google Scholar]
- 7.Baiker A. Supercritical fluids in heterogeneous catalysis. Chem Rev. 1999;99(2):453–474. doi: 10.1021/cr970090z. [DOI] [PubMed] [Google Scholar]
- 8.Hutchings GJ. Heterogeneous catalysts-discovery and design. J Mater Chem. 2009;19(9):1222–1235. [Google Scholar]
- 9.Gambardella AA, Feldberg SW, Murray RW. Electron transfer dynamics of iridium oxide nanoparticles attached to electrodes by self-assembled monolayers. J Am Chem Soc. 2012;134(13):5774–5777. doi: 10.1021/ja301212r. [DOI] [PubMed] [Google Scholar]
- 10.Katz MJ, et al. Towards solar fuels: Water splitting with sunlight and “rust”? Coord Chem Rev. 2012;256(21-22):2521–2529. [Google Scholar]
- 11.Mallouk TE. Water electrolysis: Divide and conquer. Nat Chem. 2013;5(5):362–363. doi: 10.1038/nchem.1634. [DOI] [PubMed] [Google Scholar]
- 12.Nakagawa T, Bjorge NS, Murray RW. Electrogenerated IrO(x) nanoparticles as dissolved redox catalysts for water oxidation. J Am Chem Soc. 2009;131(43):15578–15579. doi: 10.1021/ja9063298. [DOI] [PubMed] [Google Scholar]
- 13.Standridge SD, Schatz GC, Hupp JT. Distance dependence of plasmon-enhanced photocurrent in dye-sensitized solar cells. J Am Chem Soc. 2009;131(24):8407–8409. doi: 10.1021/ja9022072. [DOI] [PubMed] [Google Scholar]
- 14.Swierk JR, Mallouk TE. Design and development of photoanodes for water-splitting dye-sensitized photoelectrochemical cells. Chem Soc Rev. 2013;42(6):2357–2387. doi: 10.1039/c2cs35246j. [DOI] [PubMed] [Google Scholar]
- 15.Chen H, Blaber MG, Standridge S, DeMarco EJ, Hupp JT. Computational modeling of plasmon-enhanced light absorption in a multicomponent dye sensitized solar cell. J Phys Chem C. 2012;116(18):10215–10221. [Google Scholar]
- 16.Cornils B, Herrmann WA. Concepts in homogeneous catalysis: The industrial view. J Catal. 2003;216(1–2):23–31. [Google Scholar]
- 17.Cokoja M, Bruckmeier C, Rieger B, Herrmann WA, Kühn FE. Transformation of carbon dioxide with homogeneous transition-metal catalysts: A molecular solution to a global challenge? Angew Chem Int Ed Engl. 2011;50(37):8510–8537. doi: 10.1002/anie.201102010. [DOI] [PubMed] [Google Scholar]
- 18.Loch JA, Crabtree RH. Rapid screening and combinatorial methods in homogeneous organometallic catalysis. Pure Appl Chem. 2001;73(1):119–128. [Google Scholar]
- 19.Duan L, Tong L, Xu Y, Sun L. Visible light-driven water oxidation—from molecular catalysts to photoelectrochemical cells. Energy Environ. Sci. 2011;4(9):3296–3313. [Google Scholar]
- 20.Duan L, et al. A molecular ruthenium catalyst with water-oxidation activity comparable to that of photosystem II. Nat Chem. 2012;4(5):418–423. doi: 10.1038/nchem.1301. [DOI] [PubMed] [Google Scholar]
- 21.Dau H, et al. The mechanism of water oxidation: From electrolysis via homogeneous to biological catalysis. ChemCatChem. 2010;2:724–761. [Google Scholar]
- 22.Concepcion JJ, Tsai MK, Muckerman JT, Meyer TJ. Mechanism of water oxidation by single-site ruthenium complex catalysts. J Am Chem Soc. 2010;132(5):1545–1557. doi: 10.1021/ja904906v. [DOI] [PubMed] [Google Scholar]
- 23.Liu X, Wang F. Transition metal complexes that catalyze oxygen formation from water: 1979–2010. Coord Chem Rev. 2012;256(11–12):1115–1136. [Google Scholar]
- 24.Chen Z, Concepcion JJ, Hull JF, Hoertz PG, Meyer TJ. Catalytic water oxidation on derivatized nanoITO. Dalton Trans. 2010;39(30):6950–6952. doi: 10.1039/c0dt00362j. [DOI] [PubMed] [Google Scholar]
- 25.Chen Z, Concepcion JJ, Jurss JW, Meyer TJ. Single-site, catalytic water oxidation on oxide surfaces. J Am Chem Soc. 2009;131(43):15580–15581. doi: 10.1021/ja906391w. [DOI] [PubMed] [Google Scholar]
- 26.Concepcion JJ, Jurss JW, Hoertz PG, Meyer TJ. Catalytic and surface-electrocatalytic water oxidation by redox mediator-catalyst assemblies. Angew Chem Int Ed Engl. 2009;48(50):9473–9476. doi: 10.1002/anie.200901279. [DOI] [PubMed] [Google Scholar]
- 27.Brown DG, Schauer PA, Borau-Garcia J, Fancy BR, Berlinguette CP. Stabilization of ruthenium sensitizers to TiO2 surfaces through cooperative anchoring groups. J Am Chem Soc. 2013;135(5):1692–1695. doi: 10.1021/ja310965h. [DOI] [PubMed] [Google Scholar]
- 28.deKrafft KE, et al. Electrochemical water oxidation with carbon-grafted iridium complexes. ACS Appl Mater Interfaces. 2012;4(2):608–613. doi: 10.1021/am2018095. [DOI] [PubMed] [Google Scholar]
