

Significance
Template-dependent RNA and DNA polymerization is a vital reaction in the cell and is believed to occur exclusively in the forward direction (5′-3′), which poses significant challenges to the cell in, for example, lagging strand synthesis. Although cells are mostly limited to unidirectional polymerization, we find that reverse polymerization is structurally and chemically possible utilizing the same structural core, the conserved palm domain of canonical polymerases. The structure of a unique reverse nucleotide polymerase-tRNA complex revealed that the direction of polymerization is determined by the orientation of approach of the polynucleotide substrate. Phylogenetic analysis indicates that reverse nucleotide polymerization is a primordial activity of the polymerase family.
Keywords: Thg1-tRNA complex, crystal structure, tRNA editing
Abstract
Nucleotide polymerization proceeds in the forward (5′-3′) direction. This tenet of the central dogma of molecular biology is found in diverse processes including transcription, reverse transcription, DNA replication, and even in lagging strand synthesis where reverse polymerization (3′-5′) would present a “simpler” solution. Interestingly, reverse (3′-5′) nucleotide addition is catalyzed by the tRNA maturation enzyme tRNAHis guanylyltransferase, a structural homolog of canonical forward polymerases. We present a Candida albicans tRNAHis guanylyltransferase-tRNAHis complex structure that reveals the structural basis of reverse polymerization. The directionality of nucleotide polymerization is determined by the orientation of approach of the nucleotide substrate. The tRNA substrate enters the enzyme’s active site from the opposite direction (180° flip) compared with similar nucleotide substrates of canonical 5′-3′ polymerases, and the finger domains are on opposing sides of the core palm domain. Structural, biochemical, and phylogenetic data indicate that reverse polymerization appeared early in evolution and resembles a mirror image of the forward process.
Exclusive forward 5′-3′ elongation by DNA replication poses severe challenges to the cell. Shortening or “aging” of linear chromosomes leads to cellular senescence, which is linked to many aging-related diseases (1). Sophisticated mechanisms are found in the cell to compensate for the absence of a reverse (3′-5′) nucleotide polymerase. The multisubunit ribonucleoprotein telomerase is used to prevent shortening of chromosomes by adding DNA sequence repeats and telomeres to hinder the loss of coding DNA regions from chromosomes (2). Likewise, lagging strand synthesis involves the elaborate Okazaki fragment mechanism (3), where reverse polymerization could provide a simpler mechanism.
Despite the obvious advantages of bidirectional polymerization, it has been assumed that reverse (3′-5′) elongation was not maintained or possibly never evolved (1). Although no processive reverse polymerase has been identified, reverse nucleotide addition to the 5′-end of RNA is essential for tRNAHis maturation. Eukaryotic tRNAHis guanylyltransferase (Thg1) adds a single guanylate residue (G−1) to the 5′-end of pre-tRNAHis. G−1 is the key identity element that allows histidyl-tRNA synthetase (HisRS) to differentiate tRNAHis from the complex pool of tRNAs present in the cell (4–6). This essential identity element is encoded in the pre-tRNA genes of most bacteria and archaea and is retained during tRNA processing by unusual RNase P cleavage (7, 8). In eukaryotes, however, G−1 is not encoded in the tRNAHis gene and must be added posttranscriptionally as part of the tRNA maturation process to ensure His-tRNAHis formation and accurate decoding of all His codons in the cell.
Although enzymatic guanylylation activity has been known for 3 decades (9, 10), the respective enzyme, Thg1, was only identified in yeast more recently (11, 12). In eukaryotes, addition of G−1 to the 5′-end of pre-tRNAHis requires ATP-dependent activation of the tRNA substrate, followed by guanylylation and subsequent dephosphorylation (13) to yield mature 5′ monophosphorylated-tRNAHis (Fig. S1). Eukaryotic Thg1 specifically recognizes the anticodon of tRNAHis (14) and relies on ATP for an initial tRNA activation step, whereas archaeal-type Thg1 homologs perform the activation step with both GTP and ATP and generally appear to be less stringent in tRNA recognition (15, 16). Although the bona fide function of Thg1 in eukaryotes is undoubtedly the addition of G−1 to the 5′-end of tRNAHis, archaeal-type counterparts display additional reverse polymerase capacities in vivo and may also function as tRNA repair or editing enzymes (17, 18).
Despite detailed biochemical and structural data (11, 12, 14–16, 19–23) accumulated to date, it remains unclear why Thg1 catalyzes reverse polymerization whereas structurally similar homologs in the polymerase family are capable of only 5′-3′ polymerization. The Thg1-tRNAHis complex structure presented here reveals the molecular basis of reverse polymerization and demonstrates that the directionality of polymerization is determined by the orientation of substrate binding.
Results
Reverse Polymerization Requires Reverse Substrate Orientation.
The crystal structure of the human Thg1 (HsThg1) apoenzyme (19) showed that the catalytic core of Thg1 shares structural homology with canonical forward nucleotide polymerases, such as T7 DNA polymerase (19, 24, 25). In lieu of a cocrystal structure of Thg1 with its substrate tRNA, however, the mechanism allowing the same enzymatic core to catalyze both forward and reverse polymerization is unclear. We here present the cocrystal structure of Candida albicans Thg1 (CaThg1) in complex with tRNA (Fig. 1). Like its forward polymerase relatives, each CaThg1 subunit can be described as a hand shape, consisting of a palm domain including the catalytic core (residues 1–137) and the finger domain (residues 138–268) (19, 26) (Fig. 2B). A comparison of Thg1-tRNA with the T7 DNA polymerase-DNA structure allows a direct assessment of differences between reverse and forward polymerases, which illuminates how the shared enzymatic core can catalyze polymerization in opposite directions. Strikingly, we found that the direction of substrate approach to the catalytic core correlates with the direction of polymerization (Fig. 1). In the forward T7 DNA polymerase–DNA complex (PDB ID 1T7P) (25), the 3′-end of the primer strand is situated near a magnesium ion (Mg2+A) to promote deprotonation of 3′OH, and the triphosphate of the incoming nucleotide is coordinated to Mg2+B, which facilitates release of pyrophosphate. The substrate DNA approaches the catalytic core from the direction of Mg2+A (Fig. 1A). In comparison, the tRNA substrate in the Thg1-tRNA complex approaches the catalytic core from the opposite direction (Fig. 1B). A structural alignment of the forward and reverse palm domains shows a clear reversal of substrate binding.
Fig. 1.

Reverse polymerization is a mirror image of forward polymerization. In forward and reverse polymerases, the approach of nucleic substrate binding is concurrent with the direction of polymerization. The catalytic core and the finger domain of the polymerases are shown as blue and cyan cartoon models, respectively. Stick models indicate specific base pairing and incoming nucleotides. Magnesium ions are shown as green spheres. (A) Direction of substrate binding and domain organization of T7 DNA polymerase as a forward polymerase. (B) The direction of substrate binding and domain organization of Thg1 as a reverse polymerase. The triphosphate moiety of the ppp-tRNA model was generated based on that of ATP in the CaThg1-ATP structure.
Fig. 2.

The crystal structure of the CaThg1-tRNA complex. Shown is the structural arrangement and domain organization of Thg1 in complex with tRNA. (A) The overall structure of the CaThg1-tRNA complex consists of a CaThg1 tetramer and two tRNA molecules (tRNA1 and tRNA2). The subunits of Thg1 are colored as follows: cyan, subunit A; orange, subunit B; green, subunit C; and magenta, subunit D. tRNA1 and tRNA2 are colored yellow and light yellow, respectively. (B) The domain organization of CaThg1. The palm domain (residues 1–137) and finger domain (residues 138–268) are colored in blue and cyan, respectively. (C) One tRNA molecule is recognized by cross-subunit interactions of three Thg1 molecules. The CaThg1 tetramer is displayed as a surface model. The 2mFo-DFc map for tRNA1 is colored in blue and is contoured at 1.0 σ. (D) Dual RNA-binding surface (α5, α6, and α7) of the finger domain. The helix bundle α5–α7 of subunit D with tRNA1 (magenta) was superposed onto that of subunit A (cyan). The binding of tRNA induced the conformational change of the finger domain.
Reflecting the opposing substrate orientation, the overall domain organization of forward and reverse polymerases can also be described as a mirror image. The finger domain of forward polymerases binds the template polynucleotide strand and forms the incoming nucleotide-binding site with the palm domain (27). The finger domain of Thg1 likewise interacts with the template strand and contributes to formation of the incoming nucleotide-binding site. Although the palm domains are highly similar between forward and reverse polymerases, the respective finger domains are on opposing sides of the palm domain, accommodating the reversed approach of the polynucleotide template and substrate (Fig. 1). The overall domain organization of T7 DNA polymerase excludes a reversed substrate binding, and Thg1 cannot accommodate a forward substrate.
Four Thg1 Molecules Coordinate Two tRNA Molecules by Cross-Subunit Interaction.
Previous studies and our data show that Thg1 catalyzes guanylylation of tRNAPhe with the His anticodon GUG (tRNAPheGUG) (16) (Fig. S2 A and B). The yield of in vitro transcripts for tRNAPheGUG was ∼10-fold higher than that of tRNAHisΔG−1. Therefore, initial large-scale crystallization screening of C. albicans Thg1 (CaThg1) in complex with tRNA was carried out with tRNAPheGUG. Crystals of both artificial CaThg1-tRNAPheGUG and native CaThg1-tRNAHisΔG−1 complexes were obtained under the same conditions. Both structures were determined at resolutions of 3.6 and 4.2 Å, respectively (Table S1). The two tRNA complex structures were almost identical with an rmsd of 0.29 Å (Cα atoms of CaThg1) and 1.37 Å (P atoms of tRNA), excluding the D- and variable-loops, which do not interact with Thg1 (Fig. S2 C and D). Throughout this paper the higher-resolution CaThg1–tRNAPheGUG complex structure will be used to describe the structural features. Within the asymmetric unit of the CaThg1–tRNA complex, four CaThg1 assemble as a dimer of dimers (AB and CD). Two tRNA molecules bind to a Thg1 tetramer in a parallel orientation (Fig. 2A and Fig. S3B). Gel filtration analysis and small-angle X-ray scattering (SAXS) confirm an estimated molar ratio of 4:2 of Thg1 and tRNA in solution (Figs. S3 and S4; Table S2; SI Text).