- 29.Allongue P, et al. Covalent modification of carbon surfaces by aryl radicals generated from the electrochemical reduction of diazonium salts. J Am Chem Soc. 1997;119(1):201–207. [Google Scholar]
- 30.Engstrom RC, Strasser VA. Characterization of electrochemically pretreated glassy carbon electrodes. Anal Chem. 1984;56(2):136–141. [Google Scholar]
- 31.Abruna HD. Coordination chemistry in two dimensions: Chemically modified electrodes. Coord Chem Rev. 1988;86:245–271. [Google Scholar]
- 32.Lapides AM, et al. Stabilization of a ruthenium(II) polypyridyl dye on nanocrystalline tio2 by an electropolymerized overlayer. J Am Chem Soc. 2013;135(41):15450–15458. doi: 10.1021/ja4055977. [DOI] [PubMed] [Google Scholar]
- 33.Friebe C, Hager MD, Winter A, Schubert US. Metal-containing polymers via electropolymerization. Adv Mater. 2011;24(3):332–345. doi: 10.1002/adma.201103420. [DOI] [PubMed] [Google Scholar]
- 34.Hanson K, et al. Photostability of phosphonate-derivatized, Ru(II) polypyridyl complexes on metal oxide surfaces. ACS Appl Mater Interfaces. 2012;4(3):1462–1469. doi: 10.1021/am201717x. [DOI] [PubMed] [Google Scholar]
- 35.Chen Z, et al. Concerted O atom-proton transfer in the O-O bond forming step in water oxidation. Proc Natl Acad Sci USA. 2010;107(16):7225–7229. doi: 10.1073/pnas.1001132107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hanson K, et al. Stabilization of [Ru(bpy)2(4,4′-(PO3H2)bpy)]2+ on mesoporous TiO2 with atomic layer deposition of Al2O3. Chem Mater. 2013;25:3–5. [Google Scholar]
- 37.Son HJ, et al. Dye stabilization and enhanced photoelectrode wettability in water-based dye-sensitized solar cells through post-assembly atomic layer deposition of TiO2. J Am Chem Soc. 2013;135(31):11529–11532. doi: 10.1021/ja406538a. [DOI] [PubMed] [Google Scholar]
- 38.Losego MD, Hanson K. Stabilizing molecular sensitizers in aqueous environs. Nano Energy. 2013;2:1067–1069. [Google Scholar]
- 39.Vannucci AK, et al. Water oxidation intermediates applied to catalysis: Benzyl alcohol oxidation. J Am Chem Soc. 2012;134(9):3972–3975. doi: 10.1021/ja210718u. [DOI] [PubMed] [Google Scholar]
- 40.Hoertz PG, Chen Z, Kent CA, Meyer TJ. Application of high surface area tin-doped indium oxide nanoparticle films as transparent conducting electrodes. Inorg Chem. 2010;49(18):8179–8181. doi: 10.1021/ic100719r. [DOI] [PubMed] [Google Scholar]
- 41.Concepcion JJ, et al. Catalytic water oxidation by single-site ruthenium catalysts. Inorg Chem. 2010;49(4):1277–1279. doi: 10.1021/ic901437e. [DOI] [PubMed] [Google Scholar]
- 42.Hu H, Lu ZY, Parks JM, Burger SK, Yang WT. Quantum mechanics/molecular mechanic minimum free-energy path. J Chem Phys. 2008;128:034105. doi: 10.1063/1.2816557. [DOI] [PubMed] [Google Scholar]
- 43.Hanson K, et al. Structure-property relationships in phosphonate-derivatized RuII polypyridyl dyes on metal oxide surfaces in an aqueous environment. J Phys Chem C. 2012;116(28):14837–14847. [Google Scholar]
- 44.Parsons GN, George SM, Knez M. Progress and future directions for atomic layer deposition and ALD-based chemistry. MRS Bull. 2011;36:865–871. [Google Scholar]
- 45. Peng Q, Lewis JS, Hoertz PG, Glass JT, Parsons GN (2012) Atomic layer deposition for electrochemical energy generation and storage systems. J Vac Sci Technol, A 30(1):010803–010814.
- 46.Chen Z, Vannucci AK, Concepcion JJ, Jurss JW, Meyer TJ. Proton-coupled electron transfer at modified electrodes by multiple pathways. Proc Natl Acad Sci USA. 2011;108(52):E1461–E1469. doi: 10.1073/pnas.1115769108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Song W, et al. Photoinduced stepwise oxidative activation of a chromophore-catalyst assembly on TiO2. J. Phys. Chem. Lett. 2011;2:1808–1813. [Google Scholar]
- 48.Roecker L, et al. Instability of the oxidation catalysts ([(bpy)2(py)Ru(O)]2+) and oxo(1,10-phenanthroline)(2,2′,2”-terpyridine) ruthenium(2+) ([(trpy)(phen)Ru(O)]2+) in basic solution. Inorg Chem. 1985;24(23):3784–3791. [Google Scholar]
- 49.Ghosh PK, Brunschwig BS, Chou M, Creutz C, Sutin NJ. Thermal and light-induced reduction of the ruthenium complex cation Ru(bpy)33+ in aqueous solution. J Am Chem Soc. 1984;106:4772–4783. [Google Scholar]
- 50.Costentin C, Drouet S, Robert M, Savéant J-M. Turnover numbers, turnover frequencies, and overpotential in molecular catalysis of electrochemical reactions. Cyclic voltammetry and preparative-scale electrolysis. J Am Chem Soc. 2012;134(27):11235–11242. doi: 10.1021/ja303560c. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.