Each tRNA molecule is coordinated by three subunits of the tetramer (Fig. 2 A and C). The acceptor stem of tRNA1 is situated between subunits A and B. Both backbone structures of the acceptor stem and the TΨC arm are located near several polar residues on the rear surface of the catalytic core of subunit B (Fig. S2D). This indicates that the surface interaction is involved in the stabilization of tRNA binding in a manner analogous to the thumb domain in canonical forward polymerases (27). The guanylylation of tRNA1 occurs in subunit A and B, yet the anticodon loop is bound to subunit D, thus aiding in the correct positioning of the tRNA molecule (Fig. 2C). The CaThg1-tRNA structure revealed a dual function of the fingers domain in tRNA binding (Fig. 2D). The first RNA-binding surface, composed of α5, α6, and following loop (loop α6–α7), binds to the end of the acceptor stems’ sugar phosphate backbone. The second RNA-binding surface composed of α5, α7, and α8 forms base-specific interactions with the anticodon loop. The structural superposition of the CaThg1 subunits from the obtained structures revealed the flexibility of the finger domain, especially in the helix bundle formed by α5, α6, and α7.
Recognition of the Anticodon Leads to Correct Substrate Positioning.
Biochemical analysis showed that Thg1 specifically recognizes the anticodon of its cognate substrate tRNAHisΔG−1 (14) (Fig. S2A). Interestingly, binding of Thg1 to the tRNA leads to a major distortion of the anticodon loop (Fig. 3). In the native yeast tRNAPhe structure, anticodon loop nucleotides 34–38 form a continuous base stack with the anticodon stem (28). This stacking interaction is disrupted by binding to Thg1, resulting in a flip of anticodon bases G34, U35, and G36 out of the anticodon loop toward Thg1 (Fig. S5D). Our data show that all three anticodon bases are specifically recognized by Thg1.
Fig. 3.

CaThg1 specifically recognizes the three anticodon bases of tRNAHis. Anticodon bases G34 (A), U35 (B), and G36 (C) are tightly coordinated by the finger domain. Amino acid residues interacting with the substrate tRNA are displayed as stick models; dashed lines indicate hydrogen bonds. (D) Schematic representation of anticodon loop recognition by the finger domain. Blue and red arrows show hydrogen bond interactions between Thg1 and the anticodon loop mediated by the protein main chains and side chains, respectively. A cyan arrow indicates RNA–RNA interactions; parallel horizontal lines represent stacking interactions. (E) Mutational analysis of the interface region between the finger domain and the anticodon loop. The rate of guanylylation activity using [α-32P]GTP, wild-type CaThg1, and wild-type CatRNAHis∆G−1 is denoted as 100. The error bars show the SD of three independent experiments.
The first anticodon base, G34, is recognized by aromatic stacking of the purine ring between Phe194 and the guanine base of G37 (Fig. 3A and Fig. S6A). A tRNA G34C mutant cannot be guanylylated by Thg1, confirming a purine-specific recognition at position 34 (Fig. 3E). The importance of aromatic stacking is further emphasized by the fact that the Thg1 mutant Phe194Ala as well as a tRNA G37C mutation decreased the guanylylation activity to 10% of the wild type (Fig. 3E). The Phe194Tyr mutation, which confers the aromatic stacking interaction, had little effect on the activity that was observed. Consistently, position 37 of eukaryotic tRNAHis is well conserved as a purine base (29). Anticodon base G34 forms a guanine-specific hydrogen to U35 (Fig. 3 A and B), and Thg1 activity toward a G34A mutant is negligible, demonstrating that Thg1 specifically recognizes G34 by aromatic stacking and hydrogen bond interactions (Fig. 3D).
Thg1 recognizes the second anticodon base, U35, via hydrogen bonds with Asn202 (Fig. 3B and Fig. S6 A and B). A purine base at position 35 is excluded by steric hindrance with the loop α7–α8. U35 is further stabilized by hydrogen bonds with Asn190 and G34, and corresponding mutations (Thg1 Asn190Ala and tRNA U35C) lead to decreased guanylylation (Fig. 3E). The highly conserved Asn200 is crucial for stabilization of the anticodon loop structure (Fig. 3D) and interacts with the phosphate and ribose group of U35. A Thg1 Asn200Asp mutation completely abolishes enzyme activity (Fig. 3E).
The third anticodon base, G36, is coordinated in a groove formed by α5 and α8 (Fig. 3C and Fig. S6B). Stacking interaction between His154 (Fig. S7), as part of the eukaryotic-specific sequence motif H154INNLY (30), and G36 facilitates specific recognition of the purine base, and a corresponding His154Ala mutation decreases guanylylation activity to 50% (Fig. 3E). G36 is further coordinated via hydrogen bonds with Lys209 and Lys210 (Fig. 3C). Consequently, mutants Lys209Ala and Lys209Gln showed a decrease to 15% in guanylylation activity and Lys209Glu is inactive (Fig. 3E). In summary, binding of the GUG anticodon by the finger domain of subunit D places the acceptor stem and thus the 5′-end of the tRNA in the catalytic pocket composed of subunits A and B, demonstrating why the His anticodon is essential for catalytic activity.
Coordination of the Acceptor Stem.
Binding of the anticodon appropriately places the acceptor stem to interact with the loop between α5 and α6 of the finger domain. In subunit A, the tRNA 5′-end is positioned in the catalytic pocket composed of subunits A and B (Fig. 4A and Fig. S6C). On the “template” side of the tRNA substrate, acceptor stem bases C75 and A76 were disordered in the structure. C74 is located between the N-terminal helix α1 of subunit B and the loop α5–α6 of the finger domain of subunit A. The main chain of the loop α5–α6 interacts with the backbone of the 3′ terminus of the tRNA via hydrogen bonds with the phosphate group of C74 and the ribose group of C72. tRNA deletion mutants lacking ACCA-3′ (tRNAHisΔG−1-ΔACCA) and CCA-3′ (tRNAHisΔG−1-ΔCCA) showed decreased guanylylation activity of 30%, whereas the deletion mutant CA-3′ (tRNAHisΔG−1-ΔCA) maintained wild-type activity levels (Fig. 4B). This data indicated that C75 and A76 are unlikely to participate in the catalytic reaction.
Fig. 4.
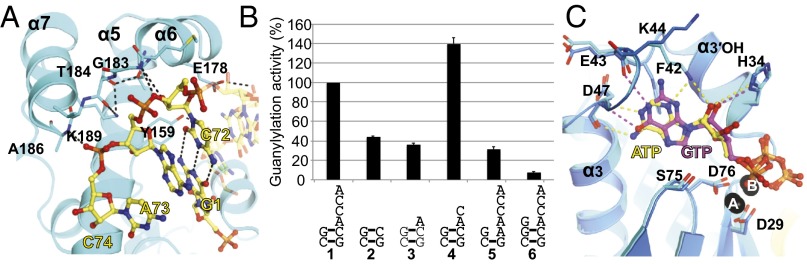
The finger domain of Thg1 tightly coordinates the 3′ RNA end. (A) Interactions between the finger domain of subunit A and the 3′-end of tRNA. The residues interacting with tRNA are shown as stick models; dashed lines indicate hydrogen bonds. (B) Guanylylation activity of 3′-end deleted variants of tRNAHis∆G−1 (lane 1); tRNAHis∆G−1-∆ACCA (lane 2), tRNAHis∆G−1-∆CCA (lane 3), tRNAHis∆G−1-∆CA (lane 4), tRNAHis∆G−1-C72A (lane 5), and tRNAHis with G−1 (lane 6). The rate of guanylylation activity using [α-32P]GTP, wild-type CaThg1, and wild-type tRNAHis∆G−1 is denoted as 100. The error bars show the SD of three independent experiments. (C) Superimposed ATP1 and GTP1 by alignment of Thg1. The yellow and magenta dashed lines indicate hydrogen bonds in the nucleotide-binding pocket with ATP and GTP, respectively.
On the “primer side” of the tRNA substrate, only weak interactions were observed between the 5′-end of tRNA and the long helix α5 of the finger domain (Fig. 4A). The highly conserved residues Tyr159 and Glu178 form hydrogen bonds with the backbone of tRNA bases G2 and G3, but no interactions were observed between the 5′-G+1 and the finger domain. The correct positioning of the primer for nucleotide addition is accomplished mainly through base pairing with the template strand and not by direct interaction of Thg1 with the primer strand. The tRNA mutant C72A, which is defective in Watson–Crick base pairing with the 5′-G+1 base, decreased guanylylation activity to the level of tRNAHisΔG−1-ΔACCA and tRNAHisΔG−1-ΔCCA (Fig. 4B). Taken together, these observations indicate that the interaction between the Thg1 and the 3′-end of tRNA and G1-C72 Watson–Crick base pairing are required to localize the 5′-G+1 of tRNA close to the catalytic core of the palm domain. Base A73, the template base for G−1 reverse polymerization, is placed within base-pairing distance of the incoming nucleotide.
Nucleotide Recognition for Adenylylation and Guanylylation.
In contrast to archaeal Thg1, which can use both ATP and GTP, eukaryotic Thg1 requires ATP for the activation step (23). To determine how eukaryotic Thg1 differentiates ATP from GTP, we solved the structures of CaThg1-ATP and CaThg1-GTP. In the ATP complex structure, two ATP molecules (ATP1 and ATP2) were observed at the catalytic pocket with clear electron densities as well as two GTP (GTP1 and GTP2) in the CaThg1-GTP structure (Fig. S8 A and B). Furthermore, in both structures, three magnesium ions (Mg2+A, Mg2+B, and Mg2+C) were observed at the same position previously identified by a manganese ion soaking in the HsThg1 structure (19). Superposition of CaThg1-GTP and CaThg1-ATP shows that the adenine base of ATP1 is more deeply embedded into the nucleotide-binding pocket than GTP1 and recognized by hydrogen bonds with the main chain of Asp47 (Fig. 4C). The base of GTP1 forms hydrogen bonds with the main-chain atoms of Glu43 and Asp47, which are also observed in the HsThg1-dGTP structure (19). Remarkably, unlike the CaThg1-GTP structure, electron density maps in the CaThg1-ATP structure clearly show that the side chain of Lys44 interacts directly with ATP1 (Fig. 4C and Fig. S8A). The mutant Lys44Ala of Saccharomyces cerevisiae Thg1 decreased the catalytic efficiency for the adenylylation step 2,000-fold (21).
Discussion
Thg1: A Mirror Image of Forward Polymerization.
In all known living organisms, nucleotide polymerization occurs exclusively in a 5′-3′ direction, adding 5′ nucleotide triphosphates to the 3′ hydroxyl group of the growing polynucleotide chain. Two divalent metal cations (Mg2+) facilitate the transfer of an electron pair from the free 3′ hydroxyl group to the α-phosphate of the incoming nucleotide. The required catalytic energy for this elongation reaction is derived from the hydrolysis of the triphosphate group of the incoming nucleotide. For reverse polymerization, the incoming nucleotide is added to the 3′-end of the polynucleotide substrate. In this reaction, the nucleotide addition to the 5′ monophosphate of the RNA requires an initial activation step, yielding a hydrolyzable di- or triphosphate. All following nucleotide additions can than be achieved by hydrolysis of the 5′ triphosphate of the previously added nucleotide. Thus, reverse polymerization requires an initial activation step, followed by chain elongation similar to forward polymerization.
In the reverse polymerase Thg1, the activation step is either an adenylylation reaction (eukaryotic Thg1) or, alternatively, a guanylylation reaction (archaeal Thg1) (16). In comparison with canonical forward polymerases, Thg1 contains an additional Mg2+ binding site, which accommodates the activation step. Mg2+A and Mg2+B participate in the initial activation step, whereas Mg2+C coordinates the incoming nucleotide-binding site for reverse polymerization. This additional nucleotide-binding site may allow template-dependent reverse polymerization without steric hindrance. With the means to accommodate the initial activation step, the structural components to adequately position the substrate polynucleotide in the reverse orientation, and the catalytic palm domain capable of carrying out extended polymerization, Thg1 contains all factors required to promote reverse nucleotide polymerization. Although most Thg1 variants seem limited to single-nucleotide addition, a few variants have been shown to promote an extended reverse polymerization (16, 18). These variants are candidates for future protein engineering toward a high-efficiency reverse polymerase, which will impact diverse areas of biotechnology and biochemical investigation including sequencing, 3′ UTR analysis, and 3′ DNA and RNA labeling.
Mechanistic Insights into Reverse Polymerization.
Following tRNA binding, reverse nucleotide addition of Thg1 requires three distinct catalytic steps: adenylylation, guanylylation, and dephosphorylation (13) (Fig. S1). By combining structural information from CaThg1-tRNA, -ATP, and -GTP complexes, we constructed the reaction model of the adenylylation and guanylylation steps (Fig. 5 and Fig. S9 A and B).
Fig. 5.
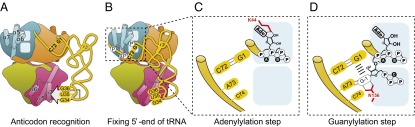
Proposed reaction model for Thg1. (A) The finger domain of Thg1 specifically recognizes the anticodon bases of tRNAHis∆G−1. (B) Next, the 5′-end of tRNAHis∆G−1 is indirectly coordinated through base pairing with the 3′-end of the acceptor stem, which interacts with the reverse side of the finger domain. (C) The tRNA 5′ phosphate group is coordinated by a magnesium ion (Mg2+A) and subsequently attacks the α-phosphate of ATP, which is activated by two magnesium ions. The hydrogen bonding between Lys44 and ATP is critical for the adenylylation of tRNAHis∆G−1. (D) The tRNA binding induces the coordination and deprotonation of 3′OH of GTP through Mg2+A. Amino acid residue Asn156 specifically recognizes the base moiety of GTP.
Preceding the adenylylation step, Thg1 binds to the substrate tRNAHis and differentiates substrate from nonsubstrate tRNA by recognition of the GUG anticodon (Fig. 5A). Previous biochemical data showed that the activating adenylylation of tRNA by Thg1 is strongly dependent on anticodon recognition; guanylylation using 5′-ppp-tRNA as the substrate, however, is not (22). Our data indicate that anticodon binding by the finger domain is crucial for correctly positioning the tRNA base 5′-G+1 into the catalytic pocket for adenylylation of the 5′-monophosphate group (Fig. 5B). Binding of the anticodon leads to a major distortion of the tRNA anticodon, which may provide the binding energy required for the activation reaction to occur.
The first catalytic step, adenylylation, is mechanistically very similar to the nucleotide addition carried out by forward polymerases (Fig. 5C). The CaThg1-ATP structure indicates that the invariant Lys44 recognizes the base and ribose of ATP. Among all forward polymerases, the two-metal-ion mechanism is conserved, with Mg2+A contributing to the positioning and deprotonation of the 3′OH of the primer strand (27). In the Thg1 reaction model, the 5′-phosphate moiety of the tRNA is located between Mg2+A and Mg2+B. Such coordination adequately positions the 5′-phosphate for attack on the ATP α-phosphate. Consistent with the reaction mechanisms deduced from the HsThg1 structure, the initial activation of tRNA is similar to the general polymerase mechanism (19).
The subsequent guanylylation is directed from the second nucleotide-binding site (ATP2/GTP2) (Fig. 5D). Although the triphosphate group of GTP is tightly coordinated between Mg2+C and conserved Arg92, Arg312, Lys10, and Lys95 (Fig. S7), the base moiety of GTP is not specifically recognized, so conceivably any nucleotide could be accommodated in the second nucleotide-binding site. This is the key finding for ongoing engineering efforts to develop a versatile reverse polymerase. We propose that, during the reaction, the 3′OH of GTP could be coordinated by Mg2+A and deprotonated to attack the activated 5′-end (triphosphate group) of the tRNA during reverse polymerization. Although the 3′OH of GTP is ∼7–10 Å from Mg2+A in the CaThg1-GTP structure, a slight rotation of GTP would allow the coordination of the GTP 3′OH to Mg2+A without steric hindrance (Fig. S9A). Interestingly, the base moiety of rotated GTP would accommodate base stacking with G+1. Base flipping of GTP would further result in the formation of hydrogen bonds of GTP with the conserved side chain Asn156 in the HIN156NLY motif (Fig. 5D and Fig. S9A). Although Bacillus thuringiensis Thg1 (BtThg1) does not feature the eukaryote-specific HINNLY motif, the structural arrangement is very similar (20), with Asn154 likely assuming the role of Asn156 in CaThg1.
Taken together, our reaction model indicates that tRNA binding and hydrogen bonding stabilize the conformational change of GTP, which allows the coordination of GTP 3′OH with Mg2+A for the subsequent nucleophilic attack of the activated tRNA, the same mechanisms used by canonical forward polymerases. The presented structure gives detailed information on substrate binding, but deciphering the complete reaction mechanism, including pyrophosphate removal, will require further structural analysis of Thg1 with nonhydrolyzable ATP, GTP, and tRNA.
Evolution of Forward and Reverse Polymerization.
A structure-based phylogeny of palm domain–containing enzymes reveals a “star” phylogeny, indicating that polymerases likely diverged very early in evolution into families with distinct functions (Fig. 6). The various palm-domain–containing proteins, including DNA and RNA polymerases, adenylyl cyclases, and reverse transcriptases, form separate phylogenetic clades that are as distantly related to each other as they are to Thg1. Thg1 is a separate group that is not recently derived from modern canonical polymerases. Thg1, or a reverse polymerase Thg1 ancestor, is thus an early invention in evolution. This is also reflected in the observation that the polymerase clades share little amino acid sequence identity. Although the palm domain core is structurally well-conserved, additional domains, such as the finger and thumb domain, were likely added to refine polymerase function including the direction of polymerization. Within most subgroups, proteins from archaea, bacteria, eukaryotes, and viruses can be found, indicating that the functions of the various families evolved before the split of these taxonomic domains (14). This agrees with the sequence-based phylogeny of Thg1 proteins, which showed that Thg1 groups in accordance with accepted taxonomy (16). It is therefore likely that a Thg1-like reverse polymerase existed in the last universal common ancestor. Indeed, an extended reverse polymerization function, as occurs in certain modern Thg1 variants (16, 18), evolved but was restricted to the tRNA maturation role, possibly because 5′-3′ polymerases developed selectively advantageous capabilities such as proofreading and effective processing. The Thg1-tRNA complex shows that reverse polymerization is a molecular mirror image of the more common 5′-3′ process, and it is conceivable that, given the ancient emergence of reverse polymerization, this activity evolved before the biological cell was fully committed to forward nucleotide polymerization for its most basic processes.
Fig. 6.

Structural phylogeny of palm domains from the reverse polymerase Thg1 and canonical forward polymerases. For the identification of palm domains, we used the classification scheme provided by the structural classification of the protein database. Structures were downloaded from the protein database and aligned using the STAMP algorithm as implemented in Visual Molecular Dynamics (VMD) (31). The palm domains display a classical star phylogeny, with palm domains clustering according to their enzymatic function. All structures within a group are labeled with the organism name. One representative structure for each group (organism underlined) is displayed and color-coded by structural conservation (from blue indicating highly conserved to red indicating little structural conservation).
Materials and Methods
The preparation of CaThg1, tRNA and their mutants, and the details of crystallization and structure determination is covered in SI Materials and Methods. Structures of the CaThg1-tRNA, CaThg1-ATP, and CaThg1-GTP complexes were solved by molecular replacement methods. Atomic coordinates and structure factors have been deposited in the Protein Data Bank under accession numbers 3WBZ (CaThg1-ATP), 3WC0 (CaThg1-GTP), 3WC1 (CaThg1-tRNAHisΔG−1), and 3WC2 (CaThg1-tRNAPheGUG). The details of SAXS experiments, gel filtration analysis, Thg1 assay, and phylogenetic tree analysis (31) are provided in SI Materials and Methods.
Supplementary Material
Acknowledgments
We thank Dr. Nobutaka Shimizu (Photon Factory), the beamline staff of beamline 41XU, and Kotayu Moriya for their help and Drs. P. O’Donoghue, Y. Liu, and C. Polycarpo for helpful insights. A.N. is a Japan Society for the Promotion of Science Postdoctoral Fellow for Research Abroad. This work was supported by Grant-in-Aid for Scientific Research (B) (24370042 to I.T. and 21370041 to M.Y.) from the Ministry of Education, Culture, Sports, Science and Technology of Japan and by the National Institute for General Medical Sciences (GM22854 to D.S.).
Footnotes
The authors declare no conflict of interest.
Data deposition: The atomic coordinates and structure factors have been deposited in the Protein Data Bank, www.pdb.org [PDB ID codes 3WBZ (CaThg1-ATP), 3WC0 (CaThg1-GTP), 3WC1 (CaThg1-tRNAHisΔG−1), and 3WC2 (CaThg1-tRNAPheGUG)].
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1321312111/-/DCSupplemental.
References
- 1.Ballanco J, Mansfield ML. A model for the evolution of nucleotide polymerase directionality. PLoS ONE. 2011;6(4):e18881. doi: 10.1371/journal.pone.0018881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Greider CW, Blackburn EH. Identification of a specific telomere terminal transferase activity in Tetrahymena extracts. Cell. 1985;43(2 Pt 1):405–413. doi: 10.1016/0092-8674(85)90170-9. [DOI] [PubMed] [Google Scholar]
- 3.Ogawa T, Okazaki T. Discontinuous DNA replication. Annu Rev Biochem. 1980;49:421–457. doi: 10.1146/annurev.bi.49.070180.002225. [DOI] [PubMed] [Google Scholar]
- 4.Giegé R, Sissler M, Florentz C. Universal rules and idiosyncratic features in tRNA identity. Nucleic Acids Res. 1998;26(22):5017–5035. doi: 10.1093/nar/26.22.5017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Himeno H, et al. Role of the extra G-C pair at the end of the acceptor stem of tRNA(His) in aminoacylation. Nucleic Acids Res. 1989;17(19):7855–7863. doi: 10.1093/nar/17.19.7855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Rosen AE, Brooks BS, Guth E, Francklyn CS, Musier-Forsyth K. Evolutionary conservation of a functionally important backbone phosphate group critical for aminoacylation of histidine tRNAs. RNA. 2006;12(7):1315–1322. doi: 10.1261/rna.78606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Burkard U, Willis I, Söll D. Processing of histidine transfer RNA precursors. Abnormal cleavage site for RNase P. J Biol Chem. 1988;263(5):2447–2451. [PubMed] [Google Scholar]
- 8.Orellana O, Cooley L, Söll D. The additional guanylate at the 5′ terminus of Escherichia coli tRNAHis is the result of unusual processing by RNase P. Mol Cell Biol. 1986;6(2):525–529. doi: 10.1128/mcb.6.2.525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cooley L, Appel B, Söll D. Post-transcriptional nucleotide addition is responsible for the formation of the 5′ terminus of histidine tRNA. Proc Natl Acad Sci USA. 1982;79(21):6475–6479. doi: 10.1073/pnas.79.21.6475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Williams JB, Cooley L, Söll D. Enzymatic addition of guanylate to histidine transfer RNA. Methods Enzymol. 1990;181:451–462. doi: 10.1016/0076-6879(90)81143-i. [DOI] [PubMed] [Google Scholar]
- 11.Gu W, Jackman JE, Lohan AJ, Gray MW, Phizicky EM. tRNAHis maturation: An essential yeast protein catalyzes addition of a guanine nucleotide to the 5′ end of tRNAHis. Genes Dev. 2003;17(23):2889–2901. doi: 10.1101/gad.1148603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Heinemann IU, et al. The appearance of pyrrolysine in tRNAHis guanylyltransferase by neutral evolution. Proc Natl Acad Sci USA. 2009;106(50):21103–21108. doi: 10.1073/pnas.0912072106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Heinemann IU, Nakamura A, O’Donoghue P, Eiler D, Söll D. tRNAHis-guanylyltransferase establishes tRNAHis identity. Nucleic Acids Res. 2012;40(1):333–344. doi: 10.1093/nar/gkr696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Jackman JE, Phizicky EM. tRNAHis guanylyltransferase adds G-1 to the 5′ end of tRNAHis by recognition of the anticodon, one of several features unexpectedly shared with tRNA synthetases. RNA. 2006;12(6):1007–1014. doi: 10.1261/rna.54706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Abad MG, Rao BS, Jackman JE. Template-dependent 3′-5′ nucleotide addition is a shared feature of tRNAHis guanylyltransferase enzymes from multiple domains of life. Proc Natl Acad Sci USA. 2010;107(2):674–679. doi: 10.1073/pnas.0910961107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Heinemann IU, Randau L, Tomko RJ, Jr, Söll D. 3′-5′ tRNAHis guanylyltransferase in bacteria. FEBS Lett. 2010;584(16):3567–3572. doi: 10.1016/j.febslet.2010.07.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Abad MG, et al. A role for tRNA(His) guanylyltransferase (Thg1)-like proteins from Dictyostelium discoideum in mitochondrial 5′-tRNA editing. RNA. 2011;17(4):613–623. doi: 10.1261/rna.2517111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Rao BS, Maris EL, Jackman JE. tRNA 5′-end repair activities of tRNAHis guanylyltransferase (Thg1)-like proteins from Bacteria and Archaea. Nucleic Acids Res. 2011;39(5):1833–1842. doi: 10.1093/nar/gkq976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hyde SJ, et al. tRNA(His) guanylyltransferase (THG1), a unique 3′-5′ nucleotidyl transferase, shares unexpected structural homology with canonical 5′-3′ DNA polymerases. Proc Natl Acad Sci USA. 2010;107(47):20305–20310. doi: 10.1073/pnas.1010436107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hyde SJ, Rao BS, Eckenroth BE, Jackman JE, Doublié S. Structural studies of a bacterial tRNA(HIS) guanylyltransferase (Thg1)-like protein, with nucleotide in the activation and nucleotidyl transfer sites. PLoS ONE. 2013;8(7):e67465. doi: 10.1371/journal.pone.0067465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Jackman JE, Phizicky EM. Identification of critical residues for G-1 addition and substrate recognition by tRNA(His) guanylyltransferase. Biochemistry. 2008;47(16):4817–4825. doi: 10.1021/bi702517q. [DOI] [PubMed] [Google Scholar]
- 22.Jackman JE, Phizicky EM. tRNAHis guanylyltransferase catalyzes a 3′-5′ polymerization reaction that is distinct from G-1 addition. Proc Natl Acad Sci USA. 2006;103(23):8640–8645. doi: 10.1073/pnas.0603068103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Smith BA, Jackman JE. Kinetic analysis of 3′-5′ nucleotide addition catalyzed by eukaryotic tRNA(His) guanylyltransferase. Biochemistry. 2012;51(1):453–465. doi: 10.1021/bi201397f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Jeruzalmi D, Steitz TA. Structure of T7 RNA polymerase complexed to the transcriptional inhibitor T7 lysozyme. EMBO J. 1998;17(14):4101–4113. doi: 10.1093/emboj/17.14.4101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Doublié S, Tabor S, Long AM, Richardson CC, Ellenberger T. Crystal structure of a bacteriophage T7 DNA replication complex at 2.2 A resolution. Nature. 1998;391(6664):251–258. doi: 10.1038/34593. [DOI] [PubMed] [Google Scholar]
- 26.Anantharaman V, Iyer LM, Aravind L. Presence of a classical RRM-fold palm domain in Thg1-type 3′- 5′ nucleic acid polymerases and the origin of the GGDEF and CRISPR polymerase domains. Biol Direct. 2010;5:43. doi: 10.1186/1745-6150-5-43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Joyce CM, Steitz TA. Function and structure relationships in DNA polymerases. Annu Rev Biochem. 1994;63:777–822. doi: 10.1146/annurev.bi.63.070194.004021. [DOI] [PubMed] [Google Scholar]
- 28.Shi H, Moore PB. The crystal structure of yeast phenylalanine tRNA at 1.93 A resolution: A classic structure revisited. RNA. 2000;6(8):1091–1105. doi: 10.1017/s1355838200000364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Chan PP, Lowe TM. GtRNAdb: A database of transfer RNA genes detected in genomic sequence. Nucleic Acids Res. 2009;37(Database issue):D93–D97. doi: 10.1093/nar/gkn787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Jackman JE, Gott JM, Gray MW. Doing it in reverse: 3′-to-5′ polymerization by the Thg1 superfamily. RNA. 2012;18(5):886–899. doi: 10.1261/rna.032300.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.O’Donoghue P, Luthey-Schulten Z. On the evolution of structure in aminoacyl-tRNA synthetases. Microbiol Mol Biol Rev. 2003;67(4):550–573. doi: 10.1128/MMBR.67.4.550-573.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